

A major goal for improving tuberculosis therapy is to identify drug regimens with improved efficacy and shorter treatment durations. Shorter therapies improve patient adherence to the antibiotic regimens, which, in turn, decreases resistance emergence.
KEYWORDS: moxifloxacin, Mycobacterium tuberculosis, pharmacodynamics
ABSTRACT
A major goal for improving tuberculosis therapy is to identify drug regimens with improved efficacy and shorter treatment durations. Shorter therapies improve patient adherence to the antibiotic regimens, which, in turn, decreases resistance emergence. Mycobacterium tuberculosis exists in multiple metabolic states. At the initiation of therapy, the bulk of the population is in log-phase growth. Consequently, it is logical to focus initial therapy on those organisms. Moxifloxacin has good early bactericidal activity against log-phase bacteria and is a logical component of initial therapy. It would be optimal if this agent also possessed activity against acid-phase and nonreplicative-persister (NRP) phenotype organisms. In our hollow-fiber infection model, we studied multiple exposures to moxifloxacin (equivalent to 200 mg to 800 mg daily) against strain H37Rv in the acid phase and against strain 18b in streptomycin starvation, which is a model for NRP-phase organisms. Moxifloxacin possesses good activity against acid-phase organisms, generating cell killing of 3.75 log10(CFU/ml) (200 mg daily) to 5.16 log10(CFU/ml) (800 mg daily) over the 28 days of the experiment. Moxifloxacin also has activity against streptomycin-starved strain 18b. The 400- to 800-mg daily regimens achieved extinction at day 28, while the no-treatment control still had 1.96 log10(CFU/ml) culturable. The lowest dose (200 mg daily) still had 0.7 log10(CFU/ml) measurable at day 28, a net kill of 1.26 log10(CFU/ml). Moxifloxacin is an attractive agent for early therapy, because it possesses activity against three metabolic states of M. tuberculosis.
INTRODUCTION
Mycobacterium tuberculosis infects 25% of the world’s population. In 2016, there were 10.4 million new cases of tuberculosis, which caused 1.7 million deaths (1). We have also seen the rise of multiple-drug-resistant (MDR) M. tuberculosis and extremely drug-resistant (XDR) M. tuberculosis (2). Such isolates are associated with markedly increased costs of therapy as well as increased rates of death (3).
Resistance emergence is attributable to multiple causes, but the prolonged duration of therapy makes attaining excellent adherence difficult, with a subsequent increase in the rate of resistance emergence among poorly adherent patients. Attainment of inadequate drug concentration-time profiles also has a negative impact on resistance emergence (4). Yet another factor influencing the likelihood of resistance emergence is the bacterial burden, with very high burdens being associated with increasing likelihoods of resistance emergence. Finally, all these and other factors interact with each other to inflate the rate of resistance emergence.
M. tuberculosis exists in multiple metabolic states. Some of the best characterized are log-phase growth, acid-phase growth, and the nonreplicative-persister (NRP) phenotype state. Bacterial burden interacts with these states. It is thought that slowly growing acid-phase M. tuberculosis and very slowly growing or nongrowing organisms in the NRP state are hardest to kill (5, 6) and contribute substantially to the long duration of therapy required to cure tuberculosis. Larger overall burdens consist mostly of log-phase organisms, but larger burdens also drive larger acid-phase and NRP-phase populations. Furthermore, resistance occurs mostly (but not exclusively) in log-phase populations. At therapy initiation, it would be optimal to employ a regimen with excellent bactericidal activity against log-phase organisms but also substantial activity against acid-phase and NRP-phase organism populations. We decided to examine moxifloxacin as one of the agents for initial therapy. Our group examined this fluoroquinolone previously in the hollow-fiber infection model (HFIM) (7–9). In those evaluations, it was clear that moxifloxacin possessed excellent early killing activity. Indeed, in the REMox trial (10), the arm that replaced isoniazid with moxifloxacin cleared the sputum significantly faster than the control arm. Consequently, this agent is attractive for inclusion in the regimen at therapy initiation; it would be even more attractive if it also possessed activity against acid-phase organisms and NRP-phase organisms. It was the purpose of these studies to examine the activity of moxifloxacin against M. tuberculosis in those metabolic states.
