

Abstract
Mass spectrometry imaging (MSI) has proven to be a valuable tool for drug and metabolite imaging in pharmaceutical toxicology studies and can reveal, for example, accumulation of drug candidates in early drug development. However, the lack of sample cleanup and chromatographic separation can hamper the analysis due to isobaric interferences. Multiple reaction monitoring (MRM) uses unique precursor ion-product ion transitions to add specificity which leads to higher selectivity. Here, we present a targeted imaging platform where desorption electrospray ionization is combined with a triple quadrupole (QqQ) system to perform MRM imaging. The platform was applied to visualize (i) lipids in mouse brain tissue sections and (ii) a drug candidate and metabolite in canine liver tissue. All QqQ modes were investigated to show the increased detection time provided by MRM as well as the possibility to perform dual polarity imaging. This is very beneficial for lipid imaging because some phospholipid classes ionize in opposite polarity (e.g., phosphatidylcholine/sphingomyelin in positive ion mode and phosphatidylserine/phosphatidylethanolamine in negative ion mode). Drug and metabolite images were obtained to show its strength in drug distribution studies. Multiple MRM transitions were used to confirm the local presence and selective detection of pharmaceutical compounds.
Drug discovery and development aims to find suitable candidates that can inhibit or activate a pathway to have a positive effect in a disease state.1 The desired effect is the potential drug being distributed to the target site and bound to the biological receptors, present for a certain amount of time at a high enough concentration.2 However, if a drug candidate or its metabolite(s) accumulates in tissue, it can result in significant toxicological effects. In addition, these effects can also derive from the presence of specific (secondary) drug metabolites. Therefore, toxicology studies are key in deciding whether a potential drug will be further developed.3 Analytical tools are thus required for species identification and localization. Current imaging technologies, like quantitative whole-body autoradiography (qWBA), examine drug distribution but require radiolabeling and therefore cannot distinguish between possible drugs and their metabolites.4
Mass spectrometry imaging (MSI) allows label-free drug distribution investigation because of its ability to map drug candidates and metabolites simultaneously.2,3,5 Matrix-assisted laser desorption/ionization (MALDI) is often combined with MSI due to its high spatial resolution, sensitivity, and throughput. However, difficulties in MALDI-MSI can arise due to inhomogeneity in matrix application and the possible interferences arising from matrix-related signals, especially in the absence of high mass resolving power. Lately, ambient liquid ionization techniques have complemented MALDI-MSI, as they do not require external matrixes to facilitate analysis. Desorption electrospray ionization (DESI)6 was introduced by Cooks and coworkers and has established itself as the main ambient tool for MSI. DESI uses an electrospray emitter to map extracted molecules directly from the cryosectioned tissue sample. The main advantages of DESI are the minimal sample preparation required prior to analysis and its compatibility with subsequent histological staining.7 DESI imaging of lipids8,9 is the most popular, but the pharmaceutical industry has gained interest in the technology10 for applications like drug blood-brain barrier permeability11 and cassette dosing.12
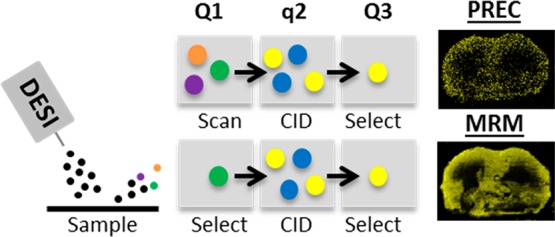
