

 Subtle changes in ligand substitution result in substantial changes in molecular structure and magnetic properties in a series of dysprosium(iii) metallocenium salts.
Subtle changes in ligand substitution result in substantial changes in molecular structure and magnetic properties in a series of dysprosium(iii) metallocenium salts.
Abstract
A series of dysprosium(iii) metallocenium salts, [Dy(CpiPr4R)2][B(C6F5)4] (R = H (1), Me (2), Et (3), iPr (4)), was synthesized by reaction of DyI3 with the corresponding known NaCpiPr4R (R = H, iPr) and novel NaCpiPr4R (R = Me, Et) salts at high temperature, followed by iodide abstraction with [H(SiEt3)2][B(C6F5)4]. Variation of the substituents in this series results in substantial changes in molecular structure, with more sterically-encumbering cyclopentadienyl ligands promoting longer Dy–C distances and larger Cp–Dy–Cp angles. Dc and ac magnetic susceptibility data reveal that these structural changes have a considerable impact on the magnetic relaxation behavior and operating temperature of each compound. In particular, the magnetic relaxation barrier increases as the Dy–C distance decreases and the Cp–Dy–Cp angle increases. An overall 45 K increase in the magnetic blocking temperature is observed across the series, with compounds 2–4 exhibiting the highest 100 s blocking temperatures yet reported for a single-molecule magnet. Compound 2 possesses the highest operating temperature of the series with a 100 s blocking temperature of 62 K. Concomitant increases in the effective relaxation barrier and the maximum magnetic hysteresis temperature are observed, with 2 displaying a barrier of 1468 cm–1 and open magnetic hysteresis as high as 72 K at a sweep rate of 3.1 mT s–1. Magneto-structural correlations are discussed with the goal of guiding the synthesis of future high operating temperature DyIII metallocenium single-molecule magnets.
Introduction
Single-molecule magnets show potential in spin-based computing and information storage, but such applications are currently precluded by low operating temperatures.1 The operating temperature is typically defined by the magnetic blocking temperature (Tb)—the temperature at which the magnetic relaxation time, τ, is equal to 100 seconds—or, less quantitatively, by the maximum temperature at which magnetic hysteresis is observed.2 Many of the most significant advances in increasing Tb for single-molecule magnets have been made with lanthanide-based systems, as the large anisotropies of the 4f elements can be exploited to achieve unparalleled thermal barriers to magnetic relaxation.3 In particular, strongly-coupled, multinuclear lanthanide complexes have dominated efforts to maximize operating temperature, as the giant-spin ground states of these molecules can suppress rapid through-barrier magnetic relaxation processes that often preclude magnetic hysteresis.4
Recently, the DyIII metallocenium salt [Dy(Cpttt)2][B(C6F5)4] (Cpttt = 1,2,4-tri(tert-butyl)cyclopentadienyl) was reported to show magnetic hysteresis at temperatures up to 60 K, as observed using a sweep rate of 2.2 mT s–1,5 a substantial increase over the previous record of 28 K.4c Significantly, this molecule represents an outlier in its class: while the majority of mononuclear single-molecule magnets show only waist-constricted magnetic hysteresis and little or no remanent magnetization due to quantum tunneling of the magnetization,6 [Dy(Cpttt)2]+ retains polarization at zero field and high temperatures.7 Magnetic and computational studies suggest that the impressive magnetic properties of [Dy(Cpttt)2]+ arise from its unique coordination environment—in particular, the rigid η5-cyclopentadienyl ligands constrain metal–ligand vibrational modes, thereby reducing pathways for through-barrier relaxation.5b This represents a new approach to limiting through-barrier magnetic relaxation and enabling higher operating temperatures. It is thus essential to develop a deeper understanding of how structure influences the magnetic properties of [Dy(CpX)2]+ complexes in order to determine how coordination environment can be tuned to further increase operating temperatures.8
The design of mononuclear 4f-element single-molecule magnets can be guided by a simple electrostatic model that compares the shape of lanthanide MJ electron distributions to the crystal field.9 A number of studies of mononuclear, D5h-symmetric complexes with strongly-donating axial ligands have demonstrated the validity of this qualitative approach, with molecules in this class possessing thermal barriers to magnetic relaxation as large as 1260 cm–1.3b–f Importantly, the electrostatic model provides a rationale for the high anisotropy engendered by the axial cyclopentadienyl ligands in [Dy(Cpttt)2]+ and outlines a clear route for improvement. Increasing the Cp–Dy–Cp angle of this bent metallocene from 152.56(7)° toward 180° and shortening the Dy–Cp bond distances should increase the axiality of the crystal field, leading to larger thermal barriers to magnetic relaxation and higher potential operating temperatures.
Equally important in the design of new [Dy(CpX)2]+ molecules are the metal–ligand vibrational modes. Phonons—vibrational modes of the lattice that couple to the electronic states of a molecule—provide the necessary energy for magnetic relaxation10 and determine viable relaxation processes at a given temperature. Computational studies indicate that high-temperature magnetic relaxation in [Dy(Cpttt)2]+ is induced by Cp–H bending modes,5b leading to the suggestion that substituting the hydrogen positions in this molecule with rigid functional groups could result in more constrained vibrations, further slowing magnetic relaxation and resulting in magnetic hysteresis at higher temperatures.
Herein, we report a series of DyIII metallocenium cation salts, [Dy(CpiPr4R)2][B(C6F5)4] (R = H (1), Me (2), Et (3), iPr (4)), in which subtle variation of the substituents on the cyclopentadienyl rings produces molecules with a large range of Dy–C distances and Cp–Dy–Cp angles. This series includes DyIII metallocenium salts of two new cyclopentadienyl ligands, CpiPr4R (R = Me, Et). By varying the substituents, it is also possible to tune the metal–ligand vibrational modes in these complexes. Modifications result in a record high thermal barrier to magnetic relaxation (Ueff) of 1468 cm–1 and a record high 100 s magnetic blocking temperature of 62 K for the methyl-substituted complex, 2. A careful comparison of the structural and magnetic properties in this series elucidates important design principles for the synthesis of further new DyIII metallocenium single-molecule magnets.