RESULTS
MICs and resistance mutational frequency.
The resistance mutational frequency was 1/106.78 CFU for H37Rv in the acid phase and 1/105.87 CFU for strain 18b in streptomycin starvation (ss-18b) (NRP phase). The selection plates contained 75 mg/liter streptomycin to allow organism growth. Henceforth, we refer to NRP-phase organisms as ss-18b; this isolate is a streptomycin auxotroph (11). The moxifloxacin MICs were 0.25 mg/liter for acid-phase H37Rv and 0.125 mg/liter for strain 18b grown in the presence of streptomycin. The MIC for the ss-18b state could not be determined.
Moxifloxacin activity against acid-phase M. tuberculosis H37Rv.
The activities of 200, 400, 600, and 800 mg exposure equivalents of moxifloxacin administered daily are shown in Fig. 1. The reduction of bacterial burden from baseline (day zero) to day 28 ranged from 3.75 log10(CFU/ml) (200 mg daily) to 5.16 log10(CFU/ml) (800 mg daily).
FIG 1.

Activity of four daily doses of moxifloxacin (MOX), plus a no-treatment control, against acid-phase (AP) Mycobacterium tuberculosis H37Rv examined in a HFIM over 28 days.
The effect of each moxifloxacin regimen on the subpopulation of M. tuberculosis with reduced susceptibilities to moxifloxacin was assessed by quantitatively culturing bacterial suspensions collected from the HFIM on agar supplemented with 3.0× MIC of moxifloxacin. No less-susceptible isolates were recovered from any arm at any time point. This is consistent with the resistance mutational frequency (1/106.78 CFU) and the initial bacterial burdens [5.56 to 5.86 log10(CFU/ml)].
Mathematical modeling of acid-phase study data.
Both system outputs (drug concentration-time profile and total bacterial burden) were modeled simultaneously for all regimens, using a variant of the model we described previously (12). As in that publication, we included a natural death rate term (Knat-death, in hours−1) because of the overall lack of increase in the bacterial burden over time in the untreated control.
The fit of the model to the data was acceptable when the predicted-observed plots and measures of bias and precision were examined. The pre-Bayesian (population) and Bayesian (individual) regressions are shown for both outputs in Fig. 2. For the pre-Bayesian regression, the mean or median parameter vector was used to generate the predicted values in the predicted-observed plot. For the Bayesian analysis, the Bayesian parameter estimates for each hollow-fiber arm were used to generate the predicted values. It was expected that the Bayesian estimates would perform better, in terms of measured bias and precision, than the pre-Bayesian estimates.
FIG 2.

Predicted-observed plots for drug (moxifloxacin) concentrations (upper) and total bacterial burdens (lower) for Mycobacterium tuberculosis H37Rv, from the pre-Bayesian regression (left) and the Bayesian regression (right).
The mean and median parameter vectors and their standard deviations are displayed in Table 1. When the Bayesian model parameters were employed for simulation, the bacterial kill attributable to 800 mg of moxifloxacin daily was 4.940 log10(CFU/ml), relative to the no-treatment control.
TABLE 1.
Mean, median, and standard deviation estimates for parameter values for the effect of moxifloxacin on acid-phase H37Rv
| Parametera | Mean | Median | Standard deviation |
|---|---|---|---|
| V (liters) | 338.8 | 351.3 | 22.44 |
| CL (liter/h) | 19.63 | 19.75 | 0.0506 |
| Kg (h−1) | 0.0350 | 0.0204 | 0.0279 |
| Kkill (h−1) | 0.677 | 0.301 | 0.430 |
| C50 (mg/liter) | 4.87 | 4.97 | 0.237 |
| Hb | 4.65 | 4.39 | 0.436 |
| POPMAX (CFU/ml) | 1.00 × 107 | 1.04 × 107 | 2.92 × 103 |
| Knat-death (h−1) | 0.0439 | 0.0321 | 0.0225 |
| IC2 (CFU/ml) | 3.00 × 105 | 3.01 × 105 | 4.68 |
V, volume of distribution; CL, clearance; Kg, first-order growth rate constant; Kkill, first-order kill rate constant; C50, moxifloxacin concentration at which the kill rate is half-maximal; H, Hill constant; POPMAX, maximal bacterial concentration; Knat-death, first-order kill rate constant independent of moxifloxacin concentration; IC2, initial condition of M. tuberculosis concentration.