Independent of the ionization source, constant technical improvements are made to improve MS detection. The integration of ion mobility separation (IMS)13,14 with MSI increases selectivity by reducing the interference from isobaric background compounds. Use of not only IMS but also high mass resolution MSI15,16 allows the separation of isobaric interference from targeted analytes, which is beneficial for drug analyses where chemical background from biological samples is a real burden. Even though the mass resolving capability of the newest high resolution instruments is significant, examples are found in literature of isobaric compounds that could not be separated.17 In consequence, a more selective tandem MS (MS/MS) approach is required to separate these isobaric compounds and is generally executed using a triple quadrupole (QqQ) instrument. Although referred to as a QqQ by mass spectrometrists, most commercially available QqQ instruments nowadays use only two quadrupole mass analyzers and, therefore, are often called tandem quadrupole (TQ) instruments.18 In consistency with the MS field, we refer to the TQ system as a QqQ instrument.19 The QqQ used for the studies described in this paper uses a first quadrupole (Q1) followed by a (nonquadrupolar) collision cell (q) for precursor fragmentation and second quadrupole (Q3) to either select or scan ions. The system can be used in different modes:20,21 MS scan (Q1 operates in RF only mode, no fragmentation, Q3 is scanning), product ion scan (Q1 is selecting, Q3 is scanning; PROD), precursor ion scan (Q1 is scanning, Q3 is selecting; PREC), neutral loss scan (Q1 is scanning and Q3 is scanning with an offset specific for a neutral fragment; NL), and multiple reaction monitoring (Q1 and Q3 are selecting; MRM). Once the precursor and product ions are known, a targeted and high-throughput MRM method can analyze multiple compounds simultaneously. In addition, the system enables dual polarity imaging in one experiment. Polarity switching has been explored previously for lipid MS imaging.22 MALDI-QqQ imaging has also improved sensitivity for drug distribution studies.23,24 Other MALDI-MRM drug applications have been focused on skin analysis25 and forensics.26,27 MRM imaging allows for quantitative MSI approaches due to its wide dynamic range.28
Here, we report the benefits of using a QqQ mass spectrometer for targeted DESI-MS imaging, which is illustrated with different sets of experiments. First, the improved specificity of MRM is compared with the QqQ scanning modes for multiple lipid classes in rat brain tissue. In addition, DESI-MRM was applied to map a drug candidate and its metabolite in canine liver. At last, lipid imaging of rat brain tissue was performed to prove the benefit of dual polarity imaging.
Experimental Section
Materials and Reagents
ULC/MS-grade water (H2O), ULC/MS-grade methanol (MeOH), ULC/MS-grade ethanol (EtOH), and 99% formic acid (FA) were purchased from Biosolve (Valkenswaard, NL). Microscopic glass slides were purchased from Thermo Scientific (Braunschweig, DE). Drug standard (compound A) was provided by Janssen R&D (Beerse, Belgium).
Tissue Collection and Preparation
Rat brain and canine liver were used in this research. MAASTRO clinic (Maastricht, NL) provided Wistar Albino Glaxo Rat brain tissues that were snap frozen in liquid nitrogen. Janssen R&D (Beerse, Belgium) provided canine liver tissues dosed with a drug candidate A at 65 mg/kg. For confidentiality reasons, structural information on the drug candidate cannot be revealed. All tissue samples were stored at −80 °C until cryosectioning. Fresh-frozen tissues were cryosectioned using a Microtome cryostat (Thermo Scientific, Braunschweig, Germany) into 12 μm-thick tissue sections and thaw mounted on microscope glass slides, stored at −80 °C, and air-dried prior to DESI-QqQ analysis.
DESI-QqQ Instrumentation
A Xevo TQ-S micro was equipped with a DESI source for the MSI analysis (Waters, Wilmslow, UK). DESI solvent (MeOH/H2O, 98/2, v/v) was supplied by a Waters ACQUITY UPLC M-class binary solvent manager at 2–5 μL/min. For positive ionization experiments, 0.1% FA was added to the DESI solvent. General parameters were: N2 nebulizing gas pressure = 3–5 bar; spray voltage = ±3–4 kV; MS source temperature = 150 °C; sampling cone voltage = 25 V. A custom-built inlet capillary was heated to 300–500 °C. Pixel sizes differed per MSI analysis from 30 to 100 μm. Mass resolution settings were optimized to unit resolution. NL/PREC/PROD scan times ranged from 0.995 to 2.496 s/pixel, and MRM dwell times ranged from 0.199 to 0.999 s/pixel. All images were acquired with Omni Spray 2-D version 2.0.1 (Prosolia, Indianapolis, IN, USA) combined with MassLynx version 4.1 (Waters, Milford, MA, USA).
