

Abstract
Histone deacetylase 6 (HDAC6), a specific histone deacetylase family member, serves an essential role in the regulation of gene expression, cell cycle progression, autophagy and apoptosis. There are numerous reports on the function of HDAC6 in cancer. However, the specific function of HDAC6 in hepatocellular carcinoma (HCC) has yet to be revealed. In the present study, the expression of HDAC6 was revealed to be downregulated in human HCC cell lines and tissues. The aberrant activation of the canonical Wnt/β-catenin signaling pathway was revealed to be involved in hepatocarcinogenesis and metastasis. It was additionally revealed that the overexpression of HDAC6 decreased the expression of β-catenin protein levels which attenuated the canonical Wnt/β-catenin signaling pathway and suppressed the proliferation of HCC cells. In addition, the upregulation of HDAC6 inhibited the epithelial-to-mesenchymal transition in HCC by increasing the E-cadherin protein levels and decreasing the N-cadherin, vimentin and matrix metalloproteinase-9 protein levels. Furthermore, HDAC6 also exerted an effect on the cell cycle arrest and the induction of apoptosis. These results demonstrated that HDAC6 functioned as a tumor suppressor in HCC by attenuating the activity of the canonical Wnt/β-catenin signaling pathway. Therefore, HDAC6 may serve as a potential therapeutic target for the treatment of HCC.
Keywords: histone deacetylase 6, Wnt, β-catenin, hepatocellular carcinoma, epithelial-to-mesenchymal transition
Introduction
Hepatocellular carcinoma (HCC) has become the second leading cause of cancer-associated mortality in men and sixth in women globally; and in 2012, of the total number of new cases of HCC globally, 50% of the cases originated from China (1). Although the correct identification of high-risk groups for liver cancer is improving and screening methods are becoming more accurate, the onset and rapid progression of HCC means that the majority of patients are diagnosed at an advanced stage of the disease, the effect of treatment is unsatisfactory and the median overall survival time has remained at <12 months (2). Therefore, further examination of HCC is required to identify novel therapeutic targets for treatment.
Histone deacetylase (HDAC) is mainly involved in histone acetylation, and serves a vital role in the structural modification of chromosomes and regulation of gene expression (3,4). HDAC6 belongs to class II of the HDAC family, has two homologous catalytic domains and is mainly localized in the cytoplasm (5). Studies have confirmed that HDAC6 serves an essential role in transcriptional regulation, cell cycle progression, autophagy and apoptosis (6–8). Wnt signaling is essential for organogeny and tumorigenesis (9,10). The aberrant activation of Wnt signaling is involved in hepatocarcinogenesis and metastasis (11,12). β-catenin, regarded as the key protein of the canonical Wnt/β-catenin signaling pathway, promotes the activation of Wnt signaling through translocating to the nucleus and attenuating Wnt signaling by degrading itself (13). In addition, the post-transcriptional modifications of β-catenin, including phosphorylation and acetylation, serve key roles in the regulation of β-catenin stability, its endocellular location and transcriptional activity (14,15). It has been revealed that β-catenin may be deacetylated by HDAC6 and associated with the activation of interferon regulatory factor 3 signaling (16). However, the function of HDAC6 in the canonical Wnt/β-catenin signaling pathway and its underlying mechanism of action in HCC cells remain unclear.
To date, there are only a few articles on HDAC6 in HCC (17–21), and the results are conflicting. Therefore, the role of HDAC6 in HCC cells requires further investigation. Consequently, the present study aimed to uncover the function of HDAC6 in HCC cells, and further understanding of its function in HCC.
Materials and methods
Cell culture and plasmid transfection
Human HCC cell lines Huh-7 and Hep3B were obtained from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China) and cultured in Dulbecco's modified Eagle's medium (DMEM; Thermo Fisher Scientific, Inc., Waltham, MA, USA) mixed with 10% fetal bovine serum (Hangzhou Sijiqing Biological Engineering Materials Co., Ltd., Hangzhou, China), 100 µg/ml streptomycin and 100 µg/ml penicillin, in a humidified incubator with 5% CO2. The HDAC6 overexpression plasmid (Shanghai GenePharma Co. Ltd., Shanghai, China) was named P3-HDAC6. HCC cells were transfected with P3-HDAC6 (4 ng) using Lipofectamine 2000 for 20 min at room temperature (Invitrogen; Thermo Fisher Scientific, Inc.) when the cells were cultured to 80–90% confluence.
Patient information and immunohistochemical staining
In the present study, the pathological tissue sections of 7 male and 3 female (n=10) patients (range, 34–65; mean, 47.5 years of age) with HCC were collected from the Department of Hepatobiliary Surgery, the Affiliated Hospital of Xuzhou Medical University (Xuzhou, China) between October 2015 and May 2016. All patients had a confirmed diagnosis of hepatocellular carcinoma by pathology. The pathological diagnosis of these patients was divided into histological grade I, grade II and grade III. The present study was ethically approved by the Ethics Committee of Xuzhou Medical University. All experiments were performed consistent with the principles of the Declaration of Helsinki. Written informed consent was obtained from the patients prior to the study.
