

Abstract
Aims
This study aimed to demonstrate that the pharmacokinetic (PK) and pharmacodynamic (PD) profile of Sandoz proposed biosimilar pegfilgrastim (LA‐EP2006) matches reference pegfilgrastim (Neulasta®) in healthy subjects. Safety and immunogenicity were also assessed.
Methods
The phase I, randomized, double‐blind, two‐period crossover study consisted of two treatment periods separated by an 8‐week washout period. Healthy subjects aged 18–45 were randomized to either proposed biosimilar/reference pegfilgrastim or reference pegfilgrastim/proposed biosimilar. Proposed biosimilar and reference pegfilgrastim were administered on Day 1 of each treatment period (single 6 mg subcutaneous injection). Blood samples for PK/PD analysis were taken predose and ≤336 h postdose. PK/PD similarity was claimed if 90% (PK) and 95% (PD) confidence intervals (CI) for geometric mean ratios of the area under the serum concentration–time curve (AUC) from time of dosing and extrapolated to infinity (AUC0–inf), or to the last measurable concentration (AUC0–last), maximum observed serum concentration (Cmax), absolute neutrophil count (ANC) area under the effect curve from the time of dosing to the last measurable concentration (AUEC0–last) and ANC maximum effect attributable to the therapy under investigation (Emax) were completely contained within the predefined margin (0.8 to 1.25).
Results
Overall, 169 subjects completed the study. PK/PD similarity was demonstrated; 90% CIs of geometric mean ratio of proposed biosimilar/reference for PK: AUC0–inf (1.0559–1.2244), AUC0–last (1.0607–1.2328), Cmax (1.0312–1.1909) and 95% CIs for PD (ANC): AUEC0–last (0.9948–1.0366), Emax (0.9737–1.0169) were completely contained within predefined margin of 0.8 to 1.25. Both biologics had similar safety profiles, were well tolerated and had low incidence of anti‐drug antibodies. No neutralizing or clinically relevant antibodies were detected.
Conclusions
PK/PD similarity of Sandoz proposed biosimilar pegfilgrastim and reference pegfilgrastim was confirmed. No clinically meaningful differences in safety, tolerability and immunogenicity were observed in healthy subjects.
Keywords: drug development < phase I, oncology < chemotherapy, pharmacodynamics, pharmacokinetics
What is Already Known about this Subject
Sandoz has developed a proposed biosimilar pegfilgrastim (LA‐EP2006).
Physicochemical and functional characterization using state‐of‐the‐art analytical procedures showed Sandoz proposed biosimilar pegfilgrastim to be highly similar to the reference pegfilgrastim (Neulasta®, Amgen).
In two confirmatory, randomized, double‐blind, phase III studies in patients with breast cancer (PROTECT‐1 and PROTECT‐2), Sandoz proposed biosimilar pegfilgrastim showed no clinically meaningful differences in efficacy and safety to reference pegfilgrastim (Neulasta®).
What this Study Adds
This phase I, randomized, double‐blind, two‐period crossover trial was conducted to demonstrate that the pharmacokinetic (PK) and pharmacodynamic (PD) profile of Sandoz proposed biosimilar pegfilgrastim (LA‐EP2006) matches reference pegfilgrastim (Neulasta®) in healthy subjects.
PK and PD similarity of Sandoz proposed biosimilar pegfilgrastim and reference pegfilgrastim was confirmed. No clinically meaningful differences in safety, tolerability and immunogenicity were observed in healthy subjects.
Introduction
Neutropenic complications are common side effects of myelosuppressive chemotherapy in patients with malignancies 1. Febrile neutropenia (FN) often requires hospitalization and can be potentially fatal due to the high risk of infection and sepsis 2, 3, and may cause chemotherapy disturbances such as dose reduction, delay or even discontinuation 1, 4.
The administration of granulocyte colony‐stimulating factors (G‐CSFs) in cancer patients undergoing chemotherapy has been shown to reduce the duration and severity of neutropenia, shorten time to absolute neutrophil count (ANC) nadir and time to ANC recovery from nadir, and reduce the incidence of FN 5, 6, 7. Current guidelines recommend prophylactic use of G‐CSFs, such as http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6968 and its long‐acting pegylated form http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6969, to reduce the risk of neutropenic complications and prevent treatment interruptions in patients receiving myelosuppressive chemotherapy 2, 3, 8. A recent meta‐analysis of randomized clinical trials (RCTs) performed in a sensitive indication compared G‐CSFs (filgrastim and pegfilgrastim) with their biosimilars, and showed no clinically meaningful differences in safety, efficacy or immunogenicity 9.
Sandoz has developed a proposed biosimilar pegfilgrastim (LA‐EP2006). Physicochemical and functional characterization using state‐of‐the‐art analytical procedures showed Sandoz proposed biosimilar pegfilgrastim to be highly similar to the reference pegfilgrastim (Neulasta®, Amgen), which has been marketed in the EU and in the USA since 2002. In two confirmatory, randomized, double‐blind, phase III studies in patients with breast cancer (PROTECT‐1 and PROTECT‐2), Sandoz proposed biosimilar pegfilgrastim showed no clinically meaningful differences in efficacy and safety to reference pegfilgrastim (Neulasta®) 10, 11, 12, 13.
Here, we report findings from a randomized, double‐blind, two‐period crossover pharmacokinetic (PK) and pharmacodynamic (PD) study (LA‐EP06–103). The study was designed to demonstrate PK and PD similarity between Sandoz proposed biosimilar pegfilgrastim and reference pegfilgrastim (Neulasta®) in healthy subjects, as well as to assess the safety and immunogenicity.
Methods
This was a phase I, single‐dose, randomized, double‐blind, two‐way crossover study conducted at two centres in the Netherlands in healthy subjects (EudraCT number 2015–003752‐51). The study was conducted in accordance with ICH Guidelines for Good Clinical Practice, the Declaration of Helsinki and applicable local regulations. The study protocol was approved by an Independent Ethics Committee. All subjects provided written informed consent before any study procedures were carried out.
Subjects
Healthy male and female subjects between the ages of 18 and 45 years (inclusive) were enrolled. The study inclusion criteria included: being physically and mentally healthy, as determined by physical examination and safety laboratory assessments; body weight ≥ 60 kg; body mass index 19.0–28.0 kg m–2 (inclusive); ANC 2–7 × 109 cells l–1 (inclusive); laboratory assessments and haematology parameters normal/within reference ranges; and nonsmoker, ex‐smoker or smoker who smoked no more than 10 cigarettes per day or equivalent. Subjects were excluded for reasons including: known prior exposure to filgrastim, pegfilgrastim, G‐CSF, or any analogue of these; history or presence of any clinically significant disease (e.g. pulmonary, haematological, hepatic, renal, gastrointestinal, cardiac, or cerebral diseases or abnormalities); positive test for anti‐drug antibodies (ADA) at screening; previous or concurrent malignancy; and abnormal vital signs or abnormal 12‐lead electrocardiogram (ECG) results. Except for medication that was required to treat adverse events (AEs; e.g. paracetamol for headache or other pain), hormonal contraceptives and hormone replacement therapy, no medication other than the study drug was allowed from 14 days before dosing until the follow‐up visit on Day 28 of Period 2. Subjects were excluded if they tested positive for ADA at Day 28 of Period 1; or if body weight differed by more than 5% between Periods 1 and 2.