Hematoxylin and Eosin (H&E) Staining
H&E staining was executed on the same tissue sections after DESI-QqQ analysis. The tissue sections were washed in successive EtOH baths (100, 96, and 70%) and deionized water for 3 min each. Hematoxylin (Merck, Darmstadt, Germany) staining was executed for 3 min followed by a gentle 3 min wash with running tap water. Eosin staining was performed for 30 s, followed by a gentle wash with running tap water for 3 min. The protocol was finalized by a 1 min EtOH wash and a 30 s xylene wash for dehydration. Coverslips were placed on the stained tissues using Entellan. A MIRAX Desk scanner (Zeiss, Gottingen, Germany) was used to acquire optical images.
Data Processing and Visualization
For all QqQ imaging experiments, individual line scans were acquired. For individual MRM transitions, all raw line scan files were converted into mzML with MSConvertGUI (Vanderbilt University, Nashville, TN, United States) and further processed by an in-house written MATLAB script (v. R2015a). To visualize NL/PREC/PROD images, raw line scan files were directly processed by a MATLAB script.
Results and Discussion
Scanning versus MRM
MRM mode was investigated and compared with several scanning modes to target multiple high and low abundant lipid classes.29 In positive ion mode, a DESI-PREC imaging experiment was executed to scan for precursors with the m/z 184 fragment ion29 corresponding to protonated phosphatidylcholine (PC) and sphingomyelin (SM) lipids. Based on the PREC spectrum (Figure S-1) 5 lipid species at variable abundance were selected. The corresponding MRM transitions were used in a DESI-MRM imaging experiment on a consecutive tissue section. The same strategy was used for the experiments in negative ion mode. First, a DESI-NL scan of 87, corresponding to deprotonated phosphatidylserine (PS) lipids, was performed.30 This was followed by a DESI-MRM imaging experiment (consecutive tissue section) of 5 corresponding MRM transitions selected from the NL spectrum (Figure S-2). Figure 1 shows the resulting MS/MS images of 10 lipid species of which 5 were obtained in positive ion mode (PC and SM, left 2 columns) and 5 in negative ion mode (PS, right 2 columns). Using MRM for MS imaging experiments significantly adds specificity to the analysis compared to MS scan. The terms specificity and selectivity are often confusingly used. Selectivity is the ability of a method to distinguish between the analyte and its sample matrix. Specificity is the ability to measure one specific analyte in the presence of its sample matrix (i.e., 100% selectivity for one specific analyte).31 With MRM imaging, the selection of the fragment ion corresponding to the precursor ion adds specificity to the analyte and, therefore, measures this compound at near 100% selectivity. This allows separation of compounds with selectivity higher than that of current high mass resolving power MS technologies. Lanekoff et al. combined nanospray DESI with multiplexed MS/MS imaging.32 In this work, MRM could separate both isobaric metabolites and isobaric lipids where current high mass resolving power MS failed. An example is the separation of the [M + Na]+ ion of phosphatidylethanolamine PE(P-16:0/22:6) at m/z 770.5095 from the [M + K]+ ion of PC 32:1 at m/z 770.5097. The use of MRM versus PREC/NL scanning modes increases the total detection time per precursor ion. For instance, in NL scanning mode, [PS(44:10)-H]− is acquired at an acquisition rate of 20,000 Da/sec over a m/z 500–1000 mass range at a scan rate of 2.496 s/pixel. This leads to a total detection time of 62.4 ms/pixel for the [PS(44:10)-H]− ion. In MRM mode, at the same total analysis time, the image of [PS(44:10)-H]− was acquired at 999 ms/pixel. This results in MRM images from low-abundant lipid species (<1% for PS(44:10)) that are poorly detected in NL scanning mode. Not only is DESI-MRM imaging more selective but also significantly reduces analysis costs relative to high mass resolving MS imaging of isobars, which can be an argument for implementation in the pharmaceutical industry.