Tissues were fixed with 10% neutral formalin for 6 h at room temperature, fixed and washed for 10 min and dipped in xylene for 5 min, gradient ethanol solution (80, 90, 95 and 100%) for 5 min at room temperature. Subsequently they were infiltrated with paraffin at 56°C. Tissue sections were cut into a thickness of ~5 µm. The sections were deparaffinized at 65°C for 2 h, and then soaked with xylene and rehydrated using a descending ethanol (80, 90, 95 and 100%) series. Tissue antigen retrieval was performed using a citrate buffer (Beyotime Institute of Biotechnology, Nantong, China). Sections were covered with 3% H2O2 for 20 min to block endogenous peroxidase activity at room temperature. Antigens were repaired by placing the sections in citrate buffer for 10 min at room temperature. The tissue sections were then incubated with HDAC6 monoclonal antibody (dilution, 1:100; cat. no. ab133539; Abcam, Shanghai, China) overnight at 4°C, treated with secondary antibody goat anti-rabbit IgG horseradish peroxidase (dilution, 1:10,000; cat. no. C10224; Anbo Biotechnology Co., Ltd. San Francisco, CA, USA) for 10 min at room temperature, stained with diaminobenzidine (OriGene Technologies, Inc., Beijing, China) diluent for 30 sec and counterstained with hematoxylin for 2 min at room temperature. Finally, sections were dehydrated using a graded alcohol series (80, 90, 95 and 100%) for ~1–2 min and mounted with a neutral balsam. Images (magnification, ×200) were captured using a Nikon DS-Ri1 microscope.
Western blot analysis
Each cell line was divided into two groups, a mock group (transfected with a blank plasmid) and a P3-HDAC6 group (transfected with P3-HDAC6 plasmid). Each group of cells were washed with cold phosphate buffered saline and lysed in 20 mM Tris (pH 7.5), 150 mM NaCl, 1% Triton X-100, sodium pyrophosphate (PBS), β-glycerophosphate, EDTA, Na3VO4, leupeptin and 1 mM phenylmethyl-sulfonyl fluoride (Beyotime Institute of Biotechnology). Lysates were centrifuged at 4°C at 12,000 × g for 5 min and the total protein was quantified using a bicinchoninic acid kit (Beyotime Institute of Biotechnology), according to the manufacturer's protocol. Total protein (50 ng) were separated by electrophoresis on SDS-PAGE (8–12% gels) and then transferred on to nitrocellulose membranes (GE Healthcare, Chicago, IL, USA). HDAC6 (dilution, 1:10,000; cat. no. ab133539), E-cadherin (dilution, 1:10,000; cat. no. ab76319), N-cadherin (dilution, 1:10,000; cat. no. ab76011), vimentin (dilution, 1:1,000; cat. no. ab92547) and matrix metalloproteinase-9 (MMP-9; dilution, 1:1,000; cat. no. ab 137867) rabbit monoclonal antibodies were purchased from Abcam (dilution, 1:10,000; cat. no. ab6721; Shanghai, China) and β-catenin (dilution, 1:10,000; cat. no. C16402), cyclin-D1 (dilution, 1:10,000; cat. no. C12333), cleaved caspase 3 (dilution, 1:10,000; cat. no. C18514) and GAPDH rabbit antibodies (dilution, 1:100,000; cat. no. AB0037) were purchased from Anbo Biotechnology Co., Ltd. (San Francisco, CA, USA). The membrane was blocked with 5% high calcium nonfat milk for >2 h at room temperature and incubated with the primary antibodies at 4°C overnight. The following day, the membrane was incubated with secondary rabbit antibody (dilution, 1: 10,000; cat. no. C10224; Anbo Biotechnology Co., Ltd. San Francisco, CA, USA) for 2 h at room temperature and rinsed with 0.05% Tris-buffered saline with Tween 20 three times for 10 min. The protein bands were visualized using DAB coloring solution (A:B 1:1) in a dark condition on ice (cat. no. AR1025; BIOSS, Beijing, China) according to the manufacturer's protocols. Membranes were then exposed to an Odyssey infrared imaging system (Odyssey; LI-COR Biosciences, Lincoln, NE, USA).
Cell proliferation assay
Each group of cells were diluted into 5×103/100 µl cell suspensions and cultured in 96-well plates, and then the cell proliferation was measured at 24, 48, 72 and 96 h using a Cell counting kit-8 (CCK-8; Dojindo Molecular Technologies, Inc., Kumamoto, Japan) assay, according to the manufacturer's protocol.
Apoptosis assay
Huh-7 cells were divided into three groups (~1×105 cells per group), the blank group (no treatment), the negative control (NC) group (transfected with the blank plasmid) and the P3-HDAC6 group (transfected with P3-HDAC6 plasmid) and used to detect the apoptosis levels with Annexin V-fluorescein isothiocyanate (FITC) Apoptosis Detection kit I (BD Biosciences, San Jose, CA, USA), according to the manufacturer's protocols. Each group of cells were washed with cold PBS, trypsinized and resuspended. Each cell suspension was added to a detecting tube and 5 µl annexin V-FITC and 5 µl propidium iodide (PI) were subsequently added in sequence. The cell suspension was incubated in the dark for 15 min at room temperature, and then 400 µl 1X binding solution was added to each tube. Finally, levels of apoptosis were detected using a flow cytometer (BD Biosciences) for 1 h and subsequently analyzed with ModFit 3.0 software (Verity Software House Inc., Topsham, ME, USA).
Flow cytometric analysis of cell cycle
The Huh-7 cell line was divided into three groups as previously described. Each group of cells (~1×105 cells per group) were harvested by trypsinization and washed with cold PBS, then fixed in 70% ethanol overnight at −20°C. On the following day, the cells were washed with cold PBS twice, incubated with 100 µl RNase A at 37°C for 30 min. Subsequently, 400 µl propidium iodide (PI) was added at 4°C in the dark for 30 min and detected using a flow cytometer (BD Biosciences). The data was analyzed by ModFit 3.0 software analysis software (Verity Software House, Inc., Topsham, ME, USA).
