

C. trachomatis infections represent a significant burden to human health. The ability to genetically manipulate Chlamydia spp. is overcoming historic confounding barriers that have impeded rapid progress in understanding overall chlamydial pathogenesis. The current state of genetic manipulation in Chlamydia spp. requires further development, including mechanisms to generate markerless gene disruption. We leveraged a stepwise Cre-lox approach to excise selection marker genes from a deleted gene locus. We found this process to be efficient, and the removal of extraneous elements resulted in the reversal of a negative polar effect on a downstream gene. This technique facilitates a more direct assessment of gene function and adds to the Chlamydia molecular toolbox by facilitating the deletion of genes within operons.
KEYWORDS: TmeA, type III secretion, FRAEM, mutagenesis, Chlamydia
ABSTRACT
As obligate intracellular bacteria, Chlamydia spp. have evolved numerous, likely intricate, mechanisms to create and maintain a privileged intracellular niche. Recent progress in elucidating and characterizing these processes has been bolstered by the development of techniques enabling basic genetic tractability. Florescence-reported allelic exchange mutagenesis (FRAEM) couples chromosomal gene deletion with the insertion of a selection cassette encoding antibiotic resistance and green fluorescent protein (GFP). Similar to other bacteria, many chlamydial genes exist within polycistronic operons, raising the possibility of polar effects mediated by insertion cassettes. Indeed, FRAEM-mediated deletion of Chlamydia trachomatis tmeA negatively impacts the expression of tmeB. We have adapted FRAEM technology by employing a gfp-bla cassette flanked by loxP sites. Conditional expression of Cre recombinase in Chlamydia tmeA containing a floxed cassette resulted in deletion of the marker and restoration of tmeB expression.
IMPORTANCE C. trachomatis infections represent a significant burden to human health. The ability to genetically manipulate Chlamydia spp. is overcoming historic confounding barriers that have impeded rapid progress in understanding overall chlamydial pathogenesis. The current state of genetic manipulation in Chlamydia spp. requires further development, including mechanisms to generate markerless gene disruption. We leveraged a stepwise Cre-lox approach to excise selection marker genes from a deleted gene locus. We found this process to be efficient, and the removal of extraneous elements resulted in the reversal of a negative polar effect on a downstream gene. This technique facilitates a more direct assessment of gene function and adds to the Chlamydia molecular toolbox by facilitating the deletion of genes within operons.
INTRODUCTION
The obligate intracellular bacterium Chlamydia trachomatis represents a significant burden to human health worldwide. Ocular infections with serovars A to C can lead to blinding trachoma, whereas genital infections mediated by serovars D to K or LGV1 to LGV3 result in typical sexually transmitted disease or lymphogranuloma venereum, respectively (1). Chlamydia spp. manifest a biphasic developmental cycle where infectious elementary bodies (EBs) and vegetative reticulate bodies (RBs) represent distinct developmental forms (2). Development occurs entirely within a parasitophorous vesicle termed an inclusion. Beginning as early as attachment and invasion, chlamydiae orchestrate changes in host cell biology to create and maintain a productive growth environment. Major affected pathways include the host cytoskeleton, membrane and vesicle trafficking, host cell survival, and immune signaling (3).
Like other Gram-negative pathogens (4), Chlamydia spp. express a type III secretion system (T3SS) that secretes and translocates effector proteins (T3SE) whose antihost activities culminate to create and maintain a hospitable intracellular niche (3). T3S activity occurs as early as invasion (5, 6) and likely continues until RBs differentiate back into EBs. Effectors that intercalate into the inclusion membrane (Incs) were first identified bioinformatically by the presence of a ca. 60-residue predicted hydrophobic domain (7) and represent the most thoroughly characterized group of chlamydial T3SEs. Surrogate T3SSs and protein localization studies have previously been leveraged to identify an additional pool of chlamydial effectors that are translocated beyond the inclusion membrane and target host proteins within the cytoplasm or organelles (3). The total number of T3S substrates likely exceeds 60 effectors (8), yet the functional role for the majority of these proteins remains to be elucidated.
Due to genetic intractability, it was historically difficult to provide definitive evidence regarding how chlamydial gene products contribute to development and pathogenesis; however, the acquisition of tools to genetically manipulate chlamydiae now provides opportunities to more efficiently reveal aspects of infection biology (reviewed in references 9 to 11). Early advances relied on elegant chemical mutagenesis strategies applied in forward and reverse genetic approaches to associate genes with particular phenotypes. The ability to coinfect chlamydial strains to exchange DNA via lateral gene transfer (LGT) was exploited for gene association studies in heavily mutagenized genomes (12). CaCl2 chemical transformation of C. trachomatis (13) with exogenous plasmid DNA has led to the development of more targeted strategies, including use of the TargeTron system (14) and fluorescence-reported allelic exchange mutagenesis (FRAEM) (15). The TargeTron system enables site-specific gene inactivation by insertion of a group II intron, whereas complete coding sequences are deleted by FRAEM and replaced with a marker cassette encoding green fluorescent protein (GFP) and BlaM. Ectopic expression of epitope-tagged gene products has also been employed to study effector localization and function (16–18). Most recently, the expression of enzymatically dead Cas9 was leveraged for conditional knockdown of targeted messages (19).
Organization of genes into polycistronic operons is a common strategy for coordinated gene expression in bacteria. For example, high-resolution RNA sequencing (RNA-seq) analyses have indicated over 1,500 operons in Escherichia coli K-12 (20). Consistent with a reductionist genome, many genes in C. trachomatis are likely also arranged in operons. Indeed, genes encoding the basal T3S apparatus are arranged within 10 different operons (21). T3SE-specific genes are distributed throughout the genome, and several of the Inc proteins are clearly encoded within operons (22). In addition, the translocated membrane-associated effectors A and B (TmeA and TmeB, respectively) are cotranscribed. The two effectors share the same T3S chaperone (23) and are secreted during invasion (24, 25). FRAEM-mediated mutagenesis revealed an active requirement for TmeA in invasion of cultured cells and in intravaginal infection of mice (26). Importantly, the tmeA strain also failed to express tmeB. Although the invasion phenotype could be complemented by transexpression of tmeA, the loss of tmeB complicates the interpretation of functional study results.
The FRAEM and TargeTron techniques require the insertion of fluorescence reporter and/or antibiotic resistance genes to detect and/or select for successful gene disruption. These insertions have the potential to alter the expression of cotranscribed genes. It would therefore be advantageous to have a mechanism of creating markerless gene deletions to avoid the possibility of polar effects. Bacteriophage Cre recombinase has been used effectively for genome editing of DNA flanked by engineered loxP recognition sequences (27). In particular, the Cre-lox system has been applied successfully in genetically tractable bacteria (28), including the intracellular pathogen Coxiella burnetii (29). Although the obligate intracellular nature and comparatively limited malleability of Chlamydia spp. present challenges, the broadly efficacious Cre-lox approach represents an attractive possibility for genome editing in Chlamydia species.