Study design
As shown in Figure 1, the study consisted of two treatment periods separated by an 8‐week washout period. Following a screening period of up to 5 weeks, subjects were randomized in a 1:1 ratio to one of two treatment sequences [Sandoz proposed biosimilar pegfilgrastim followed by reference pegfilgrastim (EU‐authorized) or reference pegfilgrastim (EU‐authorized) followed by Sandoz proposed biosimilar pegfilgrastim]. Pegfilgrastim (Sandoz proposed biosimilar or reference) was administered as a single 6 mg subcutaneous (SC) injection on Day 1 of each period, following a ≥10‐h fast (to avoid the impact of food consumption on the neutrophil count 14), into the SC tissue of the lower abdomen. A follow‐up visit was carried out 28 days after pegfilgrastim administration in each period.
Figure 1.
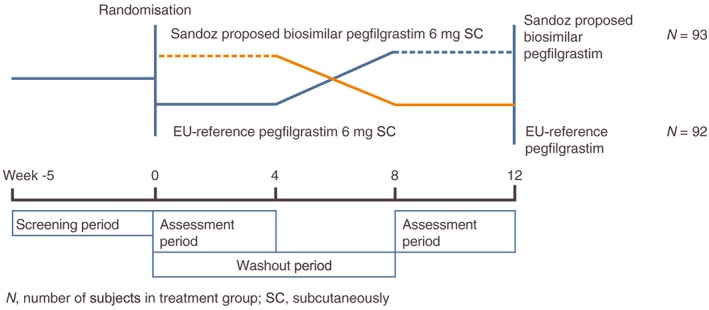
Study design
The crossover design is desirable because each subject serves as their own control, which enhances the precision of estimation of the treatment difference and results. The sample size estimation and the assessment of the study endpoints are based on intrasubject, rather than between‐subject, variability of the primary endpoints 15, 16. Baseline ANC was carefully controlled to account for the neutrophil‐mediated mechanism of pegfilgrastim clearance. The 8‐week washout period gives the blood ANC and bone marrow sufficient time to recover and is considered sufficient to avoid any potential carryover effect between the two treatment periods.
Study endpoints
The primary PK endpoint was pegfilgrastim serum concentration, evaluated by area under the serum concentration–time curve (AUC) measured from time of dosing and extrapolated to infinity (AUC0–inf) or to the last measurable concentration (AUC0–last), and maximum observed serum concentration (Cmax). The primary PD endpoint was ANC, measured by area under the effect curve (AUEC0–last) and maximum effect attributable to the therapy under investigation (Emax). Secondary PK endpoints included time to the maximum observed concentration (tmax) and elimination half‐life (t½). The secondary PD endpoint was ANC response as assessed by time to the maximum effect attributable to the therapy under investigation (tmax,E). Secondary endpoints also included safety and immunogenicity.
Study assessments
PK assessments
In each period, PK blood samples (4 ml each) were collected from subjects predose (–15 min) and then at 4, 8, 12, 24, 36, 48, 60, 72, 84, 96, 108, 120, 144, 168, 192, 216, 264 and 336 h postdose. Blood samples were collected into serum separator tubes and stored at ≤–70°C until analysis. Serum concentrations of pegfilgrastim were measured using a commercial, validated, enzyme‐linked immunosorbent assay (ELISA) kit. For determination of pegfilgrastim concentrations, the kit calibration standard was replaced by a pegfilgrastim calibration standard at concentrations ranging from 1500 to 48 000 pg ml–1. Method validation and serum sample analysis were performed in accordance with international guidelines 17, 18, 19.
PD assessments
Blood samples for PD assessment (3 ml each) were collected at the same time points as the PK blood sampling in each period. Blood samples were collected into potassium ethylenediaminetetraacetic acid tubes and kept at room temperature until analysis. ANC was measured by a validated method using a commercial flow cytometer by the local laboratory at the study site.
Safety assessments
Safety was assessed through: AE collection (incidence, severity and relationship to the study drug; coded using Novartis MedDRA version v19.1 and graded according to CTCAE v4.0); vital signs (blood pressure and pulse rate); laboratory safety tests (haematology, blood chemistry and urine); 12‐lead ECG; and physical examination (including height and body weight). AEs were recorded at every visit from Day 1 of Period 1. Vital signs were collected at screening and Days 1 (predose), 2, 3, 4, 5, 6, 7, 8, 9, 10, 12, 15 and 28 (follow‐up) of each period. Laboratory safety blood tests were completed at screening, Day –1 and Days 3, 7 and 28 (follow‐up) of each period. Physical examination and 12‐lead ECG was performed at screening, Day –1 and Day 28 (follow‐up) of each period.
Local tolerance at the injection site was assessed by subjects using a 0–100 mm visual analogue scale (VAS) and by the investigator using the injection site reaction score. The purpose of this assessment was to ensure that the pegfilgrastim was injected properly, and that there were no differences in injection site reactions between the reference and proposed biosimilar pegfilgrastim. Local tolerance assessments were performed on Days 1 (predose, 1 and 4 h postdose), 2, 3 and 7 of each period.
Immunogenicity assessment
Blood samples for immunogenicity assessment (2.5 ml each) were collected at screening and during each period on Day 1 (≤2 h predose), Day 15 and Day 28 (follow‐up). Blood samples were collected into serum separator tubes and stored at ≤–70°C until shipment for analysis. Immunogenicity, as determined by the formation of antibodies against pegfilgrastim, was evaluated with a validated ELISA assay using differently labelled pegfilgrastim for capture and detection of anti‐pegfilgrastim antibodies. The validation procedure and serum sample analysis followed international guidelines 20, 21; the sensitivity of the assay was calculated to be 4 ng ml–1 (in 100% serum). All study samples were initially analysed in a screening assay; in case of a result equal to or above the screening cut‐off point, a confirmatory/specificity assay was also performed. Binding specificity was confirmed if the assay signal depletion rate was above the confirmatory cut‐off point after spiking excess of drug.
ADA titre and specificity (i.e. ADAs directed against filgrastim or polyethylene glycol), were reported. In addition, serum samples with confirmed positive ADA were analysed for the detection of neutralizing antibodies in a cell‐based assay.
Statistical analysis
A fixed sequence hierarchical testing procedure was prespecified for the equivalence tests on primary PK and PD endpoints. The sample size for the study was determined to ensure at least 80% power for each test step. The true difference assumptions and variability estimations on primary PK and PD endpoints were based on results from a PK/PD study published by Desai et al. 15. With an estimated intrasubject coefficient of variability of 45% and an assumption of 10% true difference. The bioequivalence in AUC0–inf was the deterministic test for sample size and required 144 evaluable subjects for at least 80% power. The total sample size of 184 subjects was planned assuming a drop‐out rate of 22%.