Cell migration and invasion assay
Migration and invasion assays were performed with 24-Transwell chambers (EMD Millipore, Billerica, MA, USA). In short, Huh-7 and Hep3B (5×103 cells) were harvested and resuspended in 150 µl DMEM without serum and then loaded in the upper chamber. The lower chamber of the Transwell was filled with 600 µl DMEM supplemented with 10% FBS. The chamber contained an 8 µm pore polycarbonate membrane filter covered either without Matrigel (for the migration assay) or with Matrigel (for the invasion assay). Each cell suspension was loaded in the chamber without or with Matrigel (migration or invasion assay, respectively) and incubated at 37°C for 24 h (migration assay) or 48 h (invasion assay). The cells passed through the membrane without or with Matrigel, the upper chamber cells were removed with cotton swabs and the filtered cells were fixed with 70% methanol 20 min and stained with 0.1% crystal violet for 25 min at room temperature. They were subsequently washed with PBS and then counted under a florescent microscope (magnification, ×200, Nikon Corporation, Tokyo, Japan).
Statistical analysis
The cell proliferation assay, relative protein levels, apoptosis and cell cycle results were presented as the mean ± standard deviation from three independent experiments and analyzed using the statistical software SPSS16.0 (SPSS, Inc., Chicago, IL, USA). Differences between groups were assessed using a two-tailed Student's t-test or a one-way analysis of variance and the Least-significant Difference and Student-Neuman-Keuls post hoc test was performed by GraphPad Prism Software version 5.0 (GraphPad Software, Inc., La Jolla, CA, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
HDAC6 is downregulated in HCC cell lines and tissues
Initially, the protein expression of HDAC6 was detected in two HCC cell lines using western blot analysis and the pathological tissue sections of 10 patients with HCC using immunohistochemical staining. The protein expression of HDAC6 was downregulated in Huh-7 and Hep3B cells compared with the normal hepatic cell line Lo2 (Fig. 1A). The results of the immunohistochemical staining additionally indicated that HDAC6 was decreased in cancer tissues compared with tumor-adjacent tissues (Fig. 1B). Therefore, these results revealed that HDAC6 was downregulated in HCC cell lines and tissues.
Figure 1.

Downregulation of HDAC6 was detected in HCC cell lines and tissues. (A) Expression of HDAC6 in the HCC cell lines Hep3B and Huh-7, and the normal liver cell line Lo2 detected using western blot analysis. (B) Protein expression of HDAC6 observed in HCC tissues (magnification, ×200) and carcinoma-adjacent tissues, detected using immunohistochemical staining. HCC, hepatocellular carcinoma; HDAC6, histone deacetylase 6.
Overexpression of HDAC6 suppresses the proliferation of HCC cells via inhibiting the canonical Wnt/β-catenin signaling cascade
In order to identify the function of HDAC6, Huh-7 and Hep3B cell lines were used to investigate the effects on proliferation. It was revealed that the proliferation ability of the two types of HCC cells, Hep3B (P<0.01) and Huh-7 (P<0.05 and P<0.01), was significantly suppressed via the upregulated expression of HDAC6 using a CCK-8 assay (Fig. 2A). However, the exact mechanism of action remains unclear. The activation of the canonical Wnt/β-catenin signaling pathway regulates numerous cellular processes including tumor formation, cell proliferation and metastasis (22). A number of studies have reported that the activity of HDAC6 is associated with the acetylation, degradation and nuclear localization of β-catenin (23,24). In the present study, the protein expression of HDAC6 was upregulated following transfection with P3-HDAC6 plasmid and the β-catenin degradation was increased compared with the mock group as determined by western blot analysis (Fig. 2B). The result indicated that HDAC6 may be considered to function as a tumor suppressor through inhibiting the canonical Wnt/β-catenin signaling pathway. Subsequently, the downstream target genes Myc and Cyclin D1 were analyzed. As expected, these target genes were notably decreased in P3-HDAC6 plasmid-transfected cells (Fig. 2B). Altogether, the results indicated that the overexpression of HADC6 suppressed the proliferation of HCC cells through inhibiting the canonical Wnt/β-catenin signaling cascade.
Figure 2.

Overexpression of HDAC6 suppresses the proliferation of HCC cells and the activity of canonical Wnt/β-catenin signaling. (A) The proliferation ability of HCC cells (Hep3B and Huh-7) was analyzed using a Cell Counting Kit-8 assay. HCC cells were transfected with 4 µg P3-HDAC6 plasmid (P3-HDAC6) or an empty plasmid (Mock). Error bars represent the standard deviations of three independent experiments. *P<0.01 and #P<0.05 vs. mock. (B) The protein levels of HDAC6 and the canonical Wnt/β-catenin signaling cascade proteins (β-catenin, cyclin D1 and Myc) and cleaved caspase 3 were detected using western blot analysis for three HCC cell lines. HCC, hepatocellular carcinoma; HDAC6, histone deacetylase 6.