The goal of the work presented herein was to develop an approach for markerless gene deletion using floxed insertion cassettes and transiently expressed Cre recombinase. We focused on tmeA to test whether removal of the reporter cassette from a tmeA deletion strain would restore the expression of TmeB. A floxed-cassette tmeA mutant was generated via FRAEM, and transient production of Cre was accomplished by expression with the suicide plasmid pSUmC. The expression of Cre resulted in excision of the gfp-bla cassette, and cultivation under nonselective conditions resulted in curing of the suicide plasmid and cre. Importantly, the new tmeA mutant expressed TmeB, and the new tmeA mutant retained the previously observed invasion phenotype. Hence, the application of Cre-lox genome editing in C. trachomatis can be utilized in combination with FRAEM for markerless gene deletion. This approach provides the field with a novel technique for genetic manipulation of C. trachomatis.
RESULTS
Excision strategy and cre activity.
We hypothesized that the lack of TmeB in C. trachomatis L2 tmeA was due directly to the replacement of tmeA with the 2.1-kb gfp-bla selection marker. We chose to employ Cre-lox genome editing to investigate whether removal of the cassette could restore tmeB expression in the tmeA mutant strain. This strategy requires a mutant strain created using a suicide plasmid where a gfp-bla selection cassette, flanked by 34-bp loxP sites, replaces the target gene. The strain is then transformed with a Cre-encoding plasmid. Expression is maintained until excision of the chromosomal cassette is accomplished, yielding a single loxP scar sequence in place of the targeted gene (Fig. 1A). Transient production of Cre in Chlamydia spp. was accomplished by expression of cre on the suicide plasmid pSUmC. In addition to constitutively expressed Cre, pSU-CRE expresses AadA and mCherry for selection and detection, respectively (Fig. 1B). E. coli was used to test the functional integrity of Cre and to confirm the overall efficiency of the system. E. coli was cotransformed with pSU-CRE and ploxP-GFP expressing blaM on the plasmid backbone and a floxed cassette containing gfp. Cotransformation with pSUmC was used as a negative control. We examined 100 plated colonies by direct fluorescence 24 h after transformation. No green bacteria were observed in the presence of pSU-CRE, whereas all colonies were both red and green in the presence of empty pSUmC (Fig. 1C). To confirm excision of the cassette from ploxP-GFP, plasmid DNA was harvested from multiple E. coli isolates, and the cassette locus was PCR amplified using primers annealing to regions flanking the loxP sites. All amplicons from the pSU-CRE-transformed E. coli strains migrated at a size smaller than the pSUmC control, corresponding to loss of the gfp-containing DNA (Fig. 1D). These data indicated that the pSU-CRE-encoded recombinase was functional and that excision was efficient in E. coli.
FIG 1.

Cre recombinase excises a fluorescence cassette in E. coli. (A) Schematic representation of Cre recombinase strategy for use in Chlamydia spp. Cre expression is used to excise the GFP and β-lactamase resistance (Res) reporter genes when flanked by upstream (US) and downstream (DS) loxP sites, leaving behind a loxP scar sequence. The resulting locus is shown and contains one remaining loxP site. (B) Schematic of pSU-CRE plasmid for conditional expression of Cre in C. trachomatis. (C) E. coli colonies expressing ploxP-GFP and lacking (pSUmC) or expressing (pSU-CRE) Cre recombinase. Colonies were imaged with bright-field and fluorescence microscopy. (D) Excision of the reporter cassette was confirmed for three different transformants (1 to 3) by PCR amplification of the locus with primers annealing within the upstream and downstream flanking regions.
We next investigated the expression of Cre by chlamydiae. C. trachomatis L2 was transformed with pSU-CRE, and protein samples were harvested from monolayers infected with either wild-type (WT) or C. trachomatis/pSU-CRE for 24 h. Cre was detected via immunoblotting only in the presence of pSU-CRE (Fig. 2A). Although the inclusions formed by pSU-CRE-expressing chlamydiae appeared to be normal (not shown), we wanted to confirm that the expression of Cre had no obvious negative impact on chlamydial fitness. Chlamydia spp. expressing or lacking Cre were used to infect HeLa cells for 24 h. Cultures were then either methanol fixed and stained for inclusion area analysis or disrupted for enumeration of progeny inclusion-forming units (IFU). As expected, there was no significant difference in inclusion areas or production of progeny IFUs. Therefore, Cre is stably expressed in Chlamydia spp. and does not overtly impact chlamydial development.
FIG 2.

Cre is expressed in Chlamydia spp. and does not impact development. HeLa cells were infected for 24 h with equivalent IFUs of C. trachomatis expressing (+CRE) or lacking (-CRE) pSU-CRE. (A) Whole-culture material was probed in immunoblots with Cre-specific antibodies or anti-Hsp60 as the loading control and visualized via chemiluminescence. (B) Cultures were methanol fixed and stained for inclusion visualization using indirect immunofluorescence. Areas of 50 representative inclusions were measured and plotted individually, with the means and standard deviations shown. (C) Primary cultures were harvested, and progeny Chlamydia spp. were enumerated after secondary passage onto fresh HeLa cells.
Generation of a markerless tmeA mutant.
Our approach to create a markerless tmeA strain began by creating a new tmeA mutant. WT C. trachomatis L2 was transformed with pSUmC-tmeA-lox-gfp-bla, and FRAEM was performed as previously described (15, 30). Isolates were then subjected to cultivation in the presence of rifampin (Rif) to select Rif-resistant chlamydiae, and a clonal strain was derived by limiting dilution. The resulting strain, L2Rif tmeA-lx-gfp-bla, contained a floxed gfp-bla cassette in place of tmeA and served as the progenitor for genomic editing (Fig. 3A). We were unable to transform this strain with pSU-CRE after several attempts. We therefore leveraged lateral gene transfer (LGT) to mobilize pSU-CRE from WT L2 by coinfection with L2Rif tmeA-lx-gfp-bla and selecting for red and green inclusions that were resistant to Rif, penicillin G (PenG), and spectinomycin (Spec). Cultures were maintained in the presence of anhydrotetracycline (aTc) to promote the retention of pSU-CRE. Red-only inclusions were observed after one passage in the absence of PenG, indicating successful excision of the gfp-bla cassette. Because this process yielded a mixed population of chlamydiae (lacking or still containing the gfp-bla cassette), Rif- and Spec-resistant red-only Chlamydia spp. were clonally isolated by limiting dilution. Curing of pSU-CRE was then achieved by multiple passages in the absence of Spec and aTc. Phenotypic and PCR analyses indicated that the resulting strain, L2RRif tmeA-lx, had also been cured of the endogenous plasmid pL2 (not shown). We therefore performed coinfections with WT L2 and L2RRif tmeA-lx to restore pL2 via LGT to yield L2Rif tmeA-lx.