Experimental
General procedures
All manipulations were performed using Schlenk or glovebox techniques under an atmosphere of purified argon with rigorous exclusion of water and oxygen. All solvents were purchased from Sigma-Aldrich as anhydrous grade in Sure/Seal™ bottles, purged for several hours with purified argon and stored over activated 3 Å molecular sieves in an argon filled glovebox. A solution of LiAlH4 (1.0 M in Et2O) was purchased from Sigma-Aldrich and used as received. Anhydrous LnX3 (Ln = Dy, Y; X = Cl, I) were purchased from Alfa Aesar as Ultra Dry™ grade reagents and used as received. The halide abstraction reagent, [H(SiEt3)2][B(C6F5)4], was prepared freshly before use via a reported method.18 The salt NaCpiPr4 was synthesized via a literature route11 and dried under vacuum to remove traces of THF. Tetraisopropylfulvene and methyl-substituted tetraisopropylfulvene were prepared according to the literature12 and recrystallized several times from anhydrous ethanol at –70 °C. The obtained fulvenes were assayed via GC-MS, and purities of 86% and 85% were found for tetraisopropylfulvene and methyl-substituted tetraisopropylfulvene, respectively. The main impurity in both cases was a mixture of tetraisopropylcyclopentadiene isomers. The salt NaCpiPr5 was obtained from methyl-substituted tetraisopropylfulvene in two steps via a combination of literature methods12,13 with modifications detailed below. Methyl-substituted tetraisopropylfulvene was initially converted to the Li+ salt of pentaisopropylcyclopentadiene by the method of Dezember et al.12 and hydrolyzed in situ by careful addition of H2O to give free pentaisopropylcyclopentadiene after extraction into Et2O and solvent removal. The resulting compound was deprotonated using excess NaNH2 (1.2 equiv.) in refluxing THF with the aid of sonication via a known method.13 The salt NaCpiPr5 was extracted from the crude material (mostly a mixture of NaCpiPr4 and NaCpiPr5) with benzene and the solvent removed under vacuum. The off-white solid was dried under vacuum at 100 °C for 2 h to remove coordinated THF, washed with pentane, and dried under vacuum at room temperature to give pure NaCpiPr5 as a white powder. GC-MS experiments were performed using a Thermo Scientific Exactive GC-MS (Orbitrap) with a Thermo Scientific Trace 1310 GC using a TG-5SILMS column. NMR spectra were recorded on a Bruker Avance 300 MHz or 500 MHz spectrometer and internally referenced to the residual solvent signals. FT-IR spectra were recorded on a Perkin Elmer Avatar Spectrum 400 FTIR Spectrometer equipped with an attenuated total reflectance (ATR) attachment. Elemental analyses (C, H, N) were performed by the Microanalytical Facility at the University of California, Berkeley using a Perkin-Elmer 2400 Series II combustion analyzer. Metals analyses (Dy, Y) were performed on a Thermo Fisher Scientific iCAP 6300 Inductively Coupled Plasma-Atomic Emission Spectrometer (ICP-AES). Magnetic susceptibility measurements were collected using a Quantum Design MPMS2 SQUID magnetometer; additional details including description of magnetic measurements, sample preparation, and data fitting are included in the ESI.†
Synthesis of compounds
Synthesis of NaCpiPr4Me
Tetraisopropylfulvene (1.00 g, 4.05 mmol) (86% pure by GC-MS) and Et2O (40 mL) were combined in a 100 mL Schlenk flask to produce a bright orange solution. The solution was cooled to 0 °C and 1.0 M LiAlH4 in Et2O (12.1 mL, 12.1 mmol) was added all at once via syringe. The reaction flask was allowed to warm to room temperature and stirred for 20 h. At this point, the initial orange solution had been replaced with a grayish suspension and solvent was removed under vacuum to leave a grayish residue. This residue was slurried in 30 mL pentane for 30 min, filtered and washed with 2 × 15 mL pentane to leave a white/gray powder. The powder was taken up in 50 mL of Et2O, cooled to 0 °C, and 20 mL of H2O was added (H2O should be added very slowly at first to prevent excessive frothing), followed by 20 mL of 1.0 M HCl. This mixture was stirred vigorously for 30 min and then the layers separated. The aqueous layer was extracted with 2 × 50 mL of Et2O and the combined organic fractions were washed with 25 mL of brine, dried with MgSO4, and the solvent was removed under vacuum to leave a light yellow oil (0.70 g, 2.8 mmol of HCpiPr4Me tautomers). This oil was combined with NaNH2 (0.22 g, 5.60 mmol) and 20 mL of THF in a 50 mL Schlenk flask equipped with a reflux condenser. The reaction flask was immersed in a sonication bath filled with H2O at a temperature ranging from 40–50 °C and sonicated overnight under argon. The flask was subsequently cooled to room temperature and the solvent was removed under vacuum to leave a beige residue. This residue was transferred to an Ar-filled glovebox, re-suspended in 25 mL of THF, filtered through Celite, and the filter cake was then washed with additional THF to give an amber filtrate. Solvent was removed from the filtrate under vacuum and the resulting off-white solid heated under vacuum at 100 °C for 2 h to remove coordinated THF. This solid was then slurried in 25 mL pentane, filtered, washed with additional pentane, and dried under vacuum at room temperature to give pure NaCpiPr4Me as a white powder (0.59 g, 54% yield based on tetraisopropylfulvene). 1H NMR (THF-d8, 500 MHz, 338 K): δ = 1.24 (d, 12H, Cp–CH(CH3)2), 1.25 (d, 12H, Cp–CH(CH3)2), 2.19 (s, 3H, Cp–CH3), 3.19 (m, 4H, Cp–CH(CH3)2). 13C{1H} NMR (THF-d8, 125 MHz, 338 K): δ = 15.00 (Cp–CH3), 25.37 (Cp–CH(CH3)2), 26.19 (Cp–CH(CH3)2), 28.21 (Cp–CH(CH3)2), 28.32 (Cp–CH(CH3)2), 104.78 (–C(iPr)–C(Me)–C(iPr)–), 116.60 (–C(iPr)–C(iPr)–C(iPr)–), 118.53 (–C(Me)–C(iPr)–C(iPr)–). Calcd for C18H31Na (270.44) (%): C 79.94, H 11.55; found (%): C 77.94, H 10.32 (low percent carbon values were obtained across multiple measured samples).
Synthesis of NaCpiPr4Et
Following the general procedure outlined above for the synthesis of NaCpiPr4Me (the quantity of all solvents was doubled), NaCpiPr4Et was synthesized from methyl-substituted tetraisopropylfulvene (3.00 g, 11.5 mmol) (85% pure by GC-MS) and 1.0 M LiAlH4 in Et2O (34.4 mL, 34.4 mmol) to initially give a light yellow oil after quenching with H2O (40 mL) and 1.0 M HCl (40 mL) (2.1 g, 8.0 mmol of HCpiPr4Et tautomers). This was deprotonated in the same fashion as NaCpiPr4Me using NaNH2 (0.62 g, 16 mmol). Pure NaCpiPr4Et was isolated as a white powder (2.05 g, 63% yield based on methyl-substituted tetraisopropylfulvene). 1H NMR (THF-d8, 500 MHz, 338 K): δ = 1.02 (t, 3H, Cp–CH2CH3), 1.25 (d, 12H, Cp–CH(CH3)2), 1.26 (d, 12H, Cp–CH(CH3)2), 2.56 (q, 2H, Cp–CH2CH3), 3.14 (br, 2H, Cp–CH(CH3)2), 3.26 (sept, 2H, Cp–CH(CH3)2). 13C{1H} NMR (THF-d8, 125 MHz, 338 K): δ = 19.79 (Cp–CH2CH3), 21.78 (Cp–CH2CH3), 25.78 (Cp–CH(CH3)2), 26.12 (Cp–CH(CH3)2), 28.14 (br, Cp–CH(CH3)2), 28.51 (Cp–CH(CH3)2), 112.76 (–C(iPr)–C(Et)–C(iPr)–), 116.89 (–C(iPr)–C(iPr)–C(iPr)–), 117.79 (–C(Et)–C(iPr)–C(iPr)–). Calcd for C19H33Na (284.46) (%): C 80.22, H 11.69; found (%): C 79.91, H 11.66.
Synthesis of [Dy(CpiPr5)2][B(C6F5)4] (4)
DyI3 (0.24 g, 0.44 mmol) and NaCpiPr5 (0.33 g, 1.10 mmol) were combined in a 35 mL pressure flask containing a glass-coated magnetic stirring bar. Toluene (15 mL) was then added, and the reaction flask was covered in Al foil and heated gradually to 160 °C with vigorous stirring. After 72 h at 160 °C, the flask was allowed to cool to room temperature, the reaction mixture was filtered through Celite, and the pad was washed with additional toluene (3 × 5 mL). Solvent was removed from the filtrate under vacuum and the residue was then gently heated (50 °C) under vacuum for 1 h. The residue was taken up in pentane (25 mL), stirred for 0.5 h, filtered through Celite, and the pad washed with additional pentane (3 × 5 mL). Solvent was removed from the filtrate under vacuum and the residue was then gently heated (50 °C) under vacuum for 0.5 h to give crude Dy(CpiPr5)2I. This was dissolved with benzene (10 mL) in a 25 mL Schlenk flask equipped with a Teflon-coated magnetic stirring bar. Under vigorous stirring at room temperature, a solution of [H(SiEt3)2][B(C6F5)4] (0.36 g, 0.39 mmol) in benzene (5 mL) was added dropwise to the Dy(CpiPr5)2I solution over 5 min. After stirring for 72 h at room temperature, solvent was removed from the reaction mixture under vacuum. The residue was then slurried with pentane (20 mL) for 0.5 h to yield a powder which was collected on a medium-porosity fritted filter and washed with pentane (3 × 10 mL). This solid was dried under vacuum, dissolved in dichloromethane (4 mL), filtered with the aid of Celite through a 0.2 µm porosity PTFE syringe filter, and layered with pentane (15 mL) in a 20 mL vial. After 48 h at room temperature, well-formed crystals were obtained, isolated on a medium-porosity fritted filter, and washed consecutively with pentane (5 mL), benzene (2 × 5 mL) and pentane (5 mL). The crystallization was repeated and the twice recrystallized solid was dried under vacuum to yield 4 as orange needles (0.35 g, 57% yield based on DyI3). Calcd for C64H70BF20Dy (1392.54) (%): C 55.20, H 5.07, Dy 11.7; found (%): C 55.21, H 4.80, Dy 11.3 (For 4, a 3rd recrystallization was needed to obtain acceptable values).