Upregulation of HDAC6 attenuates the metastasis of HCC cells via the inhibition of epithelial-mesenchymal transition (EMT)
Based on former results that the degradation of β-catenin was increased following HDAC6 overexpression, the present study determined the effect of HDAC6 on the metastasis of HCC cells using invasion and migration assays. The P3-HDAC6 plasmid was transfected into HCC cell lines (Hep3B and Huh-7) for 48 h and the invasion and migration of the cell population was observed and compared with a mock group. Concurrent with the results of the cell proliferation assay, the metastasis ability of the HCC cells was significantly attenuated in the P3-HDAC6 group compared with the mock group (P<0.01; Fig. 3A and B). The process of EMT is essential for the metastasis of a malignant tumor (25,26). Thus, the protein expression of a number of EMT markers was examined, including E-cadherin, N-cadherin, vimentin and MMP-9 (Fig. 3C). E-cadherin expression was substantially increased in the P3-HDAC6 group accompanied with the reduction of the expression of N-cadherin, vimentin and MMP-9 compared with the mock group. These results revealed that HDAC6 functioned as a tumor suppressor via inhibiting the EMT process in HCC cells.
Figure 3.
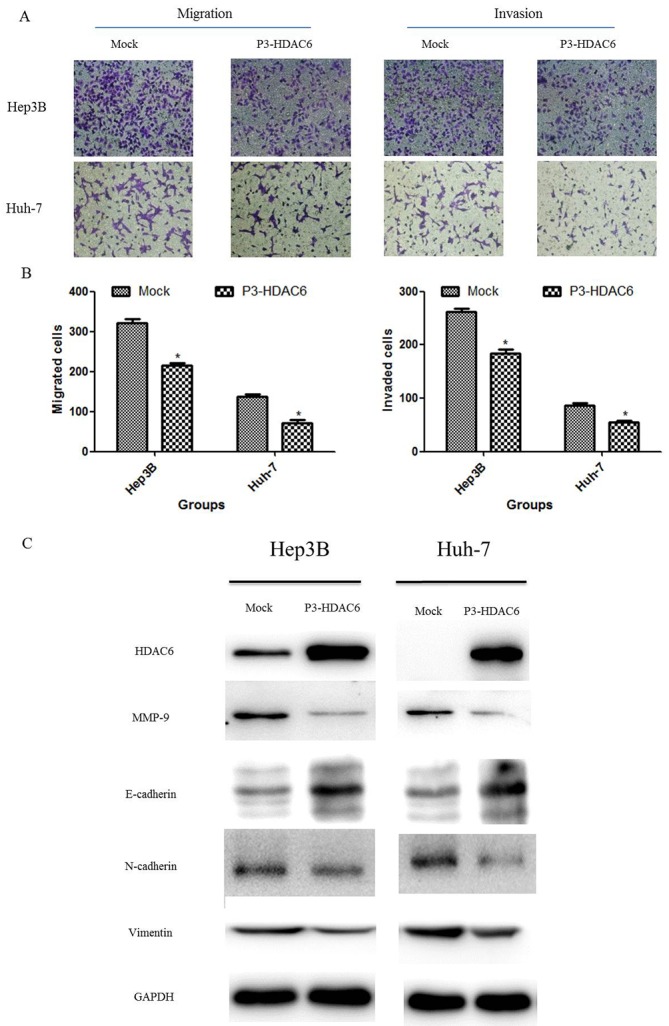
Effect of HDAC6 on the inhibition of EMT. (A) Cell migration and (B) invasion potential was decreased in the P3-HDAC6 group compared with the mock group. Photos were captured under a light microscope (magnification, ×200). Error bars represent the standard deviations of three independent experiments. *P<0.01 vs. the mock group. (C) EMT-associated proteins N-cadherin, vimentin and MMP-9 were decreased and E-cadherin was increased in hepatocellular carcinoma cells that were transfected with the P3-HDAC6 plasmid compared with the mock group. These results were repeated three times. HDAC6, histone deacetylase 6; EMT, epithelial-to-mesenchymal transition; MMP-9, matrix metalloproteinase-9.
Overexpression of HDAC6 is associated with cell cycle arrest and the induction of apoptosis in the Huh7 cell line
As presented in Fig. 2B, cyclin D1 was decreased and cell proliferation was suppressed in the P3-HDAC6 group compared with the mock group. Accordingly, the Huh-7 cell line was used to perform a cell cycle and apoptosis assay using flow cytometry. The HDAC6 overexpression group (P3-HDAC6) produced an increased number of cells at the G1 phase by 10% (Fig. 4A), and a statistically significant compared with other two groups (group NC and Blank, P<0.05). Moreover, once Huh7 cells were transfected with the P3-HDAC6 plasmid and stained using PI, a significant change in cell apoptosis rate was observed compared with the control cells (P<0.01; Fig. 4B). In addition, an increase in cleaved caspase-3 expression in the P3-HDAC6 groups of Huh-7 and Hep3B cells was observed compared with the control cells (Fig. 2B). Thus, the overexpression of was HDAC6 revealed to affect cell cycle arrest and the induction of apoptosis.
Figure 4.