FIG 3.

Construction of a markerless Chlamydia tmeA mutant. (A) Schematic representation of the strategy used to create a markerless tmeA mutant. Each intermediate is depicted with fluorescent qualities (green, GFP+; red, mCherry+; gray, no fluorescence) and antibiotic sensitivities. (B) McCoy cell cultures were infected with equal IFUs of WT, L2 tmeA, or L2Rif tmeA-lx C. trachomatis and serially passaged every 24 h in medium containing PenG. At each passage, DNA was harvested for quantitative real-time PCR to determine genome equivalents based on chlamydial 16S rRNA. (C) qPCR-based comparison of endogenous pL2 (plasmid) copy number in WT or tmeA-lx mutant chlamydiae relative to chlamydial 16S rRNA. DNA was harvested from infected McCoy cells at 24 hpi. PenGr, penicillin G resistant; PenGs, penicillin susceptible; Rifr, rifampin resistant; Specs, spectinomycin susceptible; Specr, spectinomycin resistant.
Sensitivity to PenG was assayed as an indicator for loss of the gfp-bla cassette (Fig. 3B). Genomes of Chlamydia spp. grown in antibiotic-supplemented media were enumerated over three passages. In contrast to L2 tmeA retaining gfp-bla, WT and tmeA-lx mutant Chlamydia sp. levels dropped below detection by the third passage with PenG. Finally, we confirmed the presence of pL2 via quantitative PCR (qPCR) (Fig. 3C). Levels of pL2 in the tmeA-lx mutant were comparable to the WT, indicating restoration of the endogenous plasmid. Overall, our strategy resulted in the generation of a markerless tmeA deletion strain.
We next focused directly on the tmeA locus to verify that genome editing occurred in accordance with our design. Analysis of genomic DNA via qPCR from the WT, tmeA mutant, or tmeA-lx mutant Chlamydia strains confirmed the absence of tmeA and retention of tmeB (Fig. 4A). In addition, a gfp-specific signal was detected for only the tmeA mutant, and no cre-specific signal was detected for any strain. These data confirmed the complete loss of both the gfp-bla cassette and cre in the tmeA-lx mutant. Excision of the gfp-bla cassette should reduce the locus by ca. 2.1 kb. This was tested by PCR using primers flanking the locus (Fig. 4B). The amplicons from WT and tmeA mutant Chlamydia strains migrated at the expected 1.4 and 2.9 kb, respectively. The tmeA-lx amplicon migrated at ca. 0.5 kb, corresponding to the loss gfp-bla. Finally, we directly sequenced the locus (Fig. 4C). As expected, a single loxP site was detected positioned immediately after the tmeA initiation codon and prior to the stop codon. The intervening sequence leading up to tmeB and the GTG start were not altered from those of the WT. In aggregate, our data indicate the successful generation of a markerless tmeA mutant using Cre-lox.
FIG 4.

Direct evidence of gfp-bla excision. (A) McCoy cells infected with equal IFUs of WT, L2 tmeA, or L2Rif tmeA-lx C. trachomatis were harvested at 24 hpi, and DNA was extracted for qPCR. Relative copy numbers for tmeA, tmeB, gfp, and cre were assessed by signal normalized to chlamydial 16S rRNA. ND, none detected. (B) Excision of the reporter cassette confirmed by PCR by amplifying the tmeA locus with primers in the surrounding upstream and downstream regions. Products are shown resolved in a 1.0% agarose gel. (C) The sequenced tmeAB locus from L2Rif tmeA-lx indicating the remaining loxP scar sequence (underlined). Flanking DNA appears in blue, while start codons are in green and the tmeA stop is highlighted in red. The noncanonical start codon for TmeB is also depicted.
Excision of the gfp-bla cassette reverses polar disruption of tmeB expression.
Having established a successful removal of a floxed gfp-bla cassette to yield L2Rif tmeA-lx, we tested whether or not the goal of alleviating polar effects on tmeB was achieved. RNA and protein analyses were conducted on material extracted from 24-h cultures to examine tmeB-specific products (Fig. 5). When signals were normalized for rpoD levels, approximately similar levels of tmeA- and tmeB-specific amplicons were detected in the WT. As expected, no signal was detected for tmeA and tmeB in the respective null backgrounds. Levels of tmeB appeared to be reduced in the polar tmeA mutant containing a gfp-bla cassette. However, tmeB was restored to WT levels in the tmeA-lx mutant. Previous work has indicated that ct696 is transcribed independently of tmeA and tmeB (25), and no changes in ct696 message were observed among the tested strains. Western blot data for TmeA and TmeB were consistent with reverse transcription-quantitative PCR (qRT-PCR) results (Fig. 5B). TmeB was absent in lysates from tmeA Chlamydia spp. but was restored in the tmeA-lx mutant. Hence, the remaining loxP scar sequence did not affect TmeB expression. Interestingly, TmeB appeared to be more abundant in the tmeA-lx mutant complemented in trans with tmeA.
FIG 5.