Synthesis of [Dy(CpiPr4)2][B(C6F5)4] (1)
Following the general procedure used for the synthesis of 4, [Dy(CpiPr4)2][B(C6F5)4] (1) was synthesized from DyI3 (0.21 g, 0.39 mmol), NaCpiPr4 (0.25 g, 0.98 mmol) and [H(SiEt3)2][B(C6F5)4] (0.28 g, 0.31 mmol). Subsequent crystallizations were carried out over 24 h at room temperature and 24 h at –35 °C. Compound 1 was obtained as yellow prisms (0.21 g, 41% yield based on DyI3). Calcd for C58H58BF20Dy (1308.38) (%): C 53.24, H 4.47, Dy 12.4; found (%): C 52.88, H 4.45, Dy 12.8.
Synthesis of [Dy(CpiPr4Me)2][B(C6F5)4] (2)
Following the general procedure used for the synthesis of 4, [Dy(CpiPr4Me)2][B(C6F5)4] (2) was synthesized from DyI3 (0.20 g, 0.37 mmol), NaCpiPr4Me (0.25 g, 0.92 mmol) and [H(SiEt3)2][B(C6F5)4] (0.27 g, 0.30 mmol). The first crystallization was conducted at –35 °C and the second at room temperature. Compound 2 was obtained as orange-yellow prisms (0.25 g, 50% yield based on DyI3). Calcd: C60H62BF20Dy (1336.43) (%): C 53.92, H 4.68, Dy 12.2; found (%): C 53.36, H 4.48, Dy 12.6.
Synthesis of [Dy(CpiPr4Et)2][B(C6F5)4] (3)
Following the general procedure used for the synthesis of 4, [Dy(CpiPr4Et)2][B(C6F5)4] (3) was synthesized from DyI3 (0.23 g, 0.42 mmol), NaCpiPr4Et (0.30 g, 1.05 mmol) and [H(SiEt3)2][B(C6F5)4] (0.31 g, 0.34 mmol). Crystallizations were carried out in the same manner as 4. Compound 3 was obtained as orange-yellow needles (0.31 g, 54% yield based on DyI3). Calc. C62H66BF20Dy (1364.49) (%): C 54.58, H 4.88, Dy 11.9; found (%): C 54.05, H 4.68, Dy 12.1.
Results and discussion
In designing this series of complexes, we sought tetra- and penta-substituted cyclopentadienyl ligands with sufficient steric bulk to preclude coordination of equatorial ligands, such as coordinating solvent. We thus targeted the known tetra- and pentaisopropyl substituted Cp ligands, as well as two new tetraisopropyl substituted Cp variants, all of which were obtained as the sodium salts, NaCpiPr4R (R = H, Me, Et, iPr). The tetraisopropyl substituted ligand, NaCpiPr4 was synthesized as previously described by step-wise deprotonation and alkylation of cyclopentadiene in three steps.11 Although it is possible to obtain NaCpiPr5 by further direct alkylation of NaCpiPr4, the yield for the final alkylation step is very low, producing mainly geminally-substituted isomers. Rather, NaCpiPr5 was obtained from NaCpiPr4 in four steps using the high-yielding fulvene route.12 The lithium salt acquired initially was hydrolyzed and then deprotonated with NaNH2, activated by sonication, to give the desired sodium salt.13
In order to complete a stepwise series of metallocenium salts in which the R group in CpiPr4R varies by only one methyl or methylene group in adjacent members of the series, it was necessary to synthesize the heretofore unknown tetraisopropyl substituted Cp ligand salts NaCpiPr4R (R = Me, Et). This was achieved by exploitation of the fulvene route used to prepare NaCpiPr5. Here, tetraisopropylfulvene and methyl-substituted tetraisopropylfulvene12 reacted with excess LiAlH4 and were then hydrolyzed to produce the corresponding cyclopentadiene isomer mixtures. These were then converted to the sodium salts NaCpiPr4Me and NaCpiPr4Et with sonication in the presence of excess NaNH2 (Scheme 1). In all cases, the sodium salts were preferred for their moderate solubility and ready accessibility as ether-free compounds, allowing facile workup in subsequent steps. For example, the lithium salt of CpiPr5 is obtained as the diethyl etherate and is soluble in pentane,12 whereas the sodium salt can be freed of coordinated THF by heating under vacuum and is soluble in arenes but insoluble in alkanes.
Scheme 1. Synthesis of NaCpiPr4Me and NaCpiPr4Et.

With the Cp transfer reagents in hand, we turned to the synthesis of the neutral dysprosium compounds, Dy(CpiPr4R)2X (R = H, Me, Et, iPr) (X = Cl, Br, I). The most common strategy for transfer of Cp ligands to LnIII centers involves the reaction of MCpX (commonly M = Li, Na, K) with a lanthanide halide. For Sm, Eu, and Yb, which have relatively stable divalent oxidation states, the initial reaction can occur between MCpX and LnI2, followed by oxidation to obtain the LnIII complex; an example of this approach is the well-known synthesis of 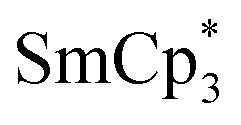 (Cp∗ = pentamethylcyclopentadienyl).14
(Cp∗ = pentamethylcyclopentadienyl).14
In many cases, the lanthanide iodide is preferred over the other halides, especially in the case of very bulky ligand environments.15 For instance, the reaction of YCp″2X (Cp″ = 1,3-bis(trimethylsilyl)cyclopentadienyl, X = Cl, I) with KCp″ in refluxing toluene to produce YCp″3 was most successful when X = I.16 Our own exploratory reactions with YCl3 and excess NaCpiPr5 (2.2 equiv.) in toluene at 130 °C indicated YCpiPr5Cl2 as the dominant product, and thus we opted for DyI3 or DyI3(THF)3 as the most promising DyIII sources. In some cases, the THF adduct allows for substitutions that are not possible with the unsolvated precursor, such as in the reaction of YbCp″2I(THF) with KCp″ to form YbCp″3.16 Conversely, the presence of THF can result in unproductive side reactions.17 In our hands, reaction of DyI3(THF)3 with NaCpiPr5 in toluene at 130 °C resulted in isolation of THF ring-opened products, as has been observed previously during attempts to synthesize 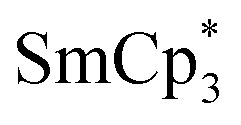 from the di-substituted SmIII salt.14 Therefore, DyI3 was used in all subsequent reactions.
from the di-substituted SmIII salt.14 Therefore, DyI3 was used in all subsequent reactions.
In general, DyI3 and a Cp transfer reagent NaCpiPr4R (R = H, Me, Et, iPr) (2.5 equiv.) were heated together at 160 °C in toluene for three days to generate the putative intermediate complexes Dy(CpiPr4R)2I (R = H, Me, Et, iPr), which were isolated from excess ligand and NaI by extraction with pentane (Scheme 2). A pressure flask was used to obtain the high temperature necessary for complete di-substitution.
Scheme 2. Synthesis of metallocene salts 1–4 and Y1–Y4.

Crystallization of Dy(CpiPr4R)2I from pentane at low temperature (–35 °C) was largely fruitless owing to the high solubility of the compounds. Instead, the crude products were used directly in the subsequent halide abstraction step. The addition of a sub-stoichiometric quantity (0.8 equiv.) of [H(SiEt3)2][B(C6F5)4]—freshly prepared before use18—to pale yellow benzene solutions of Dy(CpiPr4R)2I (R = H, Me, Et, iPr) resulted in a rapid color change to a more vibrant color ranging from yellow to orange. In most cases, yellow-orange powdery to oily precipitate was also observed, which changed over the course of several days, typically becoming more solid and increasing in volume. It is known that reactions involving [SiEt3]+ in benzene are complex, forming biphasic mixtures.18 This may be the source of the oily precipitates observed initially in some cases. It was assumed that the initial challenging reaction of DyI3 with NaCpiPr4R was not quantitative, therefore a sub-stoichiometric (versus DyI3) quantity of [H(SiEt3)2][B(C6F5)4] was employed in the reactions. Here, we wanted to avoid the presence of excess [SiEt3]+ in the reaction mixtures, which can render purification more difficult. After stirring for several days at room temperature, the desired DyIII metallocene salts [Dy(CpiPr4R)2][B(C6F5)4] (R = H (1), Me (2), Et (3), iPr (4)) were obtained as crude powders (Scheme 2) and subsequently recrystallized 2–3 times by layering dichloromethane solutions of the salts with pentane. In this way, analytically pure material and crystals suitable for X-ray diffraction analysis were obtained. After drying under reduced pressure, compounds 1–4 were obtained as yellow (1), orange-yellow (2–3) or orange crystals (4) and found to be indefinitely stable at room temperature under argon; however at ambient temperatures in air they decay rapidly with an accompanying color change to green/blue.