PK and PD treatment comparisons
The fixed sequence hierarchical step‐wise testing procedure was prespecified in the testing sequence of AUC0–inf, AUC0–last, Cmax, ANC AUEC0–last, and ANC Emax. The equivalence hypothesis test for a PK or PD endpoint was only performed if the null hypotheses, higher in the testing hierarchy, were all rejected. PK and PD similarity were claimed if the 90% confidence intervals (CI) for the geometric mean ratios of AUC0–inf, AUC0–last and Cmax, and the 95% CIs for the geometric mean ratios of ANC AUEC0–last and ANC Emax were completely contained within the equivalence margin of 0.8 to 1.25 22.
The analyses of the primary PK endpoints were based on the PK analysis set (all subjects who received study drug and completed PK sampling in both periods without a major protocol violation). PK parameters were calculated with noncompartmental methods using Phoenix™ WinNonLin® Version 6.3 (Pharsight Corporation) based on actual sample times.
Analyses of variance (ANOVA) were performed on the log‐transformed PK parameters (AUC0–inf, AUC0–last and Cmax), including treatment, sequence and period as fixed effects and subject nested within sequence as a random effect. The ratio of the adjusted geometric means was calculated using the exponentiation of the estimate of difference between the least‐squares means obtained from the analyses on the corresponding log‐transformed PK parameters. The 90% CIs for these ratios were derived for PK parameters AUC0–inf, AUC0–last and Cmax. Analysis of covariance (ANCOVA) was a sensitivity analysis, performed with the model adjusted for the additional term of baseline ANC (predose of each period) for PK analyses. An additional sensitivity analysis, based on the ANOVA model used for the primary analyses, was conducted for AUC0–inf excluding AUC0–inf values with adjusted r2 < 0.75.
The analyses of the primary PD endpoints were based on the PD analysis set (all subjects who received study drug and completed PD sampling in both periods without a major protocol violation). PD parameters were estimated from the ANC time profiles for all subjects in the PD analysis set. The calculation was performed on baseline corrected and uncorrected values of ANC using actual sample times. The linear trapezoidal calculation method was used to calculate AUEC.
The primary PD endpoints (ANC AUEC0–last and ANC Emax) were defined as the baseline‐corrected PD parameters. ANCOVA analyses were performed with the primary PD endpoints as the dependent variables. The ANCOVA model included treatment, sequence and period as fixed effects and subject nested within sequence as a random effect, and baseline ANC (predose of each period) as a covariate. ANOVA models removing baseline ANC as a covariate were also performed on primary PD endpoints as sensitivity analyses. Similar sensitivity analyses were performed for the PD parameters AUEC0–last and ANC Emax (not baseline corrected), with and without baseline ANC as a covariate.
All clinical analyses were performed using SAS version 9.4 or higher.
Safety analyses
All safety parameters were analysed descriptively for the safety population, which included all subjects who received study drug and had at least one postbaseline safety assessment.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 23.
Results
Subject demographics
A total of 185 subjects were randomized to receive either Sandoz proposed biosimilar pegfilgrastim followed by reference pegfilgrastim (n = 92; proposed biosimilar/reference) or reference pegfilgrastim followed by Sandoz proposed biosimilar pegfilgrastim (n = 93, reference/proposed biosimilar). Most subjects were white (proposed biosimilar/reference, n = 78; reference/proposed biosimilar, n = 72), with small numbers of Native American, Asian, African American, mixed race and other subjects also included in the study (total for proposed biosimilar/reference, n = 14; total for reference/proposed biosimilar, n = 20). One subject randomized to reference/proposed biosimilar did not receive study treatment and was excluded from the analysis. A total of 184 subjects were therefore included in the safety set. Overall, 169 subjects (proposed biosimilar/reference, n = 86; reference/proposed biosimilar, n = 83) received both study treatments and completed PK and PD blood sampling in Periods I and II up to the follow‐up visit without any major protocol deviations and so were included in the PK and PD analysis sets.
In total, 15 dosed subjects withdrew from the study; six in the proposed biosimilar/reference group and nine in the reference/proposed biosimilar group. Reasons for discontinuation included withdrawal of informed consent (n = 6), AEs (n = 4) and other reasons (n = 5).
Subject demographics, as recorded in Period 1, were similar between the two groups (Table 1). In addition, baseline characteristics (such as mean baseline values for body weight, white blood cells, haemoglobin and platelets) were similar at the beginning of Period 1 and Period 2 and between the two treatments in each period (Table 2). Baseline ANC values however, were slightly lower at the start of Period 2 than at the start of Period 1 for both treatments. There were no clinically significant medical history or previous medication findings in either group and no clinically relevant treatment differences in terms of concomitant medications. The most frequently reported concomitant medications were paracetamol (proposed biosimilar, n = 102; reference, n = 95), ibuprofen (proposed biosimilar, n = 43; reference, n = 42), naproxen (proposed biosimilar, n = 6; reference, n = 7) and cetirizine (proposed biosimilar, n = 2; reference, n = 2). Other concomitant medications permitted by study inclusion/exclusion criteria were reported in no more than one subject in each group.
Table 1.
Demographics (Period 1)
| Proposed biosimilar/reference (N = 92) | Reference/proposed biosimilar (N = 92 a ) | ||
|---|---|---|---|
| Age (years) | Mean | 26.30 | 27.10 |
| SD | 6.77 | 7.90 | |
| Sex n (%) | Female | 34 (37) | 33 (36) |
| Male | 58 (63) | 59 (64) | |
| BMI (kg m–2) | Mean | 24.22 | 23.51 |
| SD | 2.25 | 2.03 | |
93 subjects were randomized in this group but one did not receive study medication
N, number of subjects in treatment group; BMI, body mass index; n, number of subjects with characteristic
Table 2.
Baseline characteristics
| Period 1 | Period 2 | |||
|---|---|---|---|---|
| Sandoz proposed biosimilar pegfilgrastim (N = 92) | Reference pegfilgrastim (N = 92) | Sandoz proposed biosimilar pegfilgrastim (N = 84) | Reference pegfilgrastim (N = 86) | |
| Body weight (kg), mean (SD) | 75.86 (9.77) | 73.98 (8.85) | 74.11 (8.83) | 75.88 (10.06) |
| WBC (109 l–1), mean (SD) | 6.77 (1.33) | 6.99 (1.42) | 6.69 (1.52) | 6.24 (1.46) |
| Haemoglobin (mmol l–1), mean (SD) | 9.10 (0.76) | 9.09 (0.85) | 9.01 (0.90) | 9.03 (0.82) |
| Platelets (109 l–1), mean (SD) | 246.30 (53.96) | 251.50 (54.08) | 245.10 (48.89) | 235.80 (51.14) |
| ANC (109 l–1), mean (SD) | 2.90 (0.93) | 2.97 (0.91) | 2.70 (0.89) | 2.49 (0.83) |
N, number of subjects in treatment group; SD, standard deviation; WBC, white blood cells; ANC, absolute neutrophil count
PK and PD treatment comparisons
The primary PK and PD comparisons between Sandoz proposed biosimilar pegfilgrastim and reference pegfilgrastim demonstrated similarity between the two treatments. The 90% CIs for the geometric mean ratio of the PK parameters (AUC0–inf, AUC0–last and Cmax), and the 95% CIs for the geometric mean ratio of the PD parameters (AUEC0–last and Emax), for proposed biosimilar/reference were completely contained within the prespecified range of 0.8 to 1.25 (Figure 2). The results of all sensitivity analyses were similar to the results of the primary analysis, supporting PK and PD similarity between Sandoz proposed biosimilar pegfilgrastim and reference pegfilgrastim.