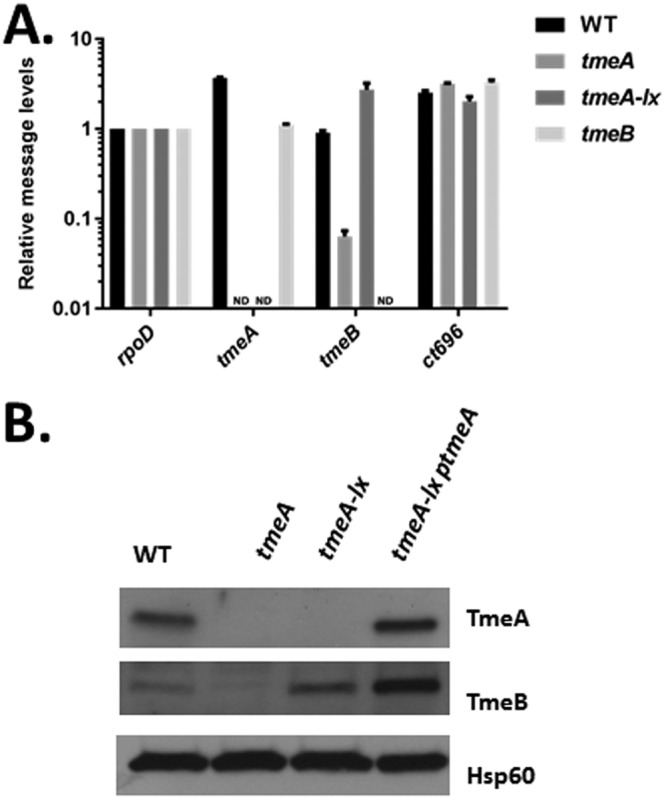
Removal of the reporter cassette relieves polar effects on tmeB. (A) The presence of transcripts downstream of tmeA was determined by reverse transcriptase (RT) quantitative PCR. Total RNA was isolated from at 24 hpi from McCoy cells infected at an MOI of 1 with WT, L2 tmeA, L2 tmeB, or L2Rif tmeA-lx. Transcripts for tmeA, tmeB, and ct696 were detected by qRT-PCR, and signals are presented after normalization to ropD. ND, none detected. (B) Equal quantities of whole-culture material from 24-h cultures infected with equal IFUs of WT, L2 tmeA, L2Rif tmeA-lx, or L2Rif tmeA-lx ptmeA were probed in immunoblots for TmeA and TmeB. Hsp60 was used as a loading control, and proteins were visualized by chemiluminescence.
We have previously shown that the elimination of tmeA manifests as a defect in invasion efficiency that correlates with a reduction in inclusion numbers in primary and secondary cultures (26). Since this strain also lacked TmeB, we could not formally exclude the possibility that the loss of this effector contributed to the observed phenotypes. To ensure valid phenotypic comparisons, we generated a Rif-resistant WT strain expressing pCompAII. Likewise, a tmeA-lx vector-only mutant was used as an isogenic control for the complementation tmeA-lx ptmeA mutant. HeLa cell cultures were infected with chlamydial strains normalized for particles (Fig. 6, left) or IFUs (Fig. 6, right). Inclusions were enumerated in particle-normalized cultures at 24 h postinfection, whereas IFU-normalized cultures were harvested and passaged onto fresh cells for enumeration of progeny EBs. In both cases, infection was significantly reduced for the tmeA-lx mutant compared to the WT. This deficiency could be reversed by complementation, although restoration of progeny yields was not as robust as those seen in primary cultures. Overall, the data are consistent with our findings that TmeA is important for chlamydial development and that this phenotype is independent of TmeB.
FIG 6.

L2Rif tmeA-lx manifests a developmental defect in tissue culture. (A) HeLa cultures were infected in triplicate with equal numbers of Chlamydia spp. (A) or IFUs (B) using L2Rif (WT) + pCompAII or L2Rif tmeA-lx expressing pCompAII (vector) or ptmeA to achieve an approximate MOI of 0.1. Cultures were methanol fixed and stained for chlamydiae (left) or processed for enumeration of progeny EBs (right) at 24 hpi. All inclusions were enumerated by fluorescence staining of chlamydiae in fixed samples, and data for direct and progeny counts are represented as mean ± standard deviation of triplicate samples. A Student t test with Welch’s correction was used to address significance (*, P < 0.02; **, P < 0.005).
DISCUSSION
An obligate intracellular existence presents obvious barriers to direct genetic manipulation of bacteria. As with Coxiella, Anaplasma, and Ehrlichia spp., the addition of a biphasic developmental cycle has further complicated progress in Chlamydia spp. (31). The ability to transform Chlamydia spp. with a stably maintained shuttle vector, however, has ushered in the ability to inactivate targeted chromosomal genes via insertion with group II introns or complete gene deletion using FRAEM (11). Both of these processes require integration of a selectable marker and/or a fluorescence reporter to recover desired strains due to (i) the numerical confines imposed by the requirement for cell culture and (ii) low-frequency events. These engineered elements have the potential to disrupt the possessivity of RNA polymerase and/or translating ribosomes, thereby impacting de novo synthesis of downstream genes. This is an important issue given the propensity of bacteria to organize genes into polycistronic operons (20). While promoter and operon structures have not been well characterized in Chlamydia spp. (32), transcriptome studies clearly indicate the presence of polycistronic messages. Deep sequencing of the C. trachomatis L2b transcriptome was not of sufficient depth to identify all transcription start sites (33), yet at least 246 polycistronic transcripts were detected using a similar approach with the closely related Chlamydia pneumoniae (34). Therefore, overcoming the possibility of polar effects inherent in the application of current gene inactivation technologies in Chlamydia spp. is needed.
C. trachomatis tmeA and tmeB represent invasion-related T3SEs that are cotranscribed as a bicistronic operon (25). TmeB levels were significantly reduced when tmeA was replaced with a gfp-bla cassette via FRAEM (15), raising the possibility of cassette-dependent polar inactivation. In this study, we have devised an approach that sequentially couples FRAEM-mediated gene deletion with Cre-lox-mediated excision of the resulting selection cassettes. This approach was applied successfully to generate a markerless deletion of tmeA that did not negatively impact tmeB expression. Although the Cre-lox system has been used in Coxiella spp. in a two-step gene deletion strategy, mutagenesis was accomplished with the benefit of an axenic medium, and the resulting deletion strains retained a drug resistance cassette (29). Our work represents the first application of Cre-lox technology for markerless gene deletion in an obligate intracellular bacterium during host cell infection.
Cre recombinase mediates the resolution of bacteriophage P1 dimers through recognition and binding to 34-bp direct repeats termed loxP sites (35). The Cre-lox system has been adapted to successfully engineer a wide diversity of genomes, and this includes the excision of selection markers from bacteria (36). For marker removal, Cre must be present/active subsequent to insertion of the floxed selection cassette and then eliminated from the genome after successful excision. One strategy developed for Mycoplasma spp. relied on conditional expression of Cre using a tet-inducible promoter (37). In this instance, Cre was encoded within the selection cassette such that it was lost concomitantly with the excision event. Importantly, the loxP scar sequence that remains after marker excision has not been observed to exert polar effects on downstream genes (29, 38). Our strategy relies on the expression of Cre in Chlamydia spp. using the conditionally replicating plasmid pSUmC (30). Cre is constitutively expressed from pSU-CRE via a blaM promoter, and the plasmid is maintained via selection with Spec and aTC to induce the expression of pgp6. mCherry is also expressed as a fluorescence marker. Given the labor-intensive requirements for genetically manipulating chlamydiae, our first step was to ensure that the pSU-CRE-encoded recombinase was active in E. coli and adequately expressed in WT C. trachomatis without interfering with development.