To obtain solid-state dilutions of 1-4, it was necessary to synthesize the diamagnetic YIII analogues, [Y(CpiPr4R)2][B(C6F5)4] (R = H (Y1), Me (Y2), Et (Y3), iPr (Y4)) (see ESI† for experimental procedures). Utilizing an analogous route to that employed for 1–4, YI3 was converted to the putative Y(CpiPr4R)2I (R = H, Me, Et, iPr) complex by reaction with the appropriate NaCpiPr4R (R = H, Me, Et, iPr) (2.5 equiv.) at high temperature, followed by iodide abstraction with [H(SiEt3)2][B(C6F5)4] (Scheme 2). The YIII metallocenium complexes were obtained as colorless (Y1) to yellow crystals (Y2–Y4) after multiple recrystallizations followed by prolonged drying under vacuum. Like the DyIII metallocenes salts, Y1–Y4 are stable under argon at ambient temperature, but decay rapidly in air as indicated by the appearance of a green/blue color.
NMR spectra were obtained for the diamagnetic YIII analogues Y1–Y4 to assess their structural dynamics in solution (Fig. S15–S26†). Y1 exhibited sharp, fully resolved signals at 25 °C in the 1H-NMR spectra. Two methine (–CH(CH3)2) septets and four methyl (–CH(CH3)2) doublets were observed, consistent with restricted Cp–iPr bond rotation giving rise to separate methyl group (inner and outer CH3) environments.19 At 25 °C, Y4 displayed somewhat broadened peaks, which sharpened at –10 °C to three methyl (–CH(CH3)2) doublets (δ = 1.07, 1.66, 1.69) and one methine (–CH(CH3)2) septet. The two closely spaced downfield doublets probably correspond to the inner methyl group of the rac and meso isomers, while the upfield doublet likely corresponds to the outer methyl group of both isomers. Similar features have been associated with rac and meso isomers for [(CpiPr5)2M] (M = Ca, Sr, Ba).20 The 13C spectrum of Y4 also indicates rac and meso isomers, with four methyl (–CH(CH3)2) and two methine (–CH(CH3)2) peaks. The 1H spectra of Y2 and Y3 vary substantially with temperature. For Y2, one methyl singlet (Cp–CH3), two overlapping methine (–CH(CH3)2) septets and broadened peaks representing the four methyl (–CH(CH3)2) environments are observed at 25 °C, however, when heated to 75 °C, the four methyl (–CH(CH3)2) doublets are sharp and fully resolved. In the case of Y3 at 25 °C, sharp peaks are observed for the ethyl (Cp–CH2CH3) group and one of the methine (–CH(CH3)2) signals, while one methine (–CH(CH3)2) group is broadened and the methyl (–CH(CH3)2) peaks are observed as a single broad resonance. Heating to 85 °C results in sharp peaks for all groups, although three of the four methyl (–CH(CH3)2) doublets are overlapping.
The solid-state structures of the metallocene salts 1–4 were elucidated by single-crystal X-ray diffraction analysis (Fig. 1). Compounds 1 and 2 crystallize with two [Dy(CpiPr4R)2][B(C6F5)4] molecules in the asymmetric unit, while compounds 3 and 4 crystallize with only one molecule in the asymmetric unit. The structural analyses were complicated by pseudomerohedral twinning for 1 and 2 and positional disorder in 2 and 4. Two of the cyclopentadienyl ligands in the structure of 2 are disordered, each over two positions, and the DyIII center in the structure of 4 displays positional disorder over four sites (see ESI† for refinement details). Twinning and disorder were found to be consistent over several tested crystal samples.
Fig. 1. Structures of the metallocenium complexes in crystals of [Dy(CpiPr4R)2][B(C6F5)4] (R = H (1), Me (2), Et (3), iPr(4)). Green and gray spheres represent Dy and C atoms, respectively; hydrogen atoms, [B(C6F5)4]– counteranions, and positional disorder are omitted for clarity.

The Dy–Cp separations, as assessed by the distances from the Dy atom to the centroid of each cyclopentadienyl ligand, lie in the ranges 2.27(1)–2.30(1) Å for 1, 2.273(3)–2.382(3) Å for 2, 2.297(4)–2.306(4) Å for 3, and 2.245(15)–2.392(16) Å for 4. Because of the disorder present in each structure and the similarity in the resulting bond distances (within statistical error), it was not possible to derive a meaningful structural trend from these distances alone. Nonetheless, a trend can be drawn from the major component in each of the crystal structures: the average Dy–Cp(centroid) distances for the major component are 2.29(1), 2.298(5), 2.302(6), and 2.340(7) Å for 1–4, respectively (Table 1). Although the average Dy–Cp(centroid) distance does not vary by more than the statistical error in some adjacent members of the series (e.g., 1 and 2), the change is statistically significant across the series as a whole. Similarly, the average Dy–C distances for the major component are 2.587(5), 2.600(3), 2.596(2), and 2.625(3) Å for 1–4, respectively. The increase of both the average Dy–Cp(centroid) and Dy–C distances suggests elongation of the interaction between the Dy atom and the cyclopentadienyl rings as the series progresses from 1 to 4. In addition, the Cp–Dy–Cp angle increases across the series, lying in the ranges 146.5(8)–148.0(8), 155.9(3)–157.3(4), 161.1(2), and 157.8(2)–167.9(15)° in 1–4, respectively (Table 1). The angle between the planes formed by the cyclopentadienyl rings follows a similar trend with values in the range of 148.1–150.5, 162.3–162.8, 165.0, and 172.5° for 1–4, respectively. While the cyclopentadienyl rings in 4 are nearly parallel, the Dy atom moves off-center, leading to a reduced Cp–Dy–Cp angle. These structural trends are consistent with the expected steric environment imposed by the cyclopentadienyl ligands; that is, more sterically-encumbered substituents prompt the two cyclopentadienyl ligands in [Dy(CpiPr4R)2]+ to be positioned further away from each other, increasing linearity and lengthening the average Cp–Dy bond distance.
Table 1. Comparison of selected structural parameters and magnetic data for [Dy(CpiPr4R)2][B(C6F5)4] (R = H (1), Me (2), Et (3), iPr (4)).
| 1 | 2 | 3 | 4 | [(Cpttt)2Dy]+ e | |
| Cp–Dy–Cp a (°) | 147.2(8) | 156.6(3) | 161.1(2) | 162.1(7) | 152.70(7) |
| Dy–Cp b (Å) | 2.29(1) | 2.298(5) | 2.302(6) | 2.340(7) | 2.316(3) |
| U eff (cm–1) | 1285 | 1468 | 1380 | 1334 | 1223 or 1277 |
| T b c (K) | 17 | 62 | 59 | 56 | 53 |
| τ tunnel (s) | 439 | 2452 | 447 | 1187 | n/a |
| T max d (K) | 32 | 72 | 66 | 66 | 60 |
| 2 K Mr (µB) | 2.6 | 4.4 | 3.9 | 4.2 | 4.2 |
| 2 K Hc (T) | 1.4 | 2.4 | 1.4 | 1.8 | 2.4 |
aAverage value for all positions in the crystal structure.
bAverage value for the highest occupancy component in the crystal structure.
c T b is defined as the 100 s magnetic blocking temperature.
d T max is the maximum hysteresis temperature.
eValues are an average of those reported in ref. 5.
The solid-state structures of the yttrium metallocene salts Y1–Y4 were also investigated by single-crystal X-ray diffraction (Fig. S31–S34†). As in 1–4, positional disorder complicated the structural analyses. Compounds Y1 and Y2 crystalize with two molecules in the asymmetric unit; both the YIII center and one cyclopentadienyl ligand display positional disorder over two sites.