Unitless.
In Table 1, the growth rate constant (Kg) values from both the mean and median parameter vectors are low (0.0350 and 0.0204 h−1, respectively), indicating the slow growth of M. tuberculosis in this metabolic state. The kill rate constant (Kkill) estimates are 10 to 20 times the Kg estimates. Finally, it is of note that the Knat-death estimates are of the same magnitude as the Kg estimates, which explains the overall lack of growth in the no-treatment control over the 28 days of the experiment.
Activity against ss-18b M. tuberculosis.
The activity of 200, 400, 600, and 800-mg exposure equivalents of moxifloxacin administered once daily is shown in Fig. 3. The 400, 600, and 800mg daily doses mediated killing of 1.96 log10(CFU/ml) at day 28, relative to the no-treatment control, when eradication was achieved. The decline in the untreated control is due to the substantial natural death rate for M. tuberculosis in this metabolic state in the HFIM. The substantial decline in the no-treatment control was greater than that seen previously in static in vitro experiments (13) but was consistent with previous experiments in the HFIM in which the restricted replication rate was induced by Wayne-Hayes level II anaerobiosis (8).
FIG 3.

Activity of four daily doses of moxifloxacin (MOX), plus a no-treatment control, against NRP-phenotype Mycobacterium tuberculosis 18b (streptomycin-starved) examined in a HFIM over 28 days.
The effect of each moxifloxacin regimen on the less-susceptible M. tuberculosis subpopulation was determined by quantitatively culturing an aliquot of the bacterial suspensions on agar supplemented with 3.0× MIC of moxifloxacin. All arms had some less-moxifloxacin-susceptible isolates recovered at baseline; this is a function of the baseline bacterial burden [6.47 to 6.65 log10(CFU/ml)] relative to the inverse of the resistance mutational frequency [1/105.87 log10(CFU)]. This is consistent with the observed recovery of 0.6 to 0.72 log10(CFU/ml) of less-susceptible organisms at baseline. We were unable to recover resistant organisms from all arms after 2 to 4 days. This is also consistent with the resistance mutational frequency and the decline in the no-treatment control over this time frame [6.53 log10(CFU/ml) at baseline and 5.62 log10(CFU/ml) at day 4]. Isolates recovered from the antimicrobial-containing plates had elevated moxifloxacin MIC values (8- to 32-fold increases).
Mathematical modeling of NRP study data.
We modeled all system outputs simultaneously for all regimens, using an expansion of the model we described previously (12). A natural death rate term (Knat-death) was included in the model because of the lack of bacterial burden increases over time in the untreated controls. In contrast to the acid-phase analysis, we were able to recover less-susceptible isolates at baseline in this experiment; therefore, we added another differential equation (and system output) to the model.
The model fit to the data was acceptable when the predicted-observed plots and measures of bias and precision were examined. The pre-Bayesian (population) and Bayesian (individual) regressions are shown for all outputs in Fig. 4.
FIG 4.

Predicted-observed plots for drug (moxifloxacin) concentrations (upper), total bacterial burdens (middle), and resistant colony counts (lower) for Mycobacterium tuberculosis 18b, from the pre-Bayesian regression (left) and the Bayesian regression (right).
The mean and median parameter vectors plus the standard deviations are displayed in Table 2. Employing the Bayesian model parameters allowed us to calculate that the bacterial kill for ss-18b attributable to 800 mg of moxifloxacin daily was 2.98 log10(CFU/ml), relative to the no-treatment control [the observed data indicated a bacterial cell kill of 1.96 log10(CFU/ml)].
TABLE 2.