Figure 2.
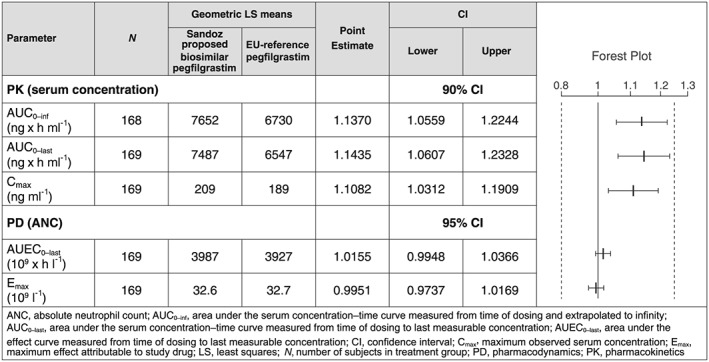
Pharmacokinetic (PK) and pharmacodynamic (PD) parameters for the comparison of Sandoz proposed biosimilar pegfilgrastim/reference pegfilgrastim
The mean pegfilgrastim serum concentration–time profiles were similar for Sandoz proposed biosimilar pegfilgrastim and reference, with slightly higher mean concentrations following Sandoz proposed biosimilar pegfilgrastim administration (Figure 3). Mean pegfilgrastim serum concentrations increased rapidly, with the maximum concentration reached after approximately 12 h. Concentrations subsequently decreased very slowly until 36 h postdose and then more rapidly from 36 h onwards. By Day 6 (120 h postdose), more than 50% of subjects had pegfilgrastim serum concentrations below the lower limit of quantification (LLOQ, i.e. 1500 ng ml–1). From Day 8 (168 h postdose), pegfilgrastim serum concentrations were below the LLOQ in all samples.
Figure 3.
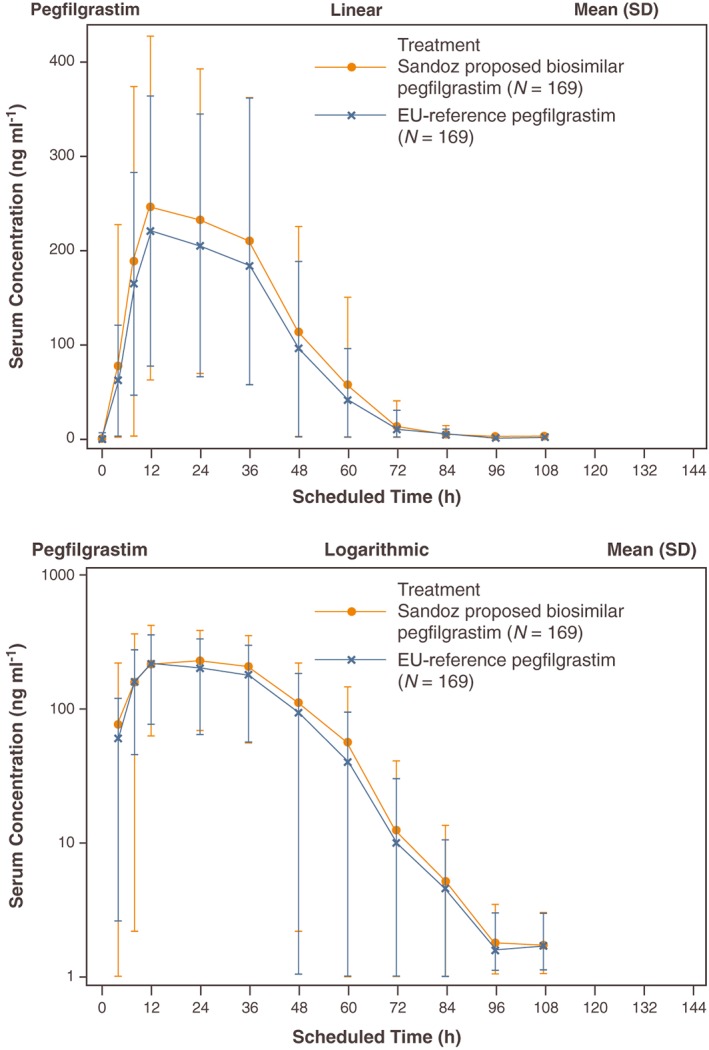
Pegfilgrastim serum concentration–time profiles (linear and logarithmic). SD, standard deviation; N, number of subjects in treatment group
The mean baseline corrected ANC time profiles were similar for the two treatments (Figure 4). Following administration of Sandoz proposed biosimilar pegfilgrastim or reference pegfilgrastim, mean baseline corrected ANC increased steadily to a maximum change from baseline of 32.2 × 109 cells l–1 for both groups after approximately 60 h. After reaching the peak, ANC decreased gradually until counts returned to baseline values around Day 15 (336 h postdose).
Figure 4.
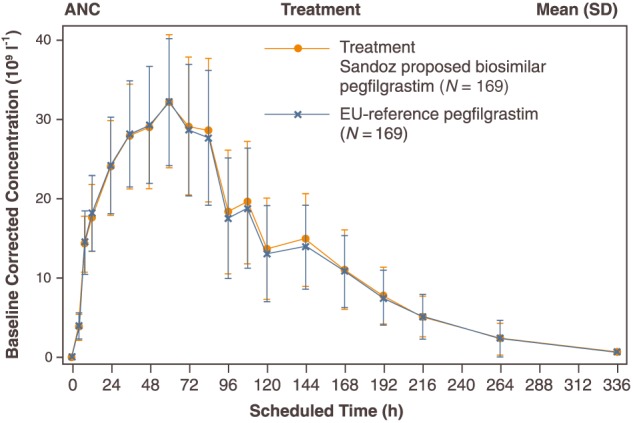
Baseline corrected absolute neutrophil count (ANC)–time profile. SD, standard deviation; N, number of subjects in treatment group
Secondary PK/PD analyses
Secondary PK and PD endpoints (tmax, t½ and tmax,E) were analysed descriptively and were similar between Sandoz proposed biosimilar pegfilgrastim and reference pegfilgrastim (Table 3). Median (range) tmax was 12.0 (4.1–60.0) h for Sandoz proposed biosimilar pegfilgrastim and 12.0 (8.0–48.0) h for reference pegfilgrastim. Mean (standard deviation [SD]) t½ was 17.9 (18.9) h for Sandoz proposed biosimilar pegfilgrastim and 18.2 (20.4) h for reference pegfilgrastim. Median (range) tmax,E was 60.0 (36.0–108.0) h for Sandoz proposed biosimilar pegfilgrastim and 60.0 (24.0–108.3) h for reference pegfilgrastim.