Similar structural trends to those seen in 1–4 are observed for the series of YIII congeners. Namely the average Cp–Y bond distance and Cp–Y–Cp angle generally increase across the series from Y1 to Y4. The average Y–Cp(centroid) distances for the major component are 2.273(10), 2.305(6), 2.294(3), and 2.331(2) Å for Y1–Y4, respectively, and the average Cp–Y–Cp angles are 145.7(5), 157.1(4), 161.3(1) and 180°. While the Dy atom in 4 moves off-center of the cyclopentadienyl rings, the Y atom in Y4 sits on an inversion center in the asymmetric unit, leading to a 180° Cp–Y–Cp angle. While both rac and meso isomers of Y4 are observed in the 1H- and 13C-NMR spectra, only the meso isomer is observed in the crystal structure.
In order to investigate the effect of the structural changes on the magnetic properties of each complex, dc magnetic susceptibility data were collected for 1–4 from 2 to 300 K under an applied magnetic field of 1 kOe (Fig. S35, S37, S39 and S41†). The room temperature χMT values for 2 and 3 are 14.17 and 14.28 emu K mol–1, respectively, agreeing well with the expected value of 14.17 emu K mol–1 for a free DyIII ion. The room temperature χMT values for 1 and 4 are slightly higher at 14.62 and 14.57 emu K mol–1, respectively, likely due to the presence of weak temperature-independent paramagnetism (TIP). In the presence of a stronger applied field, TIP should be diminished, and accordingly χMT decreases to 14.18 and 14.25 emu K mol–1 for 1 and 4, respectively, under a 10 kOe dc field (Fig. S36 and S42†). For all compounds, χMT gradually decreases as the temperature is lowered followed by a precipitous drop at low temperatures. This sudden decrease in χMT is indicative of magnetic blocking, wherein the magnetic moments of individual crystallites in the sample become pinned along a preferred axis and no longer respond to an external magnetic field.
Field-cooled and zero-field cooled measurements collected under an applied magnetic field of 1 kOe were compared for 1–4 (Fig. 2), as the temperature at which these measurements diverge (Tirrev) can provide an estimate of the magnetic blocking temperature.2 Divergence in these curves occurs at 28, 65, 60, and 60 K for 1 through 4, respectively. Such a large range in Tirrev provided an initial indication that the structural changes across this series induce major changes in the magnetic behavior. This result also suggested that compound 2 might exhibit a higher operating temperature than [Dy(Cpttt)2]+, which shows a divergence in field-cooled and zero-field cooled data at 61 K.5 However, Tirrev varies with both the applied magnetic field used to conduct the experiment and the sweep rate,2,21 and thus we turned to measurements of the magnetic relaxation time to more accurately determine the operating temperature.
Fig. 2. Field-cooled (red) and zero-field cooled (blue) magnetic susceptibility measurements collected at Hdc = 1000 Oe for 1–4 (left to right). Dashed lines mark Tirrev, the temperature at which the two plots diverge.

High-temperature magnetic relaxation in 1–4 was probed by ac magnetic susceptibility measurements from 70 to 114 K (Fig. 3, S43, S46 and S48†). Magnetic relaxation times, τ, were extracted from a simultaneous fit of in-phase (χ′) and out-of-phase (χ″) components of the magnetic susceptibility to a generalized Debye model (Fig. S44, S45, S47 and S49†). Despite the two molecules observed in the crystal structures of 1 and 2, and the positional disorder in 2 and 4, only one peak was observed in the ac susceptibility data for each compound. In this high temperature regime, Arrhenius plots of inverse temperature versus the natural log of τ are linear, indicative of a thermally-activated Orbach relaxation process (Fig. 4, red circles).22 All data could be fit to the equation τ–1 = τ0–1e–Ueff/kT, yielding thermal barriers to magnetic relaxation (Ueff) of 1285, 1468, 1380, and 1334 cm–1 for 1 through 4, respectively. These Ueff values surpass those reported for [Dy(Cpttt)2]+, 1277 or 1223 cm–1,5a,5b and the previous record of Ueff = 1260 cm–1 for [Dy(OtBu)2(C5H5N)5]+.3f
Fig. 3. In-phase (χ′M, top) and out-of-phase (χ″M, bottom) components of the ac magnetic susceptibility for 2 under zero applied dc field at frequencies ranging from 0.1–1500 Hz and temperatures from 80–114 K (2 K steps). The colored lines are guides for the eye.

Fig. 4. Arrhenius plots of magnetic relaxation time (τ) for 1–4 (left to right) extracted from ac susceptibility (red circles) and dc relaxation (blue circles) data. Black lines represent fits to the data as described in the main text and are each a sum of Orbach (dashed orange line), Raman (dashed purple line), and quantum tunneling (dashed green line) relaxation processes.

The values of Ueff for 1–4 can be rationalized through structural trends. The magnitude of Ueff should depend on both the metal–ligand bond distances and the geometry of the ligand field (i.e., axial or equatorial). Such correlations have been quantified via ab initio calculations for a hypothetical two-coordinate DyIII complex with alkyl ligands.23 In that study, it was found that U increased linearly with the L–M–L angle at low angles (90 to 150°), tapering its growth at high angles, and flattening above 170°. In contrast, U increased linearly as the M–L bond distance decreased across all bond lengths calculated.
In the series 1–4, the most bent metallocene is 1, with a Cp–Dy–Cp angle of 147.2(8)°. Complex 1 also shows the smallest thermal barrier to magnetic relaxation. Linearity increases by nearly 10° in 2 (156.6(3)°) and even further in 3 (161.1(2)°) and 4 (162.1(7)°), resulting in larger Ueff values for the high-angle set 2–4. Within these three compounds, Ueff should vary less substantially with bond angle, and instead bond distance should have the largest impact on the relaxation barrier. Indeed, Ueff increases upon going from 4 to 2, as the average Dy–C distance for the major disordered component in the crystal structure decreases.
This magneto-structural correlation is significant for the design of future DyIII metallocene cations. While steric bulk in the cyclopentadienyl ligand increases the linearity of the complex in 1–4, it also tends to promote longer Dy–C distances. These factors have opposing effects on the magnitude of Ueff, with the angle having a larger impact at low angles and the bond distance having a larger impact at high angles. Clearly, cyclopentadienyl ligands that can achieve a balance between promoting a large Cp–Dy–Cp angle and short Dy–C bond distances are worth pursuing.
Increasing the Cp–Dy–Cp angle and decreasing the Dy–C bond distances increases the axiality of the ligand field surrounding the DyIII ion in these metallocenium salts, resulting in larger thermal barriers to magnetic relaxation. This is consistent with electrostatic models that have been used to guide the synthesis of lanthanide-based single-molecule magnets.9 Studies on molecules of the type 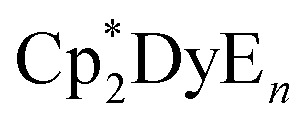 (E = equatorial ligand) have also found that increasing the axiality of the ligand field at DyIII results in larger thermal barriers, with weaker equatorial ligands leading to larger Ueff.24
(E = equatorial ligand) have also found that increasing the axiality of the ligand field at DyIII results in larger thermal barriers, with weaker equatorial ligands leading to larger Ueff.24
To probe magnetic relaxation behavior at lower temperatures, dc magnetic relaxation measurements were conducted for 1–4 from 2 to 64 K (Fig. 4, blue circles). Relaxation times were extracted from magnetization versus time plots by fitting the data with a stretched exponential function (for full details see the ESI, Fig. S50–S68 and Tables S3–S6†). The temperature dependence of τ at intermediate temperatures (20 to 64 K) is indicative of Raman relaxation, while at low temperatures the relaxation time tends toward a constant value, indicative of quantum tunneling of the magnetization. The full range of ac magnetic susceptibility and dc magnetic relaxation data could be fit to the equation τ–1 = τ0–1e–Ueff/kT + CTn + τtunnel–1 (Fig. 4 and Table 1).