Generation of a markerless deletion mutant began with a Chlamydia strain where tmeA had been replaced with a gfp-bla cassette flanked by loxP sites. pSU-CRE was then mobilized into this strain. We are not certain why the strain could not be transformed with pSU-CRE using the conventional CaCl2 method, but we suspect general low transformation efficiency in Chlamydia to be the culprit. During the course of our broader work, we have found that lateral gene transfer (LGT) can be efficiently leveraged to mobilize engineered plasmids among chlamydial strains. The exchange of genomic DNA via LGT occurs at a frequency of 10−3 to 10−4 (39), yet we find that plasmid can be transferred 10- to 100-fold more efficiently (not shown). A spontaneous Rif-resistant strain, L2Rif tmeA-lx-gfp-bla, was generated to allow selective recovery after coinfection with L2 pSU-CRE. Experiments in E. coli (Fig. 1) were consistent with Cre-mediated excision of the marker cassette being highly efficient. This was important since there was no selective pressure for excision of the cassette in Chlamydia species. Although there was also no selection to recover markerless chlamydiae, we leveraged the fluorescence reporting aspect of our constructs to monitor excision and recover appropriate strains. Indeed, we could readily visualize a minor population of red-only inclusions after a single passage of L2Rif tmeA-lx-gfp-bla/pSU-CRE. Consistent with previous observations (15), selective maintenance of an engineered plasmid in chlamydiae results in the eventual loss of endogenous pL2 during this process. Therefore, we leveraged LGT a second time to restore endogenous pL2 after curing of pSU-CRE. The final strain, L2Rif tmeA-lx, was used for further study.
Overall, our data were consistent with the marker cassette exerting a polar effect on tmeB in L2 tmeA that was alleviated by excision of gfp-bla in L2Rif tmeA-lx. Both message and protein levels for tmeB were decreased relative to the WT in L2 tmeA, raising the possibility that the presence of gfp-bla interfered with mRNA stability and/or translation. Chlamydial fitness comparisons were made using strains isogenic for Rif, since mutations conferring this resistance can impact fitness in some bacteria (40). Vector-only controls were also used given the contributions of pL2-carried genes in chlamydial infection (41) and observations that ectopic expression of fluorescent proteins can impact fitness in some bacteria (42). Similar to L2 tmeA (15), the absence of tmeA in L2Rif tmeA-lx resulted in a decrease in chlamydial infectivity. These data are therefore consistent with the proposed role of tmeA in chlamydial invasion. Although successful complementation indicated that this phenotype was due solely to TmeA, the restoration of TmeB in L2Rif tmeA-lx will greatly facilitate further study of TmeA function.
In aggregate, our data provide proof of concept that Cre-lox-mediated recombination is an effective technique for manipulation of the chlamydial genome. Due to limitations imposed by culture and genetic manipulation of Chlamydia spp., this method is necessarily somewhat laborious. It is perhaps most appropriately reserved for instances when operon-localized genes are targeted for inactivation. Other applications are clearly possible in addition to alleviation of polar effects. Alternative antibiotic resistance genes have been used to sequentially engineer a chlamydial strain harboring two inactivated genes (43). Given the limited number of effective antibiotics available for positive selection in Chlamydia spp., the Cre-lox system described herein could be exploited as a mechanism to generate multigene mutant strains. Group II introns have been widely used to disrupt chlamydial genes (11), and Cre-lox has been used to excise group II introns in other bacteria (36). However, the lack of a fluorescence reporter in group II introns would likely complicate strain recovery for Chlamydia species. We propose that Cre-lox is therefore most appropriate for use with FRAEM. Based on the wide variety of manipulations that have been accomplished in other genomes, it also possible that more generalized engineering of the C. trachomatis chromosome can be performed using Cre-lox. Inversions, insertions, and gene deletions may become possible as the chlamydial system becomes more tractable.
MATERIALS AND METHODS
Cell culture and organisms.
C. trachomatis serovar L2 (LGV 434) and derivative strains were used in these studies (Table 1). Chlamydiae were routinely maintained in either HeLa 229 epithelial cell monolayers (CCL-1.2; ATCC) or McCoy cell monolayers (CRL-1696; ATCC). All cultures were grown in RPMI 1640 medium containing 2 mM l-glutamine (Life Technologies) supplemented with 10% (vol/vol) heat-inactivated fetal bovine serum (FBS; Sigma) at 37°C in an environment with 5% CO2 and 95% humidified air. All infections were accomplished using density gradient-purified EBs (44) centrifuged onto cell monolayers at 20°C for 1 h at 900 × g. For transformation and FRAEM protocols, chlamydiae were cultivated in the presence of 600 ng/ml penicillin G (PenG; Sigma), 500 µg/ml spectinomycin (Spec; Alfa Aesar), and 1 µg/ml cycloheximide (Sigma), in addition to 50 ng/ml anhydrotetracycline (aTc) where appropriate. Rifampin (Rif)-resistant strains were generated as described previously (45) by cultivation for 4 passages in 2.5 ng/ml Rif, followed by 4 passages in 5 ng/ml Rif. Clonal isolates for all final Chlamydia strains were obtained as described by 2 sequential limiting dilution passages in 384 plates (30). Chlamydial fitness was assessed by enumeration of infectious progeny or quantitative assessment of inclusion area as described by McKuen et al. (26). Primary infections were carried out using particle- or inclusion forming unit (IFU)-normalized chlamydiae, as indicated. Penicillin sensitivity assays were carried out exactly as described by Mueller et al. (15). Escherichia coli NEB-10β (New England Biolabs) was utilized for cloning procedures and verification of Cre activity. An E. coli dam dcm deletion mutant (NEB) was used as a host to generate unmethylated plasmid DNA used to transform Chlamydia species. E. coli strains were routinely grown at 37°C in Luria-Bertani broth (Amresco) or LB agar plates supplemented with 50 μg/ml carbenicillin (Teknova) or 100 μg/ml spectinomycin, as appropriate.
TABLE 1.
C. trachomatis strains
| Strain designation | Plasmid; relevant genotype; antibiotic resistancea | Reference or source |
|---|---|---|
| L2 (WT) | pL2+; tmeA+ | ATCC |
| L2Rif | pL2+; tmeA+; Rifr | This study |
| L2R | pL2−; tmeA+ | 47 |
| L2/pSU-CRE | pL2+; tmeA+ cre+ mCherry+; Specr | This study |
| L2Rif/pCompAII | pL2−; tmeA+ gfp+ mCherry; Rifr Specr | This study |
| L2 tmeA | pL2+; tmeA absent, gfp-bla+; Penr | 15 |
| L2Rif tmeA-lx-gfp-bla | pL2+; tmeA absent, loxP+ gfp-bla+; Rifr Penr | This study |
| L2RRif tmeA-lx | pL2−; tmeA absent, loxP+; Rifr | This study |
| L2Rif tmeA-lx | pL2+; tmeA absent, loxP+; Rifr | This study |
| L2Rif/tmeA-lx pCompAII | pL2−; tmeA absent, loxP+ gfp+ mCherry+; Rifr Specr | This study |
| L2Rif/tmeA-lx pTmeA | pL2−; tmeA+ loxP+ mCherry+; Rifr Specr | This study |
| L2 tmeB | pL2+; tmeA absent, gfp-bla+; Penr | 15 |
pL2+ and pL2− refer to the presence and absence of the endogenous C. trachomatis plasmid, respectively. The coding sequences for Pgp1 to -8 are present on the engineered plasmids pCompAII, pSU-CRE, and pTmeA.