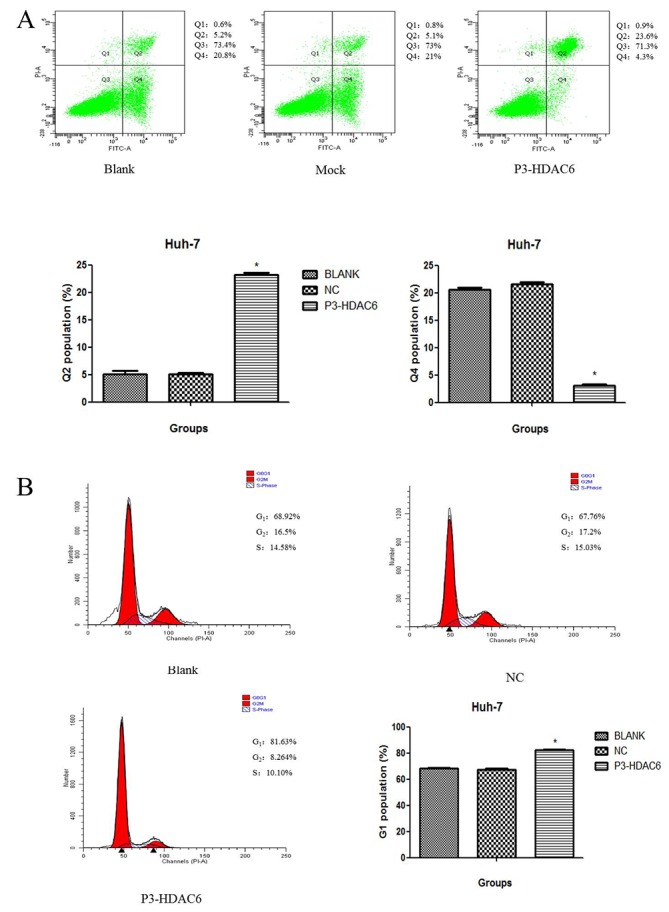
Upregulation of HDAC6 promotes cell cycle arrest and induces cell apoptosis. (A) The proportion of the G1 population was analysed in Huh7 cells using flow cytometry. Huh7 cells were tranfected with a blank plasmid (Mock), P3-HDAC6 plasmid (P3-HDAC6) or a control (Blank). (B) The proportion of apoptotic cells were measured using flow cytometry. Mean ± standard deviation was represented as error bars from three independent experiments. *P<0.01 vs. P3-HDAC6. HDAC6, histone deacetylase 6; NC, negative control.
Discussion
In the present study, it was revealed that HDAC6, a class II histone deacetylase mainly located in the cytoplasm, serves as a tumor suppressor through regulating the activation of the canonical Wnt/β-catenin signaling pathway in HCC. The protein expression of HDAC6 was downregulated in HCC cell lines (Huh-7 and Hep3B) and human liver tumor tissues. It was revealed that the overexpression of HDAC6 suppressed the proliferation and metastasis of cells in vitro by inhibiting the activity of the canonical Wnt/β-catenin signaling pathway and the transcription of downstream target genes. Upregulation of HDAC6 also suppressed the cell migratory and invasive abilities via attenuating the process of EMT in liver cancer. These results reveal a vital role for HDAC6 in hepatocarcinogenesis and progression, and provide a novel treatment method for HCC.
Acetylation modification serves an essential role in chromosome conformation and gene expression. The balance between the activity of the HDAC enzyme and histone acetyltransferase determines the degree of histone acetylation and is associated with physiological and pathological processes (27,28). A number of HDAC family members are abnormally expressed in certain tumor types and have specific functions in controlling the cell characteristics (29). Aberrant HDAC activity is involved in tumorigenesis and progression. Thus, it is difficult to research the function of HDACs. It has been suggested that HDAC6, a cytoplasmic deacetylase, is associated with the acetylation regulation of α-tubulin, Hsp90 and β-catenin and serves a key role in gene expression, and the transcription and translation processes of the aforementioned proteins (5,6,8). In addition, HDAC6 has been suggested to serve an important role in tumorigenesis and tumor progression as well as maintaining the phenotype of malignant cancer types (5,6,8). However, further investigation of the aforementioned roles of HDAC6 is required. Additionally, the expression of HDAC6 has been reported to be upregulated in human breast cancer (30), lung cancer (31) and primary acute myeloid leukemia blasts (32). However, previous studies (17–21) on the function of HDAC6 in human HCC are widely divergent as they contradict each other.
Concurrent with the results of a study by Jung et al (19), HDAC6 expression was detected to be markedly decreased in HCC cell lines and significantly (P<0.05) decreased in patients with HCC compared with their respective controls. This was revealed in a set of histopathological slides from patients with HCC and human HCC cell lines (Fig. 1A and B). These results differ from those produced in a study by Kanno et al (18). Accordingly, from these results it was hypothesized that HDAC6 functioned as a tumor suppressor during the formation of liver cancer and development. Subsequently, it was elucidated that the overexpression of HDAC6 inhibited the proliferation and metastasis of liver cancer cells (Figs. 2 and 3). Ultimately, the results of the present study suggest that HDAC6 functions as a tumor inhibitor in liver cancer.