Mean, median, and standard deviation estimates for parameter values for the effect of moxifloxacin on NRP-phase 18b
| Parametera | Mean | Median | Standard deviation |
|---|---|---|---|
| V (liters) | 359.4 | 366.6 | 34.37 |
| CL (liters/h) | 19.96 | 20.01 | 0.7932 |
| Kg-s (h−1) | 0.1310 | 0.1663 | 0.04998 |
| Kg-r (h−1) | 0.1286 | 0.1268 | 0.01349 |
| Kkill-s (h−1) | 1.786 | 1.728 | 0.2493 |
| Kkill-r (h−1) | 0.9252 | 1.098 | 0.6398 |
| C50-s (mg/liter) | 12.19 | 12.98 | 1.924 |
| C50-r (mg/liter) | 12.85 | 12.52 | 0.9446 |
| Hsb | 13.08 | 11.33 | 3.876 |
| Hrb | 12.94 | 18.76 | 7.880 |
| POPMAX (CFU/ml) | 2.357 × 108 | 1.492 × 107 | 2.760 × 108 |
| Knat-death-s (h−1) | 0.1512 | 0.1876 | 0.04798 |
| Knat-death-r (h−1) | 0.1585 | 0.1656 | 0.01431 |
| IC2 (CFU/ml) | 3.131 × 106 | 3.024 × 106 | 1.980 × 105 |
| IC3 (CFU/ml) | 7.431 | 6.085 | 2.165 |
V, volume of distribution; CL, clearance; s, susceptible subpopulation; r, resistant subpopulation; Kg, first-order growth rate constant; Kkill, first-order kill rate constant; C50, moxifloxacin concentration at which the kill rate is half-maximal; H, Hill constant; POPMAX, maximal bacterial concentration; Knat-death, first-order kill rate constant independent of moxifloxacin concentration; IC2, initial condition of total M. tuberculosis population; IC3, initial condition of resistant M. tuberculosis population.
Unitless.
The estimates of the growth rate constants for the susceptible and resistant subpopulations (Kgs and Kgr, respectively) were similar for both the mean and median parameter vectors, indicating only a minor loss of biofitness due to the resistance mechanism. The estimates of the kill rate constants did differ between the susceptible and resistant subpopulations, with the resistant subpopulation estimates being about 50 to 60% of the estimates for the susceptible subpopulation. Finally, the Knat-death values for the susceptible and resistant subpopulations were virtually identical, indicating that the physiological state did not meaningfully interact with drug susceptibility. This physiological state had a Knat-death value that was 3.5 to 6.0 times that of the acid phase, which was the explanation for the substantial decline in the no-treatment control over time.
DISCUSSION
It is critical to shorten the duration of therapy for Mycobacterium tuberculosis if we are to make progress in controlling this global infectious threat. Long durations of therapy lead to poor adherence, which, in turn, contributes to resistance. Shortening therapy should have a salutary impact on both adherence and the rate of emergence of resistance. Since MDR and XDR strains are generated by the emergence of resistance, shortening therapy would help control these more difficult-to-treat infections, save lives, and decrease costs.
In order to decrease the duration of therapy, we need to design new regimens that have substantial killing activity against multiple metabolic states of M. tuberculosis. While slowly growing or poorly growing to nongrowing subpopulations are thought to be the main roadblock to shorter therapy durations, it should be realized that, at the beginning of therapy, the greatest proportion of the total population is in log-phase growth. Hence, it is rational that, early in therapy, the choice of agents should be directed mainly at quickly reducing the log-phase population. Furthermore, resistance emergence is generated largely from this population because, after selection of a resistant clone, amplification of the clone depends on continued turnover.
Moxifloxacin is an agent with excellent bacterial killing activity against the log-phase population. We previously studied moxifloxacin against log-phase M. tuberculosis (8). In Fig. 5, we show that moxifloxacin exposures equivalent to doses of 100 mg to 800 mg daily in a HFIM generated declines in the log-phase population of about 2.5 to 3.5 log10(CFU/ml) over the first week of therapy. Consequently, we can conclude that this agent is a promising option for early inclusion in therapeutic regimens, to decrease organism loads rapidly. This has been directly demonstrated clinically (10), with the moxifloxacin-containing arm clearing sputum significantly faster. It should be noted that all exposures tested with moxifloxacin monotherapy demonstrated the emergence of resistance, even with the 800mg equivalent exposure daily, emphasizing that combination therapy is needed to suppress resistance emergence.