Table 3.
Secondary PK and PD analyses
| Parameter | N | Sandoz proposed biosimilar pegfilgrastim | Reference pegfilgrastim |
|---|---|---|---|
| PK (Serum concentration) | |||
| tmax (h), median (range) | 169 | 12.0 (4.1–60.0) | 12.0 (8.0–48.0) |
| t½ (h), mean (SD) | 168 | 17.9 (18.9) | 18.2 (20.4) |
| PD (ANC) | |||
| tmax, E (h), median (range) | 169 | 60.0 (36.0–108.0) | 60.0 (24.0–108.3) |
N, number of subjects in treatment group; PK, pharmacokinetics; tmax, time to the maximum observed serum concentration; t½, elimination half‐life; SD, standard deviation; PD, pharmacodynamics; ANC, absolute neutrophil count; tmax,E, time to the maximum effect attributable to the investigational medicinal product
Pegfilgrastim increases the production of neutrophils and neutrophil precursors, which in turn clear the compound from the blood circulation and is the predominant pathway for eliminating pegfilgrastim 24. The neutrophil response is linked to AUC, which is an indirect measure of clearance.
Safety
The safety profiles of Sandoz proposed biosimilar pegfilgrastim and reference pegfilgrastim were similar with no clinically meaningful differences in terms of safety and tolerability. The incidence of treatment‐emergent AEs (TEAEs) was the same (97% of subjects) following both Sandoz proposed biosimilar pegfilgrastim or reference pegfilgrastim administration (Table 4).
Table 4.
Safety overview of AEs
| Number of subjects with at least one | Sandoz proposed biosimilar pegfilgrastim N = 176, n (%) | Reference pegfilgrastim N = 178, n (%) |
|---|---|---|
| TEAE | 170 (97) | 172 (97) |
| Study drug‐related AE | 168 (95) | 170 (96) |
| Study drug‐related TEAE leading to study drug discontinuation | 1 (1) | 1 (1) |
| Serious TEAE | 0 | 0 |
| AE occurring ≥4 weeks after the last IMP administration leading to study drug discontinuation | 1 (1) | 1 (1) |
N, number of subjects in treatment group; n, number of subjects with event; TEAE, treatment‐emergent adverse event; AE, adverse event; IMP, investigational medicinal product
The incidence of TEAEs suspected to be related to the study drug was similar for Sandoz proposed biosimilar pegfilgrastim (95% of subjects) and reference pegfilgrastim (96% of subjects). The majority of treatment‐related TEAEs were mild in intensity across both treatment groups and no serious TEAEs were reported (Table 4). One subject in each group discontinued treatment due to TEAEs; arthralgia following Sandoz proposed biosimilar pegfilgrastim and thrombocytopenia following reference pegfilgrastim administration, both of which were mild in severity and suspected to be treatment‐related. A further two subjects discontinued treatment due to AEs occurring ≥4 weeks after the last study drug administration; back pain of moderate severity in the Sandoz proposed biosimilar pegfilgrastim group not suspected to be treatment‐related, and nasopharyngitis of mild severity in the reference pegfilgrastim group suspected to be treatment‐related.
The incidence and nature of TEAEs occurring in each group were similar, with musculoskeletal and connective tissue disorders reported the most frequently (93% of subjects following Sandoz proposed biosimilar pegfilgrastim and 90% of subjects following reference pegfilgrastim; Table 5). Following both treatments, nervous system disorders were reported by 61% of subjects. General disorders and administration site conditions (including injection site reactions) were reported by 45% (Sandoz proposed biosimilar pegfilgrastim) and 42% (reference pegfilgrastim) of subjects. The TEAEs most commonly reported by subjects were headache (57% of subjects following Sandoz proposed biosimilar pegfilgrastim and 56% of subjects following reference pegfilgrastim), bone pain (58% proposed biosimilar, 53% reference pegfilgrastim), myalgia (36% proposed biosimilar, 45% reference pegfilgrastim) and back pain (30% proposed biosimilar, 25% reference pegfilgrastim).
Table 5.
Summary of TEAEs (incidence >10%) for system organ class by treatment
|
Sandoz proposed biosimilar pegfilgrastim N = 176, n (%) |
Reference pegfilgrastim N = 178, n (%) | |
|---|---|---|
| Total number of subjects with at least one TEAE | 170 (97) | 172 (97) |
| Musculoskeletal and connective tissue disorders | 163 (93) | 161 (90) |
| Nervous system disorders | 107 (61) | 109 (61) |
| General disorders and administration site conditions | 79 (45) | 75 (42) |
| Gastrointestinal disorders | 39 (22) | 44 (25) |
| Infections and infestations | 19 (11) | 21 (12) |
| Respiratory, thoracic and mediastinal disorders | 16 (9) | 19 (11) |
N, number of subjects in treatment group; n, number of subjects with event; TEAE, treatment‐emergent adverse event
There were no clinically relevant changes observed in vital signs or ECGs or differences in local tolerability between Sandoz proposed biosimilar pegfilgrastim and reference pegfilgrastim. Most subjects had no injection site reactions; at 1‐h postdose, mild injection site reactions were reported by 22 (13%) subjects following Sandoz proposed biosimilar pegfilgrastim and 32 (18%) subjects following reference pegfilgrastim administration. Most injection site reactions were mild bruising. Mild erythema or erythema/swelling was observed in three subjects, two following reference pegfilgrastim and one following Sandoz proposed biosimilar pegfilgrastim. VAS scores indicated that most subjects had no pain at the injection site. At 1‐h postdose, mean (SD) VAS scores were at a maximum of 0.5 (2.03) following Sandoz proposed biosimilar pegfilgrastim and 1.2 (3.75) following reference pegfilgrastim administration.
Immunogenicity
Postdose, positive confirmatory ADA were observed in four subjects in Period 1: one received treatment with Sandoz proposed biosimilar pegfilgrastim and the other three received treatment with reference pegfilgrastim. Two of these subjects (one in each treatment group) tested positive for ADA at Day 28 of Period 1 and were, for safety reasons and according to the study protocol, not dosed in Period 2. Only one subject in Period 2, who received treatment with reference pegfilgrastim tested positive for ADA. None of the detected antibodies were neutralizing.
Discussion
This single‐dose, two‐way crossover study was designed to demonstrate PK and PD similarity of Sandoz proposed biosimilar pegfilgrastim and reference pegfilgrastim (Neulasta®) following a single SC 6 mg dose in healthy subjects. The safety profile and immunogenicity of Sandoz proposed biosimilar pegfilgrastim and reference pegfilgrastim were also compared. The crossover design is considered appropriate as each subject serves as their own control, enhancing the precision of the estimation of the treatment difference between the biosimilar and its reference medicine. In addition, an intrasubject rather than between‐subject, variability of the primary endpoints was used, which leads to a lower sample size 15, 16. Results from the study indicate that Sandoz proposed biosimilar pegfilgrastim is similar to the reference pegfilgrastim in terms of PK and PD parameters.