The abnormal activation of the canonical Wnt/β-catenin signaling pathway is essential for tumorigenesis and progression in a number of different tumor types, including liver cancer (22). β-catenin, the key protein of the canonical Wnt/β-catenin signaling pathway, was deacetylated during the progression of breast cancer by HDAC6, and subsequently the protein levels of acetylated β-catenin were significantly decreased (33). Stimulated by epidermal growth factor (EGF), HDAC6 is associated with the translocation of β-catenin to the caveolae membrane (23). β-catenin may be deacetylated at lysine 49 and inhibits phosphorylation at serine 45 by HDAC6 (23). HDAC6 is also involved in hindering EGF induced β-catenin nucleus localization, decreases Myc expression and suppresses tumor cell proliferation (23). Mak et al (24) reported that HDAC6, cluster of differentiation 133 (CD133) and β-catenin formed a ternary complex, and that the reduction of HDAC6 or CD133 results in raised β-catenin acetylation and degradation. Therefore, the present study assessed whether HDAC6 overexpression affects the protein levels of β-catenin in HCC cell lines. As presented in Fig. 2B, the total β-catenin levels were substantially decreased compared with the healthy controls. Furthermore, the present study detected that the canonical Wnt/β-catenin signaling pathway downstream target genes cyclin D1 and Myc were significantly (P<0.05) reduced in HCC compared with the healthy controls (Fig. 2B) and the proliferation of HCC cells was substantially suppressed compared with the controls (Fig. 2A). These results indicate that HDAC6 overexpression attenuates the activity of the canonical Wnt/β-catenin signaling via the downregulation of β-catenin in liver cancer.
EMT is essential for tumor dissemination and invasion (25). The aberrant activation of the canonical Wnt/β-catenin signaling pathway increases β-catenin and N-cadherin, decreases E-cadherin expression, enhances EMT and promotes tumor metastasis in HCC (34). In the canonical Wnt/β-catenin pathway, the degradation complex is composed of adenomatous polyposis coli, glycogen synthase kinase 3β and axin, which is associated with the phosphorylation and ubiquitin-dependent degradation of β-catenin (33,34). The accumulation of β-catenin increases nucleus localization and combines with lymphoid enhancer factor (LEF)/T-cell factor (TCF) as a transcription factor to regulate proliferation and metastasis (35). However, its effects on EMT have been gradually expounded (36). Slug or Snail, functioning as E-cadherin repressors, are also regulated by the β-catenin-LEF/TCF complex transcriptional activity (37,38). Activating the canonical Wnt/β-catenin signaling pathway is associated with EMT in a number of other tumor types (39,40). Hence, it was hypothesized that HDAC6 may modulate EMT via regulating the activity of the canonical Wnt/β-catenin signaling pathway in HCC. The association between EMT and the canonical Wnt/β-catenin signaling pathway was determined, and it was demonstrated that HDAC6 overexpression induced the degradation of β-catenin and attenuated the activity of the canonical Wnt/β-catenin signaling pathway, inhibited EMT by decreasing N-cadherin, vimentin and MMP-9 and increasing E-cadherin in HCC compared with the healthy controls (Fig. 3C). These results revealed a potential mechanism for the regulation of β-catenin expression in the HDAC6-induced inhibition of EMT in HCC. Although the specific mechanism of HDAC6-mediated degradation of β-catenin is unclear, it is overt that HDAC6 overexpression causes the suppression of the proliferation and metastasis of tumor cells via inhibition of the canonical Wnt/β-catenin signaling pathway in HCC.
In previous studies on HDAC6 in HCC, the results differ substantially between the studies. In a study by Jung et al (19), it was elucidated that HDAC6 activated the c-Jun N-terminal kinases-mediated Beclin-1 depended autophagic cell death process in HCC. It was additionally revealed that the downregulation of HDAC6 indicated the poor prognosis of patients with liver cancer. However, in a study by Kanno et al (18), it was revealed that HDAC6 was overexpressed and increased the activity of cell migration and invasion in HCC. The data of 70 patients with HCC were collected in that previous study. In the present study, it was revealed that the expression of HDAC6 was decreased in Hep3B and Huh-7 cell lines by western blot analysis, and verified in liver cancer tissues by immunohistochemical staining. Therefore, it was hypothesized that HDAC6 functioned as a tumor suppresser in HCC, although the results of the present study may differ from some previous reports. This may be caused by a difference in the cell states. In addition, it was detected that HDAC6 overexpression induced cell cycle arrest in the late G1 phase and apoptosis in Huh-7 cells (Fig. 4), decreased the protein cyclin D1 and increased cleaved casepase-3 expression (Fig. 2B). Altogether, the present study revealed that the expression of HDAC6 is downregulated in HCC, and that the aberrant expression of HDAC6 suppresses tumor cell growth and metastasis in vitro by inhibiting the activity of canonical Wnt/β-catenin signaling pathway. A further in-depth study is requires to investigate how HDAC6 induces β-catenin degradation. The present study provided evidence to identify that the HDAC6 functions as a tumor inhibitor by attenuating the activity of the canonical Wnt/β-catenin signaling pathway. Finally, the results of the present study may provide potential support for the clinical treatment of liver cancer.
Acknowledgements
The authors would like to thank Professor Wei Xu, Junnian Zheng, Hao Xu and Yuming Gu for their guidance and for assisting colleagues in the research group.
Funding
The present study was supported by the Health Department Foundation of Jiangsu province (grant no. H201322).
Availability of data and materials
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.
Authors' contributions
ZY is responsible for experimental design, experimental operation, data collation and thesis writing. WX, HX, JZ and YG are responsible for the collection of clinical pathological tissues, experimental quality control, revision and publication of research topics and papers. All authors have read and approved the final version of the manuscript.
Ethics approval and consent to participate
The present study was ethically approved by the Ethics Committee of Xuzhou Medical University (Xuzhou, China). All experiments were performed consistent with the principles of the Declaration of Helsinki. Written informed consent was obtained from the patients prior to the study.