FIG 5.
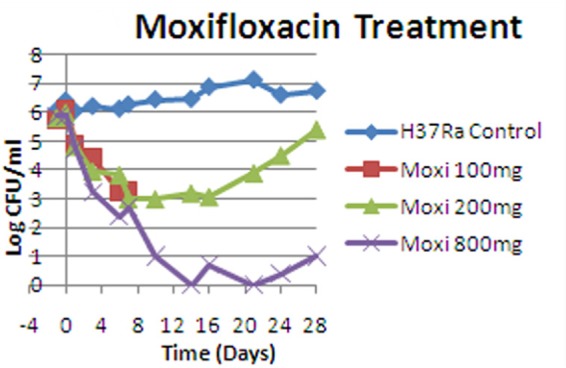
Activity of moxifloxacin (Moxi), plus a no-treatment control, against log-phase M. tuberculosis H37Rv examined in a HFIM over 28 days. (Reprinted from reference 8 under an open-access license.)
Moxifloxacin is a fluoroquinolone and acts at the level of DNA replication. This class of agents induce error-prone replication (14), even at fractions of the MIC. This process is stochastic, so that there is a likelihood of resistance emergence that is directly dependent on the number of rounds of replication. It is also clear that moxifloxacin cannot be used alone, because of the issue of resistance emergence. Consequently, combination therapy is critical to suppress resistance emergence with this drug.
Even though the vast bulk of the M. tuberculosis bacterial burden is in log phase, it would be optimal to employ agents that also have activity against acid-phase organisms and ss-18b (NRP-phase) organisms. In these experiments, we examined the activity of moxifloxacin against M. tuberculosis in these metabolic states. Figures 1 and 3 demonstrate clear activity against both acid-phase and NRP-phase organisms. For acid-phase organisms, we were surprised at the activity seen with moxifloxacin; 200 mg to 800 mg daily provided declines in bacterial burden from baseline of >3 to 5 log10(CFU/ml). This was unexpected, because lower pH is known to decrease the microbiological activity of fluoroquinolones (15, 16). We checked the pH of the system over time and the pH values were stable, as demonstrated in Fig. 6. Consequently, even in an acidic environment, clinically relevant moxifloxacin exposures produced substantial organism killing. It should also be noted that moxifloxacin generated faster bacterial killing when employed against log-phase H37Rv.
FIG 6.
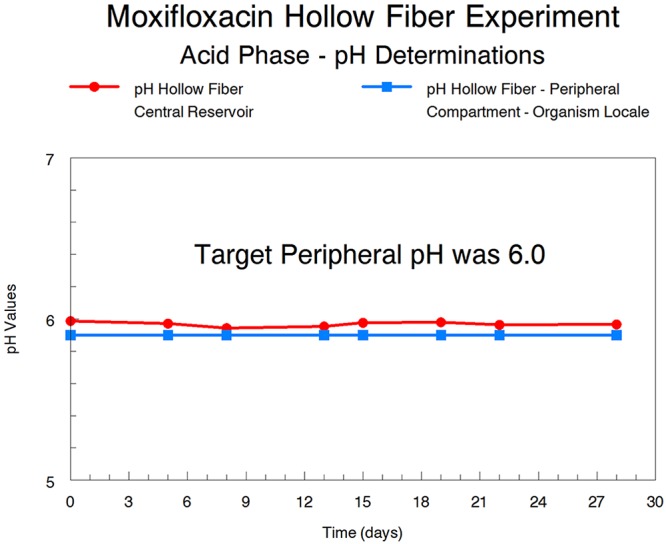
Determination of the pH of the central reservoir and peripheral compartment of the HFIM over 28 days. The peripheral compartment contained M. tuberculosis H37Rv in acid-phase growth.
As expected, no resistant isolates were recovered. First, the bacterial burden was low, relative to the inverse of the resistance mutational frequency. Second, the growth rate was quite low. Fluoroquinolones induce error-prone replication in M. tuberculosis, and the probability of a resistant mutant being created is related to the number of rounds of replication. Because the latter was low, the likelihood of resistance emergence was low; in addition, the natural death rate was the same order of magnitude as the growth rate, so that, even if a resistant clone emerged, it would have a substantial likelihood of not propagating.