The 90% CIs for the ratio of geometric means of proposed biosimilar/reference pegfilgrastim for the primary PK parameters AUC0–inf, AUC0–last and Cmax were completely contained within the predefined range of 0.8 to 1.25, demonstrating PK similarity between the two treatments. The secondary PK parameters tmax and t½ were also similar between Sandoz proposed biosimilar pegfilgrastim and reference pegfilgrastim.
Within the predefined margins of PK similarity, the mean AUCs and the mean Cmax for Sandoz proposed biosimilar pegfilgrastim were approximately 11–14% higher than for reference pegfilgrastim; however, the slightly higher exposure did not translate into any apparent differences in the PD effect or the safety profile.
The 95% CIs for the ratio of geometric means of proposed biosimilar/reference pegfilgrastim for AUEC0–last and Emax were also contained within the predefined similarity margins of 0.8 to 1.25. The secondary PD parameter tmax,E was also similar for Sandoz proposed biosimilar pegfilgrastim and reference pegfilgrastim.
While filgrastim is primarily eliminated by the kidney and neutrophils/neutrophil precursors, pegfilgrastim was designed to have reduced renal clearance. Results from a pegfilgrastim phase I clinical study demonstrated that PK and ANC profiles were similar across various renal function groups, suggesting that the kidney plays a very minor role in the elimination of pegfilgrastim 25. Neutrophil‐mediated clearance is the predominant pathway in eliminating pegfilgrastim. Pegfilgrastim increases the production of neutrophils and neutrophil precursors, which in turn clear the drug from the circulation 24. Overall, the neutrophil response is linked to AUC, which is an indirect measure of clearance.
The safety profile of Sandoz proposed biosimilar pegfilgrastim and reference pegfilgrastim were comparable; single dose administration of both treatments was safe and well tolerated in healthy subjects. The most common TEAEs were headache, bone pain, myalgia and back pain, which occurred at similar rates following Sandoz proposed biosimilar pegfilgrastim and reference pegfilgrastim. Furthermore, most of the reported TEAEs could be attributed to the primary pharmacological effect of filgrastim on the bone marrow. No clinically relevant trends were observed in any of the safety variables and local tolerability at the injection site was good, with few subjects reporting injection site reactions. The results are generally comparable to the known safety profile of the reference medicine 26, 27, 28, 29. Compared with other phase I studies of biosimilar filgrastim, our study reported a higher level of musculoskeletal and connective tissue disorders (93% with Sandoz proposed biosimilar pegfilgrastim and 90% with reference pegfilgrastim). Musculoskeletal and connective tissue disorders were reported by 46.2–65.4% and 38.5–68.0% of healthy subjects receiving 10 μg kg–1 SC biosimilar filgrastim (Nivestim™, Hospira) and reference filgrastim (Neupogen®, Amgen), respectively 30, 31.
The incidence of ADA was similarly low following administration of Sandoz proposed biosimilar pegfilgrastim and reference pegfilgrastim and no neutralizing or clinically relevant antibodies were detected. These findings confirm the low immunological potential of Sandoz proposed biosimilar pegfilgrastim and indicate that the immunological risk is no higher than for the reference pegfilgrastim. These findings are consistent with previous studies that evaluated the immunogenicity of pegfilgrastim and Sandoz proposed biosimilar pegfilgrastim 10, 13, 27, 28, 29, 32.
An important consideration is that this study was performed in healthy volunteers, and the PK/PD profile of pegfilgrastim may be different in patients with cancer. Evidence from populations of patients with breast cancer and non‐small cell lung cancer suggests that the myelosuppressive effect of chemotherapy results in increased pegfilgrastim exposure compared with chemotherapy‐naïve patients or healthy volunteers 27, 33. This continues until the onset of neutrophil recovery, supporting the role of neutrophil and neutrophil precursors in clearing pegfilgrastim 24. Also, patients with acute myeloid leukaemia are often neutropenic as a result of their disease 34 and often experience neutropenia following chemotherapy 35. This results in greater pegfilgrastim exposure compared with patients with other types of cancer undergoing chemotherapy 27, 33, 36.
Biosimilars are becoming increasingly available as biologic patents expire, creating the opportunity for biosimilar development 37. Several oncology biosimilars are currently under development, offering the potential for sustainable cancer care by improving treatment affordability and fostering competition. This can help increase patient access to biological treatments, with the aim of improved clinical outcomes. Although biosimilar development is more complex than the development of small molecule drugs, the totality‐of‐evidence approach, on which their development centres, is now well established 37. As a part of this totality‐of‐evidence approach, the development of a biosimilar medicine requires an extensive nonclinical and clinical development programme. PK/PD similarity, comparable clinical efficacy, safety and immunogenicity to the reference medicine need to be demonstrated. Regarding Sandoz proposed biosimilar pegfilgrastim, results from two landmark phase III RCTs have been published, demonstrating therapeutic equivalence and comparability in terms of safety and immunogenicity between reference pegfilgrastim and the proposed biosimilar medicine 10, 13. This is supported by the results of a recent meta‐analysis that reported no difference between G‐CSFs and their biosimilars in terms of efficacy and safety 9. The PK/PD results reported in this study are therefore a key comparison in the development of the Sandoz proposed biosimilar pegfilgrastim.
In view of the expiry of the EU and US basic patents for Neulasta®, several other proposed biosimilars of pegfilgrastim are currently under development, with the results from phase I and phase III studies already published. The results of the current study are in line with findings from other phase I studies demonstrating PK/PD similarity between proposed pegfilgrastim biosimilar medicines and reference pegfilgrastim (Neulasta®) 15, 38, 39, 40, 41. The PK/PD similarity of a proposed pegfilgrastim biosimilar (B12019, Cinfa Biotech) and the reference medicine was confirmed in a double‐blind, single‐dose, two‐way crossover study in 172 healthy subjects. Furthermore, there were no clinically relevant differences in the safety profiles of B12019 and the reference medicine, and no anti‐drug or neutralizing antibodies were detected following either treatment 38. A follow‐up, multidose, randomized, double‐blind, three‐period, two‐sequence cross‐over study, also performed in healthy subjects, confirmed the comparability of B12019 with the reference at a reduced dose of 3 mg in terms of PK/PD parameters, safety and immunogenicity 39. The immunogenicity profile also did not show any comparable differences to the reference. A single‐dose, randomized, two‐way crossover study in 66 healthy subjects was conducted to assess the PK/PD similarity of Apotex's proposed pegfilgrastim biosimilar and the reference medicine. All PK and PD parameters were similar for the two medicines, and no clinically meaningful safety or immunological differences between the proposed biosimilar and reference pegfilgrastim were reported 15. The PK/PD similarity of CHS‐1701 (Coherus BioSciences, Inc.), a proposed pegfilgrastim biosimilar, to the reference medicine was demonstrated in a single‐blind, three‐sequence crossover study in 122 healthy subjects. The PK and PD bioequivalence criteria were met, and the two medicines were shown to have comparable safety profiles 40. Finally, the PK/PD profile of MYL‐1401H (Mylan GmbH), a proposed pegfilgrastim biosimilar, was evaluated in a double‐blind, three‐way crossover trial pegfilgrastim, which enrolled 216 subjects. Results confirmed equivalence of the PK/PD profile of MYL‐1401H with the reference pegfilgrastim medicines (EU‐Neulasta® and US‐Neulasta®) and no relevant differences in safety were reported 41. MYL‐1401H has also been investigated in a phase III trial 42. This phase III multicentre, randomized, double‐blind study included 194 patients with newly diagnosed breast cancer who were eligible to receive docetaxel/doxorubicin/cyclophosphamide chemotherapy. Patients were randomized to receive MYL‐1401H or EU‐reference pegfilgrastim (Neulasta®) on Day 2 of each cycle. MYL‐1401H demonstrated equivalent efficacy and a similar safety profile to the reference medicine.