Hundred-second magnetic blocking temperatures for 1–4 were extracted from the dc magnetic relaxation data, yielding Tb = 17, 62, 59, and 56 K for 1 through 4, respectively. The values for complexes 2–4 all surpass the 100 s blocking temperature of 53 K measured for [Dy(Cpttt)2]+.5 Indeed, the value for 2 represents the highest magnetic blocking temperature yet measured for a single-molecule magnet. While τ = 100 s has been chosen as the benchmark relaxation time for comparing blocking temperatures, 2 shows longer magnetic relaxation times than [Dy(Cpttt)2]+ across the entire temperature range measured (2–114 K).
Ab initio spin dynamics calculations performed on [Dy(Cpttt)2]+ suggest that magnetic relaxation is predominantly moderated by local molecular vibrations.5b In particular, four C–H bending modes of the Cpttt ligand were identified as giving rise to the initial MJ = 15/2 to 13/2 transition that leads to Orbach relaxation. These calculations led to the proposal that changing ring hydrogens in Cpttt to a different substituent could lead to changes in magnetic relaxation time, a hypothesis that has yet to be experimentally verified.5b Substituting the ring hydrogen in 1 with alkyl substituents in 2–4 results in up to a 45 K increase in the blocking temperature. This result clearly illustrates that substitution of the cyclopentadienyl ligands in dysprosium metallocene cations is a powerful tool for tuning magnetic relaxation and Tb.
While changes in ligand substitution modify molecular vibrations, molecular structure and anisotropy are also affected. It is therefore likely that a variety of factors influence changes in Tb. Clearly, spin dynamics calculations are worth pursuing to determine if the contributions of molecular vibrations to magnetic relaxation in this series can be generalized to other molecules in this class. It is worth noting that the 100 s blocking temperature for 1–4 occurs in the regime in which Raman relaxation dominates. While spin dynamics calculations can provide insight into the contribution of molecular vibrations to high-temperature, Orbach relaxation, models have not yet been developed for through-barrier relaxation processes such as Raman relaxation or quantum tunneling of the magnetization.5b As such, developing a more quantitative magneto-structural correlation for the operating temperature in this series will require further development of theories on the impact of molecular vibrations on through-barrier magnetic relaxation. A well-characterized series like 1–4 that varies only slightly in substitution, but substantially in molecular structure and magnetic relaxation could lay the experimental groundwork for such theoretical studies. Insights from these calculations could be significant to the design of new cyclopentadienyl ligands to maximize operating temperatures in DyIII metallocene cations.
The rates of quantum tunneling of the magnetization, τtunnel, extracted from dc relaxation measurements are among the slowest reported for mononuclear single-molecule magnets. This result is consistent with the drastically reduced quantum tunneling of the magnetization for [Dy(Cpttt)2]+.5 While the reduction of quantum tunneling in [Dy(Cpttt)2]+ has been ascribed to the high axiality of the ligand field (which gives rise to nearly pure MJ states in the lowest eight Kramers doublets5a), this axiality cannot fully account for the observed behavior. Other highly anisotropic mononuclear single-molecule magnets, such as [Dy(OtBu)2(C5H5N)5]+, possess nearly pure MJ ground doublets and show fast quantum tunneling of the magnetization.3f
Another possible explanation for the reduced quantum tunneling in [Dy(Cpttt)2]+ is the constrained metal–ligand vibrational modes.5b,25 A lack of low-energy, first-coordination sphere metal–ligand vibrations in [Dy(Cpttt)2]+—such as those created by the weakly bound, equatorial pyridine ligands in [Dy(OtBu)2(C5H5N)5]+—may limit the phonons available for magnetic relaxation at low temperatures. Limiting low-energy phonons has been shown to slow magnetic relaxation at low fields for Ho atoms on an MgO surface,26 as well as for TbPc2 (Pc2– = phthalocyanine dianion) molecules on carbon nanotubes.27 The hypothesis that local molecular coordination is responsible for limiting quantum tunneling in [Dy(Cpttt)2]+ is supported by an increase in the zero-field step in magnetic hysteresis measurements performed on solution samples, which differ from bulk samples in accessible metal–ligand vibrational modes.5b
The trend in τtunnel for 1–4 deviates from that observed for Tb; namely, compounds 1 and 3 show the fastest quantum tunneling of the magnetization, while 2 and 4 show much larger τtunnel values (Table 1). As the rate of quantum tunneling can be influenced by intermolecular dipole interactions, largely dictated by crystal packing,28 magnetically dilute samples of 1–4 were synthesized in order to enable a more accurate comparison of τtunnel.
Dilute samples 1–4@Y were prepared by co-crystallization of 1–4 in a 1 : 9 ratio with the analogous YIII compound Y1–Y4. Dc magnetic susceptibility measurements of the dilute samples matched those of 1–4 at temperatures above 50 K. Relaxation times extracted from dc magnetic relaxation measurements of 1–4@Y also matched those determined for 1–4 at the highest temperatures measured for each compound. This consistency suggests that the local molecular structure of 1–4—responsible for the high-temperature magnetic relaxation dynamics—is unchanged upon dilution, despite differences in the structures of Y1–Y4. The value of τtunnel, extracted from dc magnetic relaxation measurements of 1–4@Y at 2 K, differs from the undiluted samples, increasing to 1783, 11 200, 1125, and 1956 s, respectively. While the values of τtunnel for 1–4@Y are substantially larger than those determined for the undiluted samples, the same overall trend is observed for the series, with the exception that 1@Y shows slower tunneling than 3@Y.
The trend in τtunnel breaks from that observed for the 100 s blocking temperature. While 2@Y shows the slowest quantum tunneling, 1@Y shows a value of τtunnel close to that of 4@Y and larger than that observed for 3@Y. Substitution of the ring hydrogen in 1 with alkyl substituents in 2–4 does not have a consistent influence on the rate of quantum tunneling, as it did on the rate of Raman relaxation, which dictates Tb. This underscores the need to develop a deeper understanding of the role of molecular vibrations in through-barrier magnetic relaxation. Though molecular symmetry has been shown to influence magnetic relaxation in single-molecule magnets,29 there is no apparent magneto-structural correlation between molecular symmetry and quantum tunneling of the magnetization or blocking temperature in the series 1–4, which all possess C1 symmetry.
Magnetic hysteresis data were collected for 1–4 at several temperatures in the range 2–72 K by sweeping the field between 7 and –7 T. For fields below 2 T, a magnetic field sweep rate of 3.1(4) mT s–1 was employed, similar to that used in hysteresis measurements of [Dy(Cpttt)2]+.5 A step is observed at zero field at 2 K, resulting in remanent magnetization values (Mr) of 2.6, 4.4, 3.9, and 4.2 µB for complexes 1 through 4. The magnitude of the remanent magnetization and the coercive field (Hc) for each complex mirrors the trend in τtunnel (Fig. 5a, Table 1).
Fig. 5. (a) Magnetic hysteresis data for 1–4 collected at 2, 10, 20 and 30 K with a sweep rate of 3.1(4) mT s–1 for H < 2 T and 13.2(2) mT s–1 for H > 2 T. (b) Expanded view of the region near zero field at high-temperatures.

The hysteresis loops remain open at temperatures as high as 32, 72, 66 and 66 K for 1 through 4, respectively (Fig. 5b). The maximum temperatures in the case of complexes 2–4 are higher than the 60 K maximum determined for [Dy(Cpttt)2]+,5 and this observation is consistent with the trend observed in Tb. Notably, the maximum hysteresis temperature for compound 2 is the highest that has yet been reported for a single-molecule magnet, approaching the temperature of liquid nitrogen (77 K).
Conclusions
The metallocene cation salts [Dy(CpiPr4R)2][B(C6F5)4] (R = H (1), Me (2), Et (3), iPr (4)) were synthesized by metathesis of DyI3 and the corresponding NaCpiPr4R salt, followed by iodide abstraction with [H(SiEt3)2][B(C6F5)4]. Thorough characterization of this series provides insight into the impact of molecular structure on magnetic properties in DyIII metallocenium complexes. Single-crystal X-ray diffraction studies reveal that more sterically encumbered cyclopentadienyl ligands promote longer average Dy–C distances and larger Cp–Dy–Cp angles. As demonstrated by the record thermal barrier to magnetic inversion of 1468 cm–1 determined for compound 2, a balance must be struck between achieving a large Cp–Dy–Cp angle and short Dy–C distances in order to maximize anisotropy.