We also examined strain 18b in the NRP phase with moxifloxacin in a 28-day experiment. The results are displayed in Fig. 3. As noted previously for linezolid, there was substantial decline in values for the no-treatment control, which started at 6.53 log10(CFU/ml) and declined to 1.96 log10(CFU/ml) at day 28 (12). Three of the exposures (400 mg, 600 mg, and 800 mg daily) reached an extinction event at day 28. The 200-mg dose still had 0.7 log10(CFU/ml) at that point, providing a log kill value for the 200-mg dose of 1.26 log10(CFU/ml). The other doses (400, 600, and 800 mg) all achieved extinction at day 28, compared to 1.96 log10(CFU/ml) observed for the no-treatment control at that time. This effect was still substantial but not as impressive as that we saw with linezolid (12). It should also be noted that there were less-susceptible organisms present in this experiment at baseline and for a few days thereafter, because of the starting inoculum employed, relative to the inverse of the resistance mutational frequency. The NRP-phase organisms must be grown in the presence of streptomycin, and the organisms are thus in log phase. The streptomycin is then withdrawn, with the organisms transitioning to NRP phase. The numbers of less-susceptible clones were low [the largest single measurement was 1.18 log10(CFU/ml) on day 1, with moxifloxacin at 200 mg per day]. Such clones die off quickly due to moxifloxacin and/or their own natural death rate. The differing numbers may be due to Poisson sampling (at low counts, the distribution of organisms in a sample follows a Poisson distribution, and the probability of obtaining a specific number of organisms is a function of the relative sample size), given the low numbers and the sample volume. No less-susceptible isolates were identified after day 4.
Moxifloxacin demonstrates acceptable activity against both acid-phase M. tuberculosis and the ss-18b isolate. This activity is coupled with excellent killing activity against log-phase organisms (8). Consequently, moxifloxacin represents an appealing agent to employ early in a regimen, with the goal of substantially shortening therapy. Because of the high likelihood of resistance emergence when moxifloxacin is employed as monotherapy, combination with another drug (or drugs) is mandatory. The drug combination should ideally interact in a synergistic or at least additive fashion. Having antagonism for any of the metabolic states, particularly NRP-phase organisms (8), might have caused the failure to attain the endpoint of shortened therapy in the REMox trial (10). Further, the combination employed should be orthogonal with respect to resistance emergence, so that the combination would minimize the number of less-susceptible clones. As we mentioned previously (12), it might be beneficial to switch therapy completely after 4 to 6 weeks of the initial regimen. We have examined a follow-on regimen that is aimed primarily at NRP-phase organisms and acid-phase organisms but still possesses substantial log-phase activity (17); this combination is linezolid plus bedaquiline, with additive interaction. It remains to be proven that the resistance mechanisms are not overlapping.
For the early regimen, given our findings here, we are going to evaluate the combination of moxifloxacin plus pretomanid. Pretomanid has been part of the bedaquiline/pretomanid/linezolid/moxifloxacin regimen (TB-PRACTECAL regimen; ClinicalTrials registration no. NCT02589872), for which good activity has been demonstrated against MDR tuberculosis. If the interaction is either synergistic or additive for all three metabolic states, then we will have a regimen that is worthy of further evaluation with the goal of substantially shortening therapy duration for Mycobacterium tuberculosis.
MATERIALS AND METHODS
The methods were drawn from our previous publication (12).
Bacteria.
Mycobacterium tuberculosis strains H37Rv (ATCC 27294) and 18b (11) (kindly provided by Stewart Cole) were used. Stocks of the bacteria were stored at −80°C. For each experiment, an aliquot of the bacterial stock was inoculated into filter-capped T-flasks containing 7H9 Middlebrook broth supplemented with 0.05% Tween 80 and 10% albumin, dextrose, and catalase (ADC). The culture was incubated for 4 to 5 days at 37°C in 5% CO2, on a rocking platform, to achieve log-phase growth. For strain 18b (a streptomycin auxotroph), 75 mg/liter streptomycin was added to the medium to allow growth. To generate acid-phase growth, the following procedures were used.