DNA methods.
All PCR-based amplifications for cloning were performed using Q5 high-fidelity DNA polymerase (NEB) from plasmid purified from E. coli using the Monarch plasmid miniprep kit (NEB). All primers were custom DNA oligonucleotides from Integrated DNA Technologies (see Table S1 in the supplemental material), and final constructs were confirmed by DNA sequencing of engineered regions. The allelic exchange plasmid pSU-ΔtmeA-lox-gfp-bla was generated by the addition of loxP sites flanking the bla-gfp cassette. Flanking loxP sites were added via insertion PCR (iPCR) by amplification of the previously described pUC18Δctl0063 (15) using the loxP-blagfp-F and loxP-blagfp-R primers. DNA containing the floxed gfp-bla cassette flanked by chlamydial DNA 3 kb up- and downstream of the deleted tmeA sequence was amplified with HOMRR@pSUmC-F and HOMRR@pSUmC-R primers. The amplicon was then used in iPCR with pSUmC to yield pSUmC-tmeA-loxP. pSU-CRE was constructed first by PCR amplifying the complete CRE coding sequence from pSF-CMV-CRE (Sigma) using primers CRE@pUC18A-F and CRE@pUC18A-R. The amplicon was used in an iPCR reaction to replace bla in pUC18A (30) with the CRE-encoding DNA. Cre and upstream aadA were then PCR amplified using CRE-aadA@pSUmC-F and CRE-aadA@pSUmC-R and mobilized into pSUmC via iPCR. A bla promoter (Pbla) was inserted upstream of cre via iPCR amplification of pSU-aadA-CRE with primers loxP-blagfp-F and loxP-blagfp-R. The resulting amplicon was kinased, blunt-end ligated, and transformed into E. coli to yield pSU-CRE.
Cre activity in E. coli was assessed by cotransformation of E. coli with pUC18 containing a floxed gfp cassette and pSUmC or pSU-CRE. Transformants were screened by fluorescence and PCR with sense and antisense screening primers (Table S1). PCR screening of chlamydial loci with specific primer sets (Table S1) was accomplished by harvesting whole-culture DNA using 0.5 N NaOH-mediated lysis, as described previously (46). For quantitative PCR, iTaq Universal SYBR green supermix (Bio-Rad) and gene-specific primers (Table S1) were employed. For assessment of gene expression, the Aurum total RNA minikit (Bio-Rad) was used to isolate RNA from McCoy cultures infected at a multiplicity of infection (MOI) of 1. Subsequent generation of cDNA was achieved using the QuantiTect reverse transcription kit (Qiagen), and cDNAs were amplified with iTaq Universal SYBR green supermix (Bio-Rad) and gene-specific primers (Table S1).
Genetic manipulation of chlamydiae.
Transformation of Chlamydia spp. via CaCl2 and allelic exchange mutagenesis via FRAEM were accomplished as described previously (30). Briefly, 2 µg of unmethylated pSU-CRE or pSUmC-tmeA-lox-gfp-bla was used to transform WT C. trachomatis L2, and transformants were selected with Spec or Pen, respectively. Generation of the tmeA-lx mutant was accomplished by first cultivating pSUmC-tmeA-lox-gfp-bla transformants in the absence of aTc for multiple passages, followed by clonal isolation of Pen-resistant GFP-expressing chlamydiae. The deletion of tmeA was confirmed by PCR screening with the surroundctl0063-F and surroundctl0063-R primers (Table S1) and direct DNA sequencing. The intermediate strain, L2 tmeA-lox-gfp-bla, was cultivated in Rif as described above, and spontaneous Rif-resistant chlamydiae were isolated. pSU-CRE was mobilized into these chlamydiae by lateral gene transfer. L2Rif tmeA-lox-gfp-bla chlamydiae were used with WT expressing pSU-CRE to coinfect McCoy cells at a 10:1 ratio. Cultures were maintained for two 48-h passages in the presence of aTc and cycloheximide but in the absence of antibiotic selection. Five serial passages (48 h each) were then performed in the presence of Rif, Spec, aTc, and cycloheximide. Rif- and Spec-resistant chlamydiae expressing mCherry but not GFP were isolated by limiting dilution. Strains were cultivated for five serial passages in Rif and cycloheximide, followed by clonal isolation of Rif-resistant chlamydiae lacking mCherry. The absence of gfp and bla was confirmed via PCR and penicillin sensitivity assays, respectively. The absence of tmeA was confirmed as described above via PCR and DNA sequencing. Endogenous pL2 was restored via lateral gene transfer by coinfection with C. trachomatis L2 tmeA (15) and L2RRif tmeA-lx . The final L2Rif tmeA-lx strain was isolated by limiting dilution from cultures exposed to Rif. A spontaneously Rif-resistant WT strain was also isolated and, along with the tmeA-lx mutant, was transformed with pCompAII (15) using Spec selection for the production of vector-only controls. Complementation of the tmeA mutation was accomplished by transformation of tmeA-lx with pTmeA (26).
Microscopy.
E. coli colonies were imaged at ×20 using bright-field or epifluorescence microscopy. Chlamydial inclusions were routinely visualized via GFP or mCherry fluorescence in live cultures or via indirect immunofluorescence of methanol-fixed cultures using Hsp60 (Santa Cruz), followed by secondary antibody conjugated to Alexa Fluor 594 (Invitrogen). For quantitative studies, whole-well fluorescence images were acquired using the Cell Insight CX5 platform (Thermo Scientific) and analyzed using HSC Studio version 6.6 (Thermo Scientific).
Immunoblotting.
Whole-culture material was harvested 24 house postinfection (hpi) monolayers infected at a multiplicity of infection (MOI) of 1. Proteins were precipitated in 10% (vol/vol) trichloroacetic acid in phosphate-buffered saline (PBS) and solubilized using 3× Laemmli buffer. Proteins were then resolved by 12% (vol/vol) SDS-PAGE gels and transferred to Immobilon-P membranes (Millipore). Cre recombinase (Cell Signaling), TmeA (24), TmeB (25), or Hsp60-specific antibodies were used to probe the blots. Proteins were visualized by probing with horseradish peroxidase-conjugated secondary antibodies and chemiluminescence detection using Amersham ECL Plus (GE Healthcare UK Limited).