Figure 1.

TIC images acquired in scanning and MRM modes. Left two columns show images acquired in positive ion mode, and the right two columns show images acquired in negative ion mode. In the left two columns, DESI-PREC images (scan time 0.995 s) are compared with DESI-MRM images (dwell time 0.199 s/MRM) of the same species. The right two columns show the DESI-NL images (scan time 2.496 s) and their related DESI-MRM images (dwell time 0.999 s/MRM). Pixel size is 100 μm. H&E images are displayed at the bottom of each column.
Confirmation of Spatial Localization of Drug and Metabolites
Due to the additional specificity of DESI-MRM imaging, this technology is a suitable tool to pharmaceutical studies. The canine liver tissues used for this MRM imaging application are obtained from an investigation of a possible drug candidate for prostate cancer: Compound A with a molecular weight of 515 g/mol ([A+H]+ ion at m/z 516) which is metabolized into the demethylated metabolite M1 at 501 g/mol ([M1+H]+ ion at m/z 502). Based on preliminary experiments performed in Janssen R&D, a NL spectrum of 418 was obtained followed by a PROD spectrum of compound A. This information was used in the selection of two specific MRM transitions for each compound (m/z 516 → m/z 98, m/z 516 → m/z 488; m/z 502 → m/z 84, m/z 502 → m/z 474). To confirm spatial distribution, all MRM transitions are used to map the drug candidate and its metabolite. Figure 2 shows two MRM images of compound A (Figure 2a and 2b) and two of metabolite M1 (Figure 2c and 2d). All MRM images reveal significant accumulation of A and M1 in tissue lesions, which are visible in the H&E images and highlighted by arrows in Figure 2e. Additional MALDI-MSI experiments were executed and display similar distribution of A and M1 (Figure S-3) to complement the DESI-MRM data. The accumulation of compound A correlates with histological lesions previously annotated by the pathologist and indicates severe fibrosis, which resulted in the exclusion of compound A from further development. It is worth mentioning that the occurrence of ion suppression and/or enhancement due to the presence of bile acids in the bile duct may influence the detected levels of compound A.33 However, qualitative MSI provides pharmaceutical researchers with unique understanding of drug distribution.4 This qualitative information benefits significantly from selective MS detection. Our data shows that MRM imaging can not only be used for this selective detection of drugs and metabolites but also for spatial confirmation (e.g., accumulation at the same location) by monitoring multiple MRM transitions per compound.
Figure 2.

Spatial distribution of compounds A (a and b) and M1 (c and d) by two MRM transitions. H&E image (e) shows tissue lesions (pointed by arrows) in the canine liver. Spatial resolution is 50 μm. Dwell time/MRM is 0.299 s.