As the PD marker, ANC is a clinically relevant surrogate marker of efficacy in clinical practice; the similar PD profiles reported here support the findings of clinical equivalence from the phase III efficacy and safety studies of Sandoz proposed biosimilar pegfilgrastim and reference pegfilgrastim in cancer patients 10, 11, 12, 13.
Conclusions
The PK and PD similarity of Sandoz proposed biosimilar pegfilgrastim and reference pegfilgrastim was confirmed as the 90% CIs (PK) and 95% CIs (PD) of the geometric mean ratio of proposed biosimilar/reference were completely contained within the predefined range of 0.8 to 1.25. There were no clinically meaningful differences in the safety, tolerability and immunogenicity between Sandoz proposed biosimilar pegfilgrastim and reference pegfilgrastim.
Competing Interests
There are no competing interests to declare.
Editorial support was provided by Fiona Goodwin of Spirit Medical Communications Ltd, supported by Sandoz GmbH, Kundl, Austria. Final approval of the content of the manuscript rested solely with the scientific authors.
This study was supported by Sandoz GmbH, Austria.
Nakov, R. , Gattu, S. , Wang, J. , Velinova, M. , Schaffar, G. , and Skerjanec, A. (2018) Proposed biosimilar pegfilgrastim shows similarity in pharmacokinetics and pharmacodynamics to reference pegfilgrastim in healthy subjects. Br J Clin Pharmacol, 84: 2790–2801. 10.1111/bcp.13731.
References
- 1. Weycker D, Li X, Barron R, Li Y, Reiner M, Kartashov A, et al Risk of chemotherapy‐induced febrile neutropenia with early discontinuation of pegfilgrastim prophylaxis in US clinical practice. Support Care Cancer 2016; 24: 2481–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Smith TJ, Bohlke K, Lyman GH, Carson KR, Crawford J, Cross SJ, et al Recommendations for the use of WBC growth factors: American society of clinical oncology clinical practice guideline update. J Clin Oncol 2015; 33: 3199–3212. [DOI] [PubMed] [Google Scholar]
- 3. Aapro M, Boccia R, Leonard R, Camps C, Campone M, Choquet S, et al Refining the role of pegfilgrastim (a long‐acting G‐CSF) for prevention of chemotherapy‐induced febrile neutropenia: consensus guidance recommendations. Support Care Cancer 2017; 25: 3295–3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lyman GH, Yau L, Nakov R, Krendyukov A. A systematic literature review of overall survival and delivered dose intensity in cancer patients receiving chemotherapy and G‐CSF in randomized control trials. Blood 2017; 130: 3424. [Google Scholar]
- 5. Wang L, Baser O, Kutikova L, Page JH, Barron R. The impact of primary prophylaxis with granulocyte colony‐stimulating factors on febrile neutropenia during chemotherapy: a systematic review and meta‐analysis of randomized controlled trials. Support Care Cancer 2015; 23: 3131–3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Crawford J, Ozer H, Stoller R, Johnson D, Lyman G, Tabbara I, et al Reduction by granulocyte colony‐stimulating factor of fever and neutropenia induced by chemotherapy in patients with small‐cell lung cancer. N Engl J Med 1991; 325: 164–170. [DOI] [PubMed] [Google Scholar]
- 7. Pfeil AM, Allcott K, Pettengell R, von Minckwitz G, Schwenkglenks M, Szabo Z. Efficacy, effectiveness and safety of long‐acting granulocyte colony‐stimulating factors for prophylaxis of chemotherapy‐induced neutropenia in patients with cancer: a systematic review. Support Care Cancer 2015; 23: 525–545. [DOI] [PubMed] [Google Scholar]
- 8. NCCN Clinical Practice Guidelines in Oncology. Myeloid Growth Factors Version 2.2018.
- 9. Botteri E, Krendyukov A, Curigliano G. Comparing granulocyte colony‐stimulating factor filgrastim and pegfilgrastim to its biosimilars in terms of efficacy and safety: a meta‐analysis of randomised clinical trials in breast cancer patients. Eur J Cancer 2018; 89: 49–55. [DOI] [PubMed] [Google Scholar]
- 10. Blackwell K, Donskih R, Jones CM, Nixon A, Vidal MJ, Nakov R, et al A comparison of proposed biosimilar LA‐EP2006 and reference pegfilgrastim for the prevention of neutropenia in patients with early‐stage breast cancer receiving myelosuppressive adjuvant or neoadjuvant chemotherapy: pegfilgrastim randomized oncology (supportive care) trial to evaluate comparative treatment (PROTECT‐2), a phase III, randomized, double‐blind trial. Oncologist 2016; 21: 789–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Blackwell K, Gascon P, Jones CM, Nixon A, Krendyukov A, Nakov R, et al Pooled analysis of two randomized, double‐blind trials comparing proposed biosimilar LA‐EP2006 with reference pegfilgrastim in breast cancer. Ann Oncol 2017; 28: 2272–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Harbeck N, Gascon P, Jones CM, Nixon A, Krendyukov A, Nakov R, et al Proposed biosimilar pegfilgrastim (LA‐EP2006) compared with reference pegfilgrastim in Asian patients with breast cancer: an exploratory comparison from two phase III trials. Future Oncol 2017; 13: 1385–1393. [DOI] [PubMed] [Google Scholar]
- 13. Harbeck N, Lipatov O, Frolova M, Udovitsa D, Topuzov E, Ganea‐Motan DE, et al Randomized, double‐blind study comparing proposed biosimilar LA‐EP2006 with reference pegfilgrastim in breast cancer. Future Oncol 2016; 12: 1359–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Koscielniak BK, Charchut A, Wójcik M, Sztefko K, Tomasik PJ. Impact of fasting on complete blood count assayed in capillary blood samples. Lab Med 2017; 48: 357–361. [DOI] [PubMed] [Google Scholar]
- 15. Desai K, Catalano T, Rai G, Misra P, Shah N. Confirmation of biosimilarity in a pharmacokinetic/pharmacodynamic study in healthy volunteers for an analytically highly similar pegfilgrastim. Clin Pharmacol Drug Dev 2016; 5: 354–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ofori‐Asenso R, Agyeman AA. Understanding cross over and parallel group studies in drug research. Precision Medicine 2015; 2: e1046. [Google Scholar]
- 17. Committee for Medicinal Products for Human Use (CHMP), European Medicines Agency (2012) Guideline on bioanalytical method validation. EMEA/CHMP/EWP/192217/2009. London, UK.