Patient consent for publication
All patients gave informed consent for publication.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 3.Grozinger CM, Hassig CA, Schreiber SL. Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc Natl Acad Sci USA. 1999;96:4868–4873. doi: 10.1073/pnas.96.9.4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: Causes and therapies. Nat Rev Cancer. 2001;1:194–202. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- 5.Seidel C, Schnekenburger M, Dicato M, Diederich M. Histone deacetylase 6 in health and disease. Epigenomics. 2015;7:103–118. doi: 10.2217/epi.14.69. [DOI] [PubMed] [Google Scholar]
- 6.Valenzuela-Fernández A, Cabrero JR, Serrador JM, Sánchez-Madrid F. HDAC6: A key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol. 2008;18:291–297. doi: 10.1016/j.tcb.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 7.Zheng Q, Wang X. Autophagy and the ubiquitin-proteasome system in cardiac dysfunction. Panminerva Med. 2010;52:9–25. [PMC free article] [PubMed] [Google Scholar]
- 8.Aldana-Masangkay GI, Sakamoto KM. The role of HDAC6 in cancer. J Biomed Biotechnol. 2011;2011:875824. doi: 10.1155/2011/875824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bienz M, Clevers H. Linking colorectal cancer to Wnt signaling. Cell. 2000;103:311–320. doi: 10.1016/S0092-8674(00)00122-7. [DOI] [PubMed] [Google Scholar]
- 10.Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 11.Chen C, Xue Y, Zhang D, Xu W, Xu H, Yao H, Pei D, Gu Y. Short hairpin RNA silencing of TGF-βRII and FZD-7 synergistically suppresses proliferation and metastasis of hepatocellular carcinoma cells. Oncol Lett. 2016;11:2039–2046. doi: 10.3892/ol.2016.4208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tian Y, Mok MT, Yang P, Cheng AS. Epigenetic activation of Wnt/β-catenin signaling in NAFLD-associated hepatocarcinogenesis. Cancers (Basel) 2016;8:E76. doi: 10.3390/cancers8080076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nelson WJ, Nusse R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science. 2004;303:1483–1487. doi: 10.1126/science.1094291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, Zhang Z, Lin X, He X. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell. 2002;108:837–847. doi: 10.1016/S0092-8674(02)00685-2. [DOI] [PubMed] [Google Scholar]
- 15.Ikeda S, Kishida S, Yamamoto H, Murai H, Koyama S, Kikuchi A. Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3beta and beta-catenin and promotes GSK-3beta-dependent phosphorylation of beta-catenin. EMBO J. 1998;17:1371–1384. doi: 10.1093/emboj/17.5.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu J, Coyne CB, Sarkar SN. PKC alpha regulates Sendai virus-mediated interferon induction through HDAC6 and β-catenin. EMBO J. 2011;30:4838–4849. doi: 10.1038/emboj.2011.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ding G, Liu HD, Huang Q, Liang HX, Ding ZH, Liao ZJ, Huang G. HDAC6 promotes hepatocellular carcinoma progression by inhibiting P53 transcriptional activity. FEBS Lett. 2013;587:880–886. doi: 10.1016/j.febslet.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 18.Kanno K, Kanno S, Nitta H, Uesugi N, Sugai T, Masuda T, Wakabayashi G, Maesawa C. Overexpression of histone deacetylase 6 contributes to accelerated migration and invasion activity of hepatocellular carcinoma cells. Oncol Rep. 2012;28:867–873. doi: 10.3892/or.2012.1898. [DOI] [PubMed] [Google Scholar]
- 19.Jung KH, Noh JH, Kim JK, Eun JW, Bae HJ, Chang YG, Kim MG, Park WS, Lee JY, Lee SY, et al. Histone deacetylase 6 functions as a tumor suppressor by activating c-Jun NH2-terminal kinase-mediated beclin 1-dependent autophagic cell death in liver cancer. Hepatology. 2012;56:644–657. doi: 10.1002/hep.25699. [DOI] [PubMed] [Google Scholar]
- 20.Bae HJ, Jung KH, Eun JW, Shen Q, Kim HS, Park SJ, Shin WC, Yang HD, Park WS, Lee JY, Nam SW. MicroRNA-221 governs tumor suppressor HDAC6 to potentiate malignant progression of liver cancer. J Hepatol. 2015;63:408–419. doi: 10.1016/j.jhep.2015.03.019. [DOI] [PubMed] [Google Scholar]
- 21.Lv Z, Weng X, Du C, Zhang C, Xiao H, Cai X, Ye S, Cheng J, Ding C, Xie H, et al. Downregulation of HDAC6 promotes angiogenesis in hepatocellular carcinoma cells and predicts poor prognosis in liver transplantation patients. Mol Carcinog. 2016;55:1024–1033. doi: 10.1002/mc.22345. [DOI] [PubMed] [Google Scholar]
- 22.Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434:843–850. doi: 10.1038/nature03319. [DOI] [PubMed] [Google Scholar]