Dual Polarity MS Imaging
In addition to the improved specificity, another benefit of using a QqQ mass spectrometer is the ability to perform fast dual polarity experiments. Figure 3 shows two applications of dual polarity MRM imaging: lipid imaging in rat brain and drug/metabolite imaging in canine liver. In the lipid imaging application, the images of four MRM transitions of which two transitions (Figures 3b and c) were acquired in positive ionization mode, and two transitions (Figures 3d and e) were obtained in negative ionization mode in one experiment. Even though shown in MRM mode, the dual polarity capacity also applies to the scanning modes of the QqQ. This means that lipid classes can be evaluated based on both positive and negative charged head groups and/or acyl chains in one single experiment. In the pharmaceutical application, compounds A and M1 are mapped (Figures 3g and h) in positive ionization mode, and two bile acids are targeted (Figures 3i and j) in negative ionization mode. The main advantage of dual polarity MS imaging is that the information acquired originates from the same tissue section and, therefore, significantly reduces analysis time. Previous untargeted dual polarity MSI investigations34 using Orbitrap mass spectrometers have shown that polarity switching takes up to multiple seconds to reach the expected mass accuracy. The benefit of saving analysis time got lost because this one experiment takes twice as long. Nazari and coworkers35 improved the polarity switching capability of the Orbitrap MS to a 96 ms switching time. Our QqQ MSI experiments have very limited elongation of analysis time because the polarity switching time is 15 ms. Ellis et al.22 showed the benefit of dual polarity lipid imaging in a classification model throughout tumor tissue using MALDI-MSI. The use of dual polarity MALDI imaging has the limitation that a MALDI matrix has to be applied, which can cause potential inferences, risks delocalization, and requires sample preparation time. DESI imaging does not require an external matrix to facilitate ionization and, therefore, does not suffer from these limitations. Taylor and coworkers reported the investigation of ion suppression of olanzapine on mouse brain where DESI ionization suffered from significantly less ion suppression than MALDI ionization.36 Depending on the chemical structure of the drug candidate, this makes DESI dual polarity imaging of potentially more interest for the pharmaceutical industry over MALDI dual polarity imaging. In pharmaceutical sciences, the possibility of drugs and its metabolites ionizing in different polarities is also very probable. For instance, a drug that ionizes in positive mode will ionize in negative mode after glucuronide or sulfate conjugation.37 In addition, possible endogenous markers for drug candidates could also ionize best in the opposite ionization mode (e.g., bile acids for bile duct cancer drugs). This is also shown in our drug/metabolite application in which two possible drug candidates and two bile acids are imaged and show colocalization. The ability to map drug candidates, drug metabolites and endogenous metabolites in one experiment is crucial in further targeted imaging analyses and, therefore, DESI dual polarity imaging can play an important role.
Figure 3.

Dual polarity DESI-MRM imaging performed in one experiment. In rat brain, four different DESI-MRM images of lipids were acquired, of which two in positive ion mode (b and c; dwell time 0.193 s/MRM) and two in negative ion mode (d and e; dwell time 0.193 s/MRM). In positive ion mode, two MRM transitions target the PC headgroup, while two different acyl chains are targeted in negative ion mode. Spatial resolution is 100 μm. In canine liver, two MRM transitions target compound A and M1 in positive ion mode (g and h; dwell time 0.485 s/MRM), and two MRM transitions image bile acids in negative ion mode (i and j; dwell time 0.498 s/MRM) that are colocalized with A and M1. Spatial resolution is 30 μm. H&E images of rat brain and canine liver are shown in (a) and (f), respectively.
Conclusion
This work demonstrates a novel targeted DESI imaging platform by using a QqQ mass analyzer combined with a targeted MS/MS-based data acquisition approach. The opportunity to perform dual polarity imaging combined with DESI is very advantageous for lipid and metabolite imaging because it does not rely on MALDI matrixes. QqQ scanning modes were compared with MRM showing increased ion detection time of MRM. This opens doors for the separation of isobars and certain isomers which cannot be separated with current high mass resolving power MSI technologies. Although this targeted platform requires fragmentation knowledge, the MSI field will significantly benefit from this as its applications are increasing. Our platform shows its strength in toxicology studies by selective imaging of drugs and metabolites in tissue. Spatial distribution is confirmed by monitoring the presence of pharmaceutical compounds using multiple MRM transitions of the same precursor ion. DESI-MRM imaging allows for selective imaging without the need for expensive MSI instrumentation.
MSI can suffer from lower sensitivity compared to techniques that combine mass spectrometry with a separation step prior to ionization.38 The use of MRM in the MS imaging field is very promising because improved selectivity is needed to visualize more compounds without the interference of surrounding isobars.