Compounds 1–4 provide experimental support for the hypothesis that modifying cyclopentadienyl ring substituents can tune metal–ligand vibrational modes, impacting magnetic relaxation.5b In the series investigated here, substituent modification notably results in a 45 K increase in the magnetic blocking temperature across the series, with complex 2 exhibiting the highest 100 s blocking temperature and hysteresis temperature yet measured for a single molecule magnet. Expanding the magneto-structural correlations presented in this study with ab initio calculations is a clear next step towards developing generalizable design principles for the synthesis of [Dy(CpX)2]+ single molecule magnets that function well above 77 K.
A report of a dysprosium(iii) metallocenium salt, [(CpiPr5)(Cp*)Dy][B(C6F5)4], showing a 100 s magnetic blocking temperature of 65 K and open magnetic hysteresis loops as high as 80 K was published on the same date as this work. The magneto-structural correlations outlined here can help to explain the magnetic properties observed in [(CpiPr5)(Cp*)Dy][B(C6F5)4]: the sterically encumbered CpiPr5 ligand promotes a high Cp–Dy–Cp angle of 162.5°, while the less-hindered Cp* allows for short Dy–Cp(centroid) bond distances of 2.296(1) and 2.284(1) Å for the CpiPr5 and Cp* ligand, respectively. The combination of a large Cp–Dy–Cp angle and short Dy–Cp bond distances enables a large thermal barrier to magnetic relaxation of Ueff = 1541 cm–1 and a 100 s magnetic blocking temperature 3 K higher than that observed for 2.30
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This work was supported by the Naval Air Warfare Center, Weapons Division (NAWCWD) PL-219 program and NSF grant CHE-1800252. We thank the National Science Foundation Graduate Research Fellowship Program for support of CAG. Single-crystal X-ray diffraction data for 2–4 were collected at Beamline 12.2.1 at the Advanced Light Source. This research used resources of the Advanced Light Source, which is a DOE Office of Science User Facility under contract no. DE-AC02-05CH11231. BGH and KRM would like to thank Satoshi Yamashita (Research Center for Structural Thermodynamics, Osaka University) for conducting initial magnetic measurements on compound 4, and Professor Joel Miller (University of Utah) for helpful discussions. We also thank Dr Lawrence C. Baldwin for NMR assistance, Alicia M. Hughes for GC-MS, Gregory Ostrom for metals analysis and Dr Katie R. Meihaus for editorial assistance.
Footnotes
†Electronic supplementary information (ESI) available: IR and NMR spectroscopy, synthesis of Y1–Y4, and magnetic characterization. Structures of compounds 1–4 are available under CCDC 1865386–1865389 and compounds Y1–Y4 under CCDC 1873921–1873924. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc03907k
References
- (a) Natterer F. D., Yang K., Paul W., Willke P., Choi T., Greber T., Heinrich A. J., Lutz C. P. Nature. 2017;543:226. doi: 10.1038/nature21371. [DOI] [PubMed] [Google Scholar]; (b) Leuenberger M. N., Loss D. Nature. 2001;410:789. doi: 10.1038/35071024. [DOI] [PubMed] [Google Scholar]; (c) Ardavan A., Rival O., Morton J. J., Blundell S. J., Tyryshkin A. M., Timco G. A., Winpenny R. E. P. Phys. Rev. Lett. 2007;98:057201. doi: 10.1103/PhysRevLett.98.057201. [DOI] [PubMed] [Google Scholar]; (d) Mannini M., Pineider F., Danieli C., Totti F., Sorace L., Sainctavit P., Arrio M. A., Otero E., Joly L., Cezar J. C., Cornia A., Sessoli R. Nature. 2010;468:417. doi: 10.1038/nature09478. [DOI] [PubMed] [Google Scholar]; (e) Urdampilleta M., Nguyen N. V., Cleuziou J. P., Klyatskaya S., Ruben M., Wernsdorfer W. Int. J. Mol. Sci. 2011;12:6656. doi: 10.3390/ijms12106656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatteschi D., Sessoli R. and Villain J., Molecular Nanomagnets, Oxford University Press, Oxford, 2006. [Google Scholar]
- (a) Ishikawa N., Sugita M., Ishikawa T., Koshihara S.-Y., Kaizu Y. J. Am. Chem. Soc. 2003;125:8694. doi: 10.1021/ja029629n. [DOI] [PubMed] [Google Scholar]; (b) Chen Y.-C., Liu J.-L., Ungur L., Liu J., Li Q.-W., Wang L. F., Ni Z.-P., Chibotaru L. F., Chen X.-M., Tong M.-L. J. Am. Chem. Soc. 2016;138:2829. doi: 10.1021/jacs.5b13584. [DOI] [PubMed] [Google Scholar]; (c) Gupta S. K., Rajeshkumar T., Rajaraman G., Murugavel R. Chem. Sci. 2016;7:5181. doi: 10.1039/c6sc00279j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Gregson M., Chilton N. F., Ariciu A.-M., Tuna F., Crowe I. F., Lewis W., Blake A. J., Collison D., McInnes E. J. L., Winpenny R. E. P., Liddle S. T. Chem. Sci. 2016;7:155. doi: 10.1039/c5sc03111g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Liu J., Chen Y.-C., Liu J.-L., Vieru V., Ungur L., Jia J.-H., Chibotaru L. F., Lan Y., Wernsdorfer W., Gao S., Chen X.-M., Tong M.-L. J. Am. Chem. Soc. 2016;138:5441. doi: 10.1021/jacs.6b02638. [DOI] [PubMed] [Google Scholar]; (f) Ding Y.-S., Chilton N. F., Winpenny R. E. P., Zheng Y.-Z. Angew. Chem., Int. Ed. 2016;55:16071. doi: 10.1002/anie.201609685. [DOI] [PubMed] [Google Scholar]; (g) Blagg R. J., Ungur L., Tuna F., Speak J., Comar P., Collison D., Wernsdorfer W., McInnes E. J. L., Chibotaru L. F., Winpenny R. E. P. Nat. Chem. 2013;5:673. doi: 10.1038/nchem.1707. [DOI] [PubMed] [Google Scholar]; (h) Meng Y.-S., Xu L., Xiong J., Yuan Q., Liu T., Wang B.-W., Gao S. Angew. Chem., Int. Ed. 2018;57:4673. doi: 10.1002/anie.201801223. [DOI] [PubMed] [Google Scholar]; (i) Woodruff D. N., Winpenny R. E. P., Layfield R. A. Chem. Rev. 2013;113:5110. doi: 10.1021/cr400018q. [DOI] [PubMed] [Google Scholar]
- (a) Rinehart J. D., Fang M., Evans W. J., Long J. R. Nat. Chem. 2011;3:538. doi: 10.1038/nchem.1063. [DOI] [PubMed] [Google Scholar]; (b) Rinehart J. D., Fang M., Evans W. J., Long J. R. J. Am. Chem. Soc. 2011;133:14236. doi: 10.1021/ja206286h. [DOI] [PubMed] [Google Scholar]; (c) Demir S., Gonzalez M. I., Darago L. E., Evans W. J., Long J. R. Nat. Commun. 2017;8:2144. doi: 10.1038/s41467-017-01553-w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Demir S., Zadrozny J. M., Nippe M., Long J. R. J. Am. Chem. Soc. 2012;134:18546. doi: 10.1021/ja308945d. [DOI] [PubMed] [Google Scholar]; (e) Demir S., Jeon I.-R., Long J. R., Harris T. D. Coord. Chem. Rev. 2015;289:149. [Google Scholar]