- 18. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (2005) ICH Harmonised Tripartite Guideline. Validation of analytical procedures: text and methodology Q2(R1).
- 19. US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Veterinary Medicine (CVM; 2001) Guidance for industry. Bioanalytical method validation. May 2001. Rockville, MD.
- 20. Committee for Medicinal Products for Human Use (CHMP), European Medicines Agency (2015) Guideline on immunogenicity assessment of biotechnology‐derived therapeutic proteins. EMEA/CHMP/BMWP/14327/2006 Rev. 1. London, UK.
- 21. US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER), Center for Devices and Radiological Health (CDRH; 2016) Draft Guidance for Industry. Assay development and validation for immunogenicity testing of therapeutic protein products. April 2016. Rockville, MD
- 22. Gascon P, Fuhr U, Sörgel F, Kinzig‐Schippers M, Makhson A, Balser S, et al Development of a new G‐CSF product based on biosimilarity assessment. Ann Oncol 2010; 21: 1419–1429. [DOI] [PubMed] [Google Scholar]
- 23. Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 2018; 46: D1091–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yang BB, Kido A. Pharmacokinetics and pharmacodynamics of pegfilgrastim. Clin Pharmacokinet 2011; 50: 295–306. [DOI] [PubMed] [Google Scholar]
- 25. Yang BB, Kido A, Salfi M, Swan S, Sullivan JT. Pharmacokinetics and pharmacodynamics of pegfilgrastim in subjects with various degrees of renal function. J Clin Pharmacol 2008; 48: 1025–1031. [DOI] [PubMed] [Google Scholar]
- 26. Bondarenko I, Gladkov OA, Elsaesser R, Buchner A, Bias P. Efficacy and safety of lipegfilgrastim versus pegfilgrastim: a randomized, multicenter, active‐control phase 3 trial in patients with breast cancer receiving doxorubicin/docetaxel chemotherapy. BMC Cancer 2013; 13: 386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Holmes FA, Jones SE, O'Shaughnessy J, Vukelja S, George T, Savin M, et al Comparable efficacy and safety profiles of once‐per‐cycle pegfilgrastim and daily injection filgrastim in chemotherapy‐induced neutropenia: a multicenter dose‐finding study in women with breast cancer. Ann Oncol 2002; 13: 903–909. [DOI] [PubMed] [Google Scholar]
- 28. Holmes FA, O'Shaughnessy JA, Vukelja S, Jones SE, Shogan J, Savin M, et al Blinded, randomized, multicenter study to evaluate single administration pegfilgrastim once per cycle versus daily filgrastim as an adjunct to chemotherapy in patients with high‐risk stage II or stage III/IV breast cancer. J Clin Oncol 2002; 20: 727–731. [DOI] [PubMed] [Google Scholar]
- 29. Green MD, Koelbl H, Baselga J, Galid A, Guillem V, Gascon P, et al A randomized double‐blind multicenter phase III study of fixed‐dose single‐administration pegfilgrastim versus daily filgrastim in patients receiving myelosuppressive chemotherapy. Ann Oncol 2003; 14: 29–35. [DOI] [PubMed] [Google Scholar]
- 30. Waller CF, Bronchud M, Mair S, Challand R. Pharmacokinetic profiles of a biosimilar filgrastim and Amgen filgrastim: results from a randomized, phase I trial. Ann Hematol 2010; 89: 927–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Waller CF, Bronchud M, Mair S, Challand R. Comparison of the pharmacodynamic profiles of a biosimilar filgrastim and Amgen filgrastim: results from a randomized, phase I trial. Ann Hematol 2010; 89: 971–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yang BB, Morrow PK, Wu X, Moxness M, Padhi D. Comparison of pharmacokinetics and safety of pegfilgrastim administered by two delivery methods: on‐body injector and manual injection with a prefilled syringe. Cancer Chemother Pharmacol 2015; 75: 1199–1206. [DOI] [PubMed] [Google Scholar]
- 33. Johnston E, Crawford J, Blackwell S, Bjurstrom T, Lockbaum P, Roskos L, et al Randomized, dose‐escalation study of SD/01 compared with daily filgrastim in patients receiving chemotherapy. J Clin Oncol 2000; 18: 2522–2528. [DOI] [PubMed] [Google Scholar]
- 34. Mayani H. Human preleukemia: cellular, molecular and clinical aspects. Arch Med Res 1993; 24: 317–325. [PubMed] [Google Scholar]
- 35. Iqbal M, Phan M, Machiorlatti M, Vesely SK, Morton JM, Holter J, et al Impact of duration of neutropenia and lymphopenia on AML patients undergoing induction chemotherapy. Blood 2016; 128: 5178. [Google Scholar]
- 36. Sierra J, Szer J, Kassis J, Herrmann R, Lazzarino M, Thomas X, et al A single dose of pegfilgrastim compared with daily filgrastim for supporting neutrophil recovery in patients treated for low‐to‐intermediate risk acute myeloid leukemia: results from a randomized, double‐blind, phase 2 trial. BMC Cancer 2008; 8: 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Krendyukov A, Schiestl M, Höbel N, Aapro M. Clinical equivalence with G‐CSF biosimilars: methodologic approach in a (neo) adjuvant setting in non‐metastatic breast cancer. Support Care Cancer 2018; 26: 33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Roth K, Gastl B, Lehnick D. Demonstration of pharmacokinetic and pharmacodynamic equivalence in healthy volunteers for B12019, a new proposed pegfilgrastim biosimilar. Blood 2016; 128: 5079. [Google Scholar]
- 39. Roth K, Meyer N, Wessels H, Hoefler J, Jankowsky R. Comparability of pharmacodynamics and immunogenicity of B12019, a proposed pegfilgrastim biosimilar to Neulasta. Blood 2017; 130: 1002. [Google Scholar]
- 40. Glaspy JA, O'Connor PG, Tang H, Finck B. Randomized, single‐blind, crossover study to assess the pharmacokinetic and pharmacodynamic bioequivalence of CHS‐1701 to pegfilgrastim in healthy subjects. J Clin Oncol 2017; 35 (Suppl. 15): e21693. [Google Scholar]
- 41. Waller CF, Tiessen RG, Lawrence TE, Shaw A, Liu MS, Sharma R, et al A pharmacokinetics and pharmacodynamics equivalence trial of proposed pegfilgrastim biosimilar, MYL‐1401H vs EU neulasta® and US neulasta® . Ann Oncol 2016; 27 (Suppl. 6): 1451P. [DOI] [PubMed] [Google Scholar]
- 42. Waller CF, Blakeley C, Pennella E, Bronchud M, Berzoy O, Voitko N, et al Phase 3 efficacy and safety trial of proposed pegfilgrastim biosimilar MYL‐1401H vs EU‐neulasta® in the prophylaxis of chemotherapy‐induced neutropenia. Ann Oncol 2016; 27 (Suppl. 6): 1433O. [DOI] [PMC free article] [PubMed] [Google Scholar]