Acknowledgments
This research was financially supported by the Dutch Province of Limburg as part of the LINK program and the Integrated Technology Strategy (ITS) initiative program of Janssen Pharmaceutica NV. We are very grateful to Brenda Bakker (M4I) and Rianne Biemans (MAASTRO) for providing the rat brain tissues. We thank Filip Cuyckens (Janssen Pharmaceutica) for fruitful discussions on metabolite identification and Marjolein van Heerden (Janssen Pharmaceutica) for pathological expertise.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.8b03857.
Precursor ion scan spectrum of m/z 184 acquired from rat brain, neutral loss scan spectrum of 87 acquired from rat brain, and H&E and MALDI-MSI images of A and M1 of canine liver tissue (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Nilsson A.; Goodwin R. J.; Shariatgorji M.; et al. Anal. Chem. 2015, 87, 1437–1455. 10.1021/ac504734s. [DOI] [PubMed] [Google Scholar]
- Prideaux B.; Stoeckli M. J. Proteomics 2012, 75, 4999–5013. 10.1016/j.jprot.2012.07.028. [DOI] [PubMed] [Google Scholar]
- Sugihara Y.; Watanabe K.; Vegvari A. Bioanalysis 2016, 8, 575–588. 10.4155/bio-2015-0020. [DOI] [PubMed] [Google Scholar]
- Swales J. G.; Hamm G.; Clench M. R.; Goodwin R. J.A., et al. Int. J. Mass Spectrom. 2018. 10.1016/j.ijms.2018.02.007 [DOI] [Google Scholar]
- Greer T.; Sturm R.; Li L. J. Proteomics 2011, 74, 2617–2631. 10.1016/j.jprot.2011.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takats Z.; Wiseman J. M.; Cooks R. G. J. Mass Spectrom. 2005, 40, 1261–1275. 10.1002/jms.922. [DOI] [PubMed] [Google Scholar]
- Eberlin L. S.; Liu X.; Ferreira C. R.; et al. Anal. Chem. 2011, 83, 8366–8371. 10.1021/ac202016x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberlin L. S.; Ferreira C. R.; Dill A. L.; et al. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2011, 1811, 946–960. 10.1016/j.bbalip.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C.; Dill A. L.; Eberlin L. S.; et al. Mass Spectrom. Rev. 2013, 32, 218–243. 10.1002/mas.21360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiseman J. M.; Ifa D. R.; Zhu Y., et al. Proceedings of the National Academy of Sciences; 2008; Vol. 105, pp 18120–18125. [DOI] [PMC free article] [PubMed]
- Vallianatou T.; Strittmatter N.; Nilsson A.; et al. NeuroImage 2018, 172, 808–816. 10.1016/j.neuroimage.2018.01.013. [DOI] [PubMed] [Google Scholar]
- Swales J. G.; Tucker J. W.; Strittmatter N.; et al. Anal. Chem. 2014, 86, 8473–8480. 10.1021/ac502217r. [DOI] [PubMed] [Google Scholar]
- Sans M.; Feider C. L.; Eberlin L. S. Curr. Opin. Chem. Biol. 2018, 42, 138–146. 10.1016/j.cbpa.2017.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skraskova K.; Claude E.; Jones E. A.; et al. Methods 2016, 104, 69–78. 10.1016/j.ymeth.2016.02.014. [DOI] [PubMed] [Google Scholar]
- Anderson D. M.; Ablonczy Z.; Koutalos Y.; et al. J. Am. Soc. Mass Spectrom. 2014, 25, 1394–1403. 10.1007/s13361-014-0883-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss A.; Smith D. F.; Reschke B. R.; et al. Proteomics 2014, 14, 1283–1289. 10.1002/pmic.201300306. [DOI] [PubMed] [Google Scholar]