Statistical analysis.
All presented data are representative of a minimum of three experiments. Unless otherwise noted, quantitative data were generated from experiments containing triplicate biological samples and duplicate technical replicates. Calculation of standard deviation of the mean and assessment via Student’s t test statistical analyses were performed using Prism 6, version 6.04 (GraphPad Software, Inc., La Jolla, CA).
Supplementary Material
ACKNOWLEDGMENTS
We thank M. Clouse and K. Wolf for critical reading of the manuscript and S. Ouellette for thoughtful conversations.
This work was supported by Public Health Service grants from the National Institutes of Health, NIAID (grants AI065530 and AI124649), to K. A. Fields.
Footnotes
For a commentary on this article, see https://doi.org/10.1128/JB.00590-18.
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00479-18.
REFERENCES
- 1.Schachter J. 1999. Infection and disease epidemiology, p 139–169. In Stephens RS. (ed), Chlamydia: intracellular biology, pathogenesis, and immunity. ASM Press, Washington, DC. [Google Scholar]
- 2.AbdelRahman Y, Belland R. 2005. The chlamydial developmental cycle. FEMS Microbiol Rev 29:949–959. doi: 10.1016/j.femsre.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 3.Elwell C, Mirrashidi K, Engel J. 2016. Chlamydia cell biology and pathogenesis. Nat Rev Microbiol 14:385–400. doi: 10.1038/nrmicro.2016.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galán JE, Lara-Tejero M, Marlovits TC, Wagner S. 2014. Bacterial type III secretion systems: specialized nanomachines for protein delivery into target cells. Annu Rev Microbiol 68:415–438. doi: 10.1146/annurev-micro-092412-155725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fields K, Mead D, Dooley C, Hackstadt T. 2003. Chlamydia trachomatis type III secretion: evidence for a functional apparatus during early-cycle development. Mol Microbiol 48:671–683. doi: 10.1046/j.1365-2958.2003.03462.x. [DOI] [PubMed] [Google Scholar]
- 6.Valdivia R. 2008. Chlamydia effector proteins and new insights into chlamydial cellular microbiology. Curr Opin Microbiol 11:53–59. doi: 10.1016/j.mib.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 7.Bannantine J, Griffiths R, Viratyosin W, Brown W, Rockey D. 2000. A secondary structure motif predictive of protein localization to the chlamydial inclusion membrane. Cell Microbiol 2:35–47. doi: 10.1046/j.1462-5822.2000.00029.x. [DOI] [PubMed] [Google Scholar]
- 8.Betts HJ, Wolf K, Fields KA. 2009. Effector protein modulation of host cells: examples in the Chlamydia spp. arsenal. Curr Opin Microbiol 12:81–87. doi: 10.1016/j.mib.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 9.Brothwell JA, Muramatsu MK, Zhong G, Nelson DE. 2018. Advances and obstacles in the genetic dissection of chlamydial virulence. Curr Top Microbiol Immunol 412:133–158. doi: 10.1007/82_2017_76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McClure EE, Chavez ASO, Shaw DK, Carlyon JA, Ganta RR, Noh SM, Wood DO, Bavoil PM, Brayton KA, Martinez JJ, McBride JW, Valdivia RH, Munderloh UG, Pedra JHF. 2017. Engineering of obligate intracellular bacteria: progress, challenges and paradigms. Nat Rev Microbiol 15:544–558. doi: 10.1038/nrmicro.2017.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rahnama M, Fields KA. 2018. Transformation of Chlamydia: current approaches and impact on our understanding of chlamydial infection biology. Microbes Infect S1286-4579(18)30006-6. doi: 10.1016/j.micinf.2018.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kokes M, Dunn JD, Granek JA, Nguyen BD, Barker JR, Valdivia RH, Bastidas RJ. 2015. Integrating chemical mutagenesis and whole-genome sequencing as a platform for forward and reverse genetic analysis of Chlamydia. Cell Host Microbe 17:716–725. doi: 10.1016/j.chom.2015.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Y, Kahane S, Cutcliffe LT, Skilton RJ, Lambden PR, Clarke IN. 2011. Development of a transformation system for Chlamydia trachomatis: restoration of glycogen biosynthesis by acquisition of a plasmid shuttle vector. PLoS Pathog 7:e1002258. doi: 10.1371/journal.ppat.1002258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson CM, Fisher DJ. 2013. Site-specific, insertional inactivation of incA in Chlamydia trachomatis using a group II intron. PLoS One 8:e83989. doi: 10.1371/journal.pone.0083989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mueller KE, Wolf K, Fields KA. 2016. Gene deletion by fluorescence-reported allelic exchange mutagenesis (FRAEM) in Chlamydia trachomatis. mBio 7:e01817-15. doi: 10.1128/mBio.01817-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Agaisse H, Derré I. 2013. A C. trachomatis cloning vector and the generation of C. trachomatis strains expressing fluorescent proteins under the control of a C. trachomatis promoter. PLoS One 8:e57090. doi: 10.1371/journal.pone.0057090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wickstrum J, Sammons LR, Restivo KN, Hefty PS. 2013. Conditional gene expression in Chlamydia trachomatis using the Tet system. PLoS One 8:e76743. doi: 10.1371/journal.pone.0076743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bauler LD, Hackstadt T. 2014. Expression and targeting of secreted proteins from Chlamydia trachomatis. J Bacteriol 196:1325–1334. doi: 10.1128/JB.01290-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ouellette SP. 2018. Feasibility of a conditional knockout system for Chlamydia based on CRISPR interference. Front Cell Infect Microbiol 8:59. doi: 10.3389/fcimb.2018.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Conway T, Creecy JP, Maddox SM, Grissom JE, Conkle TL, Shadid TM, Teramoto J, San Miguel P, Shimada T, Ishihama A, Mori H, Wanner BL. 2014. Unprecedented high-resolution view of bacterial operon architecture revealed by RNA sequencing. mBio 5:e01442-14. doi: 10.1128/mBio.01442-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hefty PS, Stephens RS. 2007. Chlamydial type III secretion system is encoded on ten operons preceded by sigma 70-like promoter elements. J Bacteriol 189:198–206. doi: 10.1128/JB.01034-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scidmore-Carlson M, Shaw E, Dooley C, Fischer E, Hackstadt T. 1999. Identification and characterization of a Chlamydia trachomatis early operon encoding four novel inclusion membrane proteins. Mol Microbiol 33:753–765. doi: 10.1046/j.1365-2958.1999.01523.x. [DOI] [PubMed] [Google Scholar]