- (a) Guo F.-S., Day B. M., Chen Y.-C., Tong M.-L., Mansikkamäki A., Layfield R. A. Angew. Chem., Int. Ed. 2017;56:11445. doi: 10.1002/anie.201705426. [DOI] [PubMed] [Google Scholar]; (b) Goodwin C. A. P., Ortu F., Reta D., Chilton N. F., Mills D. P. Nature. 2017;548:439. doi: 10.1038/nature23447. [DOI] [PubMed] [Google Scholar]
- (a) Pedersen K. S., Dreiser J., Weihe H., Sibille R., Johannesen H. V., Sørensen M. A., Nielsen B. E., Sigrist M., Mutka H., Rols S., Bendix J., Piligkos S. Inorg. Chem. 2015;54:7600. doi: 10.1021/acs.inorgchem.5b01209. [DOI] [PubMed] [Google Scholar]; (b) Liddle S. T., van Slageren J. Chem. Soc. Rev. 2015;44:6655. doi: 10.1039/c5cs00222b. [DOI] [PubMed] [Google Scholar]; (c) Ding Y.-S., Yu K.-Y., Reta D., Ortu F., Winpenny R. E. P., Zheng Y.-Z., Chilton N. F. Nat. Commun. 2018;9:3134. doi: 10.1038/s41467-018-05587-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Another notable exception is [Er(COT)2]–, which shows open magnetic hysteresis loops to 10 K: Meihaus K. R., Long J. R., J. Am. Chem. Soc., 2013, 135 , 17952 . [DOI] [PubMed] [Google Scholar]
- For studies on the influence of molecular structure on the magnetic properties of other classes of lanthanide sandwich complexes, see: ; (a) Jiang S.-D., Wang B.-W., Sun H.-L., Wang Z.-M., Gao S. J. Am. Chem. Soc. 2011;133:4730. doi: 10.1021/ja200198v. [DOI] [PubMed] [Google Scholar]; (b) Jiang S.-D., Liu S.-S., Zhou L.-N., Wang B.-W., Wang Z.-M., Gao S. Inorg. Chem. 2012;51:3079. doi: 10.1021/ic202511n. [DOI] [PubMed] [Google Scholar]; (c) Latendresse T. P., Bhuvanesh N. S., Nippe M. J. Am. Chem. Soc. 2017;139:8058. doi: 10.1021/jacs.7b01499. [DOI] [PubMed] [Google Scholar]; (d) Latendresse T. P., Vieru V., Wilkins B. O., Bhuvanesh N. S., Chibotaru L. F., Nippe M. Angew. Chem., Int. Ed. 2018;57:8164. doi: 10.1002/anie.201804075. [DOI] [PubMed] [Google Scholar]; (e) Harriman K. L. M., Korobkov I., Murugesu M. Organometallics. 2017;36:4515. [Google Scholar]; (f) Ungur L., LeRoy J. J., Korobkov I., Murugesu M., Chibotaru L. F. Angew. Chem., Int. Ed. 2014;53:4413. doi: 10.1002/anie.201310451. [DOI] [PubMed] [Google Scholar]
- (a) Rinehart J. D., Long J. R. Chem. Sci. 2011;2:2078. [Google Scholar]; (b) Chilton N. F., Collison D., McInnes E. J. L., Winpenny R. E. P., Soncini A. Nat. Commun. 2013;4:2551. doi: 10.1038/ncomms3551. [DOI] [PubMed] [Google Scholar]; (c) Sievers J. Z. Z. Phys. B: Condens. Matter, Phys. B. 1982;45:289. [Google Scholar]; (d) Ungur L., Chibotaru L. F. Phys. Chem. Chem. Phys. 2011;13:20086. doi: 10.1039/c1cp22689d. [DOI] [PubMed] [Google Scholar]; (e) Chilton N. F., Goodwin C. A. P., Mills D. P., Winpenny R. E. P. Chem. Commun. 2015;51:101. doi: 10.1039/c4cc08312a. [DOI] [PubMed] [Google Scholar]
- Lunghi A., Totti F., Sessoli R., Sanvito S. Nat. Commun. 2017;8:14620. doi: 10.1038/ncomms14620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weismann D., Saurenz D., Boese R., Blaser D., Wolmershauser G., Sun Y., Sitzmann H. Organometallics. 2011;30:6351. [Google Scholar]
- Dezember T., Sitzmann H. Z. Naturforsch. B. 1997;52:911. [Google Scholar]
- Sitzmann H. Z. Naturforsch. B. 1989;44:1293. [Google Scholar]
- Evans W. J., Forrestal K. J., Leman J. T., Ziller J. W. Organometallics. 1996;15:527. [Google Scholar]
- Jaroschik F., Nief F., Le Goff X. F., Ricard L. Organometallics. 2007;26:1123. [Google Scholar]
- Coles M. P., Hitchcock P. B., Lappert M. F., Protchenko A. V. Organometallics. 2012;31:2682. [Google Scholar]
- Schumann H., Glanz M., Hemling H., Gorlitz F. H. J. Organomet. Chem. 1993;462:155. [Google Scholar]
- Connelly S. J., Kaminsky W., Heinekey D. M. Organometallics. 2013;32:7478. [Google Scholar]
- (a) Burkey D. J., Hanusa T. P. Organometallics. 1995;14:11. [Google Scholar]; (b) Burkey D. J., Hays M. L., Duderstadt R. E., Hanusa T. P. Organometallics. 1997;16:1465. [Google Scholar]
- Sitzmann H., Dezember T., Ruck M. Angew. Chem., Int. Ed. 1998;37:3113. doi: 10.1002/(SICI)1521-3773(19981204)37:22<3113::AID-ANIE3113>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Bar A. K., Kalita P., Singh M. K., Rajaraman G., Chandrasekhar V. Coord. Chem. Rev. 2018;367:163. [Google Scholar]
- Finn C. B., Orbach R., Wolf W. P. Proc. Phys. Soc. 1961;77:261. [Google Scholar]
- Chilton N. F. Inorg. Chem. 2015;54:2097. doi: 10.1021/acs.inorgchem.5b00089. [DOI] [PubMed] [Google Scholar]
- (a) Meng Y.-S., Zhang Y.-Q., Wang Z.-M., Wang B.-W., Gao S. Chem.–Eur. J. 2016;22:12724. doi: 10.1002/chem.201601934. [DOI] [PubMed] [Google Scholar]; (b) Day B. M., Guo F.-S., Layfield R. A. Acc. Chem. Res. 2018;51:1880. doi: 10.1021/acs.accounts.8b00270. [DOI] [PubMed] [Google Scholar]; (c) Pugh T., Chilton N. F., Layfield R. A. Angew. Chem., Int. Ed. 2016;55:11082. doi: 10.1002/anie.201604346. [DOI] [PubMed] [Google Scholar]
- (a) Escalera-Moreno L., Baldoví J. J., Gaita-Ariño A., Coronado E. Chem. Sci. 2018;9:3265. doi: 10.1039/c7sc05464e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ortu F., Reta D., Ding Y.-S., Goodwin C. A. P., Gregson M. P., McInnes E. J. L., Winpenny R. E. P., Zheng Y.-Z., Liddle S. T., Mills D. P., Chilton N. F., ChemRxiv, 2018. 10.26434/chemrxiv.6790568.v1 , , preprint . [Google Scholar]
- Donati F., Rusponi S., Stepanow S., Wäckerlin C., Singha A., Persichetti L., Baltic R., Diller K., Patthey F., Fernandes E., Dreiser J., Šljivančanin ž., Kummer K., Nistor C., Gambardella P., Brune H. Science. 2016;352:318. doi: 10.1126/science.aad9898. [DOI] [PubMed] [Google Scholar]
- Ganzhorn M., Klyatskaya S., Ruben M., Wernsdorfer W. Nat. Nanotechnol. 2013;8:165. doi: 10.1038/nnano.2012.258. [DOI] [PubMed] [Google Scholar]
- (a) Garanin D. A., Chudnovsky E. M. Phys. Rev. B: Condens. Matter Mater. Phys. 1997;56:11102. [Google Scholar]; (b) Leuenberger M. N., Loss D. Phys. Rev. B: Condens. Matter Mater. Phys. 2000;61:1286. [Google Scholar]
- (a) Liu J.-L., Chen Y.-C., Tong M.-L. Chem. Soc. Rev. 2018;47:2431. doi: 10.1039/c7cs00266a. [DOI] [PubMed] [Google Scholar]; (b) Sørensen M. A., Hansen U. B., Perfetti M., Pedersen K. S., Bartolomé E., Simeoni G. G., Mutka H., Rols S., Jeong M., Zivkovic I., Retuerto M., Arauzo A., Bartolomé J., Piligkos S., Weihe H., Doerrer L. H., van Slageren J., Rønnow H. M., Lefmann K., Bendix J. Nat. Commun. 2018;9:1292. doi: 10.1038/s41467-018-03706-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F.-S., Day B. M., Chen Y.-C., Tong M.-L., Mansikkamäki A., Layfield R. A. Science. 2018 doi: 10.1126/science.aav0652. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.