- 23.Li Y, Zhang X, Polakiewicz RD, Yao TP, Comb MJ. HDAC6 is required for epidermal growth factor-induced beta-catenin nuclear localization. J Biol Chem. 2008;283:12686–12690. doi: 10.1074/jbc.C700185200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mak AB, Nixon AM, Kittanakom S, Stewart JM, Chen GI, Curak J, Gingras AC, Mazitschek R, Neel BG, Stagljar I, Moffat J. Regulation of CD133 by HDAC6 promotes β-catenin signaling to suppress cancer cell differentiation. Cell Rep. 2012;2:951–963. doi: 10.1016/j.celrep.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vasko V, Espinosa AV, Scouten W, He H, Auer H, Liyanarachchi S, Larin A, Savchenko V, Francis GL, de la Chapelle A, et al. Gene expression and functional evidence of epithelial-to-mesenchymal transition in papillary thyroid carcinoma invasion. Proc Natl Acad Sci USA. 2007;104:2803–2088. doi: 10.1073/pnas.0610733104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Todaro M, Iovino F, Eterno V, Cammareri P, Gambara G, Espina V, Gulotta G, Dieli F, Giordano S, De Maria R, Stassi G. Tumorigenic and metastatic activity of human thyroid cancer stem cells. Cancer Res. 2010;70:8874–8885. doi: 10.1158/0008-5472.CAN-10-1994. [DOI] [PubMed] [Google Scholar]
- 27.Salminen A, Kauppinen A, Kaarniranta K. AMPK/Snf1 signaling regulates histone acetylation: Impact on gene expression and epigenetic functions. Cell Signal. 2016;28:887–895. doi: 10.1016/j.cellsig.2016.03.009. [DOI] [PubMed] [Google Scholar]
- 28.Park G, Tan J, Garcia G, Kang Y, Salvesen G, Zhang Z. Regulation of histone acetylation by autophagy in Parkinson disease. J Biol Chem. 2016;291:3531–3540. doi: 10.1074/jbc.M115.675488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Witt O, Deubzer HE, Milde T, Oehme I. HDAC family: What are the cancer relevant targets? Cancer Lett. 2009;277:8–21. doi: 10.1016/j.canlet.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 30.Lee JY, Kuo CW, Tsai SL, Cheng SM, Chen SH, Chan HH, Lin CH, Lin KY, Li CF, Kanwar JR, et al. Inhibition of HDAC3- and HDAC6-promoted survivin expression plays an important role in SAHA-induced autophagy and viability reduction in breast cancer cells. Front Pharmacol. 2016;7:81. doi: 10.3389/fphar.2016.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lim JA, Juhnn YS. Isoproterenol increases histone deacetylase 6 expression and cell migration by inhibiting ERK signaling via PKA and Epac pathways in human lung cancer cells. Exp Mol Med. 2016;48:e204. doi: 10.1038/emm.2015.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hackanson B, Rimmele L, Benkißer M, Abdelkarim M, Fliegauf M, Jung M, Lübbert M. HDAC6 as a target for antileukemic drugs in acute myeloid leukemia. Leuk Res. 2012;36:1055–1062. doi: 10.1016/j.leukres.2012.02.026. [DOI] [PubMed] [Google Scholar]
- 33.Wang SH, Li N, Wei Y, Li QR, Yu ZP. β-catenin deacetylation is essential for WNT-induced proliferation of breast cancer cells. Mol Med Rep. 2014;9:973–978. doi: 10.3892/mmr.2014.1889. [DOI] [PubMed] [Google Scholar]
- 34.Ye Y, Long X, Zhang L, Chen J, Liu P, Li H, Wei F, Yu W, Ren X, Yu J. NTS/NTR1 co-expression enhances epithelial-to-mesenchymal transition and promotes tumor metastasis by activating the Wnt/β-catenin signaling pathway in hepatocellular carcinoma. Oncotarget. 2016;7:70303–70322. doi: 10.18632/oncotarget.11854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang W, Yan HX, Chen L, Liu Q, He YQ, Yu LX, Zhang SH, Huang DD, Tang L, Kong XN, et al. Wnt/beta-catenin signaling contributes to activation of normal and tumorigenic liver progenitor cells. Cancer Res. 2008;68:4287–4295. doi: 10.1158/0008-5472.CAN-07-6426. [DOI] [PubMed] [Google Scholar]
- 36.Yuan Z, Yu X, Ni B, Chen D, Yang Z, Huang J, Wang J, Chen D, Wang L. Overexpression of long non-coding RNA-CTD903 inhibits colorectal cancer invasion and migration by repressing Wnt/β-catenin signaling and predicts favorable prognosis. Int J Oncol. 2016;48:2675–2685. doi: 10.3892/ijo.2016.3447. [DOI] [PubMed] [Google Scholar]
- 37.Baldwin LA, Hoff JT, Lefringhouse J, Zhang M, Jia C, Liu Z, Erfani S, Jin H, Xu M, She QB, et al. CD151-α3β1 integrin complexes suppress ovarian tumor growth by repressing slug-mediated EMT and canonical Wnt signaling. Oncotarget. 2014;5:12203–12217. doi: 10.18632/oncotarget.2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zou W, Zou Y, Zhao Z, Li B, Ran P. Nicotine-induced epithelial-mesenchymal transition via Wnt/β-catenin signaling in human airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2013;304:L199–L209. doi: 10.1152/ajplung.00094.2012. [DOI] [PubMed] [Google Scholar]
- 39.Liu CC, Cai DL, Sun F, Wu ZH, Yue B, Zhao SL, Wu XS, Zhang M, Zhu XW, Peng ZH, Yan DW. FERMT1 mediates epithelial-mesenchymal transition to promote colon cancer metastasis via modulation of β-catenin transcriptional activity. Oncogene. 2017;36:1779–1792. doi: 10.1038/onc.2016.339. [DOI] [PubMed] [Google Scholar]
- 40.Liu ZJ, Liu HL, Zhou HC, Wang GC. TIPE2 inhibits hypoxia-induced Wnt/β-catenin pathway activation and EMT in glioma cells. Oncol Res. 2016;24:255–261. doi: 10.3727/096504016X14666990347356. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.