Acid-phase growth of H37Rv Mycobacterium tuberculosis.
M. tuberculosis in acid-phase growth was generated by transferring log-phase M. tuberculosis (propagated in 7H9 broth with 0.05% Tween and 10% ADC) into T-flasks containing 7H9 broth acidified to pH 6.0 with citric acid. After approximately 7 days of incubation at 37°C in 5% CO2 on a rocking platform, the replicating bacteria were transferred to hollow-fiber systems that had been preconditioned with prewarmed ADC broth adjusted to pH 6.0 with citric acid. The medium in the hollow-fiber systems was continuously replaced with fresh acidified medium over the course of the 28-day experiments. The bacteria inoculated into the hollow-fiber systems were incubated at 37°C in 5% CO2, and serial optical density measurements (wavelength, 600 nm) were conducted over time, to confirm that the bacteria were replicating in the acidified medium, prior to the start of an experiment. The pH of the medium in which the bacteria were being incubated and the pH of the fresh acidified medium that was infused into the hollow-fiber systems were measured throughout the experiments.
Mycobacterium tuberculosis ss-18b strain.
The M. tuberculosis ss-18b strain was generated by washing log-phase 18b organisms (that had been propagated in 7H9 broth with 75 mg/liter streptomycin, 0.05% Tween, and 10% ADC) 3 times in phosphate-buffered saline (PBS) with 0.05% Tween, to remove the streptomycin. The washed 18b bacteria were resuspended in 7H9 broth without streptomycin and were transferred into T-flasks. After approximately 7 days of incubation at 37°C in 5% CO2 on a rocking platform, the ss-18b bacteria were transferred to hollow-fiber systems that had been preconditioned with prewarmed ADC broth.
Drug.
Pharmaceutical-grade moxifloxacin was purchased from CuraScript (Orlando, FL) as a solution for injection and was stored at room temperature in the dark. Prior to experimental use, moxifloxacin was diluted to the desired concentrations in sterile deionized water.
Susceptibility testing and mutational frequency determination.
Moxifloxacin susceptibility studies were conducted with log-phase M. tuberculosis H37Rv and 18b, using the absolute serial dilution method, on 7H10 agar plus 10% oleic acid, albumin, dextrose, and catalase (OADC) (with 75 mg/liter streptomycin for Mycobacterium tuberculosis 18b). Briefly, 104 CFU of H37Rv in log-phase growth was plated on Middlebrook 7H10 agar (Becton, Dickinson Microbiology Systems, Sparks, MD), supplemented with 10% OADC (Becton, Dickinson Microbiology Systems), containing 2-fold dilutions of moxifloxacin. The cultures were incubated at 37°C in 5% CO2. After 4 weeks of incubation, the MICs were determined by identifying the lowest drug concentration at which there was no bacterial growth on the agar plate. For the absolute serial dilution method, the MIC was read as the lowest concentration of drug for which there was no growth on the agar plate. For susceptibility studies using acid-phase M. tuberculosis, MICs for moxifloxacin were determined on 7H10 agar (without OADC) that had been adjusted to pH 6.0 with citric acid.
The mutational frequency of the H37Rv strain and the 18b strain were evaluated using methods that are described elsewhere (9). Briefly, H37Rv and 18b (streptomycin-supplemented) cultures in log-phase growth were inoculated onto plates containing Middlebrook 7H10 agar plus 10% Middlebrook OADC, with moxifloxacin at a concentration equivalent to 3.0× the MIC. The mutational frequency was identified after 4 weeks of incubation at 37°C in 5% CO2.
HFIM 28-day studies using clinically relevant drug exposures for acid-phase Mycobacterium tuberculosis.
The goals of the continuous infusion studies were to delineate the time course of killing of M. tuberculosis in acid-phase growth by moxifloxacin, when clinically relevant exposures of the compound were examined, and to determine whether resistance to moxifloxacin could develop and whether resistance amplification could be suppressed with large exposures. The experimental arms for the studies using acid-phase M. tuberculosis H37Rv were as follows: arm A, no-treatment growth control; arms B to E, moxifloxacin administered once daily, with the area under the concentration-time curve (AUC) at steady