- 23.Chen Y-S, Bastidas RJ, Saka HA, Carpenter VK, Richards KL, Plano GV, Valdivia RH. 2014. The Chlamydia trachomatis type III secretion chaperone Slc1 engages multiple early effectors, including TepP, a tyrosine-phosphorylated protein required for the recruitment of CrkI-II to nascent inclusions and innate immune signaling. PLoS Pathog 10:e1003954. doi: 10.1371/journal.ppat.1003954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hower S, Wolf K, Fields KA. 2009. Evidence that CT694 is a novel Chlamydia trachomatis T3S substrate capable of functioning during invasion or early cycle development. Mol Microbiol 72:1423–1437. doi: 10.1111/j.1365-2958.2009.06732.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mueller KE, Fields KA. 2015. Application of β-lactamase reporter fusions as an indicator of protein secretion during infections with the obligate intracellular pathogen Chlamydia trachomatis. PLoS One 10:e0135295. doi: 10.1371/journal.pone.0135295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McKuen MJ, Mueller KE, Bae YS, Fields KA. 2017. Fluorescence-reported allelic exchange mutagenesis reveals a role for Chlamydia trachomatis TmeA in invasion that is independent of host AHNAK. Infect Immun 85:e00640-17. doi: 10.1128/IAI.00640-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yarmolinsky M, Hoess R. 2015. The legacy of Nat Sternberg: the genesis of Cre-lox technology. Annu Rev Virol 2:25–40. doi: 10.1146/annurev-virology-100114-054930. [DOI] [PubMed] [Google Scholar]
- 28.Leibig M, Krismer B, Kolb M, Friede A, Götz F, Bertram R. 2008. Marker removal in staphylococci via Cre recombinase and different lox sites. Appl Environ Microbiol 74:1316–1323. doi: 10.1128/AEM.02424-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beare PA, Larson CL, Gilk SD, Heinzen RA. 2012. Two systems for targeted gene deletion in Coxiella burnetii. Appl Environ Microbiol 78:4580–4589. doi: 10.1128/AEM.00881-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mueller KE, Wolf K, Fields KA. 2017. Chlamydia trachomatis transformation and allelic exchange mutagenesis. Curr Protocol Microbiol 45:11A.13.11–11A.13.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beare P, Sandoz K, Omsland A, Rockey D, Heinzen R. 2011. Advances in genetic manipulation of obligate intracellular bacterial pathogens. Front Cell Infect Microbiol 2:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tan M. 2012. Temporal gene regulation during the chlamydial developmental cycle, p 149–169. In Tan M, Bavoil P (ed), Intracellular pathogens I: Chlamydiales. ASM Press, Washington, DC. [Google Scholar]
- 33.Albrecht M, Sharma CM, Reinhardt R, Vogel J, Rudel T. 2010. Deep sequencing-based discovery of the Chlamydia trachomatis transcriptome. Nucleic Acids Res 38:868–877. doi: 10.1093/nar/gkp1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Albrecht M, Sharma CM, Dittrich MT, Müller T, Reinhardt R, Vogel J, Rudel T. 2011. The transcriptional landscape of Chlamydia pneumoniae. Genome Biol 12:R98. doi: 10.1186/gb-2011-12-10-r98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van Duyne GD. 2015. Cre recombinase. Microbiol Spectr 3:MDNA3-0014-2014. [DOI] [PubMed] [Google Scholar]
- 36.Enyeart PJ, Chirieleison SM, Dao MN, Perutka J, Quandt EM, Yao J, Whitt JT, Keatinge‐Clay AT, Lambowitz AM, Ellington AD. 2013. Generalized bacterial genome editing using mobile group II introns and Cre-lox. Mol Syst Biol 9:685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mariscal AM, González-González L, Querol E, Piñol J. 2016. All-in-one construct for genome engineering using Cre-lox technology. DNA Res 23:263–270. doi: 10.1093/dnares/dsw015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pomerantsev AP, Camp A, Leppla SH. 2009. A new minimal replicon of Bacillus anthracis plasmid pXO1. J Bacteriol 191:5134–5146. doi: 10.1128/JB.00422-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.DeMars R, Weinfurter J. 2008. Interstrain gene transfer in Chlamydia trachomatis in vitro: mechanism and significance. J Bacteriol 190:1605–1614. doi: 10.1128/JB.01592-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Melnyk AH, Wong A, Kassen R. 2015. The fitness costs of antibiotic resistance mutations. Evol Appl 8:273–283. doi: 10.1111/eva.12196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhong G. 2017. Chlamydial plasmid-dependent pathogenicity. Trends Microbiol 25:141–152. doi: 10.1016/j.tim.2016.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Knodler LA, Bestor A, Ma C, Hansen-Wester I, Hensel M, Vallance BA, Steele-Mortimer O. 2005. Cloning vectors and fluorescent proteins can significantly inhibit Salmonella enterica virulence in both epithelial cells and macrophages: implications for bacterial pathogenesis studies. Infect Immun 73:7027–7031. doi: 10.1128/IAI.73.10.7027-7031.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lowden N, Yeruva L, Johnson C, Bowlin A, Fisher D. 2015. Use of aminoglycoside 3' adenyltransferase as a selection marker for Chlamydia trachomatis intron-mutagenesis and in vivo intron stability. BMC Res Notes 8:570. doi: 10.1186/s13104-015-1542-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scidmore MA. 2005. Cultivation and laboratory maintenance of Chlamydia trachomatis. Curr Protocol Microbiol Chapter 11:Unit 11A.1. [DOI] [PubMed] [Google Scholar]
- 45.DeMars R, Weinfurter J, Guex E, Lin J, Potucek Y. 2007. Lateral gene transfer in vitro in the intracellular pathogen Chlamydia trachomatis. J Bacteriol 189:991–1003. doi: 10.1128/JB.00845-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kari L, Goheen MM, Randall LB, Taylor LD, Carlson JH, Whitmire WM, Virok D, Rajaram K, Endresz V, McClarty G, Nelson DE, Caldwell HD. 2011. Generation of targeted Chlamydia trachomatis null mutants. Proc Natl Acad Sci U S A 108:7189–7193. doi: 10.1073/pnas.1102229108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Olivares-Zavaleta N, Whitmire W, Gardner D, Caldwell HD. 2010. Immunization with the attenuated plasmidless Chlamydia trachomatis L2(25667R) strain provides partial protection in a murine model of female genitourinary tract infection. Vaccine 28:1454–1462. doi: 10.1016/j.vaccine.2009.11.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.