- Laskin J.; Lanekoff I. Anal. Chem. 2016, 88, 52–73. 10.1021/acs.analchem.5b04188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niessen W. M.; Falck D. In Analyzing Biomolecular Interactions by Mass Spectrometry; Wiley, 2015. [Google Scholar]
- Takeda H.; Izumi Y.; Takahashi M. J. Lipid Res. 2018, 59, 1283. 10.1194/jlr.D083014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brügger B. Annu. Rev. Biochem. 2014, 83, 79–98. 10.1146/annurev-biochem-060713-035324. [DOI] [PubMed] [Google Scholar]
- Schwudke D.; Oegema J.; Burton L.; et al. Anal. Chem. 2006, 78, 585–595. 10.1021/ac051605m. [DOI] [PubMed] [Google Scholar]
- Ellis S. R.; Cappell J.; Potocnik N. O. Analyst 2016, 141, 3832. 10.1039/C6AN00169F. [DOI] [PubMed] [Google Scholar]
- Prideaux B.; Dartois V.; Staab D.; et al. Anal. Chem. 2011, 83, 2112–2118. 10.1021/ac1029049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopfgartner G.; Varesio E.; Stoeckli M. Rapid Commun. Mass Spectrom. 2009, 23, 733–736. 10.1002/rcm.3934. [DOI] [PubMed] [Google Scholar]
- Boudon S. M.; Morandi G.; Prideaux B.; et al. J. Am. Soc. Mass Spectrom. 2014, 25, 1803–1809. 10.1007/s13361-014-0947-3. [DOI] [PubMed] [Google Scholar]
- Poetzsch M.; Baumgartner M. R.; Steuer A. E.; Kraemer T.; et al. Drug Test. Anal. 2015, 7, 143–149. 10.1002/dta.1674. [DOI] [PubMed] [Google Scholar]
- Porta T.; Grivet C.; Kraemer T.; et al. Anal. Chem. 2011, 83, 4266–4272. 10.1021/ac200610c. [DOI] [PubMed] [Google Scholar]
- Porta T.; Lesur A.; Varesio E.; et al. Anal. Bioanal. Chem. 2015, 407, 2177–2187. 10.1007/s00216-014-8315-5. [DOI] [PubMed] [Google Scholar]
- Hsu F. F.; Turk J. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2009, 877, 2673–2695. 10.1016/j.jchromb.2009.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu F. F.; Turk J. J. Am. Soc. Mass Spectrom. 2005, 16, 1510–1522. 10.1016/j.jasms.2005.04.018. [DOI] [PubMed] [Google Scholar]
- Vessman J. J. Pharm. Biomed. Anal. 1996, 14, 867–869. 10.1016/0731-7085(95)01679-1. [DOI] [PubMed] [Google Scholar]
- Lanekoff I.; Burnum-Johnson K.; Thomas M.; et al. Anal. Chem. 2013, 85, 9596–9603. 10.1021/ac401760s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis S. R.; Bruinen A. L.; Heeren R. M. A. Anal. Bioanal. Chem. 2014, 406, 1275–1289. 10.1007/s00216-013-7478-9. [DOI] [PubMed] [Google Scholar]
- Korte A. R.; Lee Y. J. J. Am. Soc. Mass Spectrom. 2013, 24, 949–955. 10.1007/s13361-013-0613-1. [DOI] [PubMed] [Google Scholar]
- Nazari M.; Muddiman D. C. Analyst 2016, 141, 595–605. 10.1039/C5AN01513H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor A.; Dexter A.; Bunch J. Anal. Chem. 2018, 90, 5637. 10.1021/acs.analchem.7b05005. [DOI] [PubMed] [Google Scholar]
- Benedetti M. S.; Whomsley R.; Poggesi I.; et al. Drug Metab. Rev. 2009, 41, 344–390. 10.1080/10837450902891295. [DOI] [PubMed] [Google Scholar]
- Lamont L.; Baumert M.; Ogrinc Potocnik N.; et al. Anal. Chem. 2017, 89, 11143–11150. 10.1021/acs.analchem.7b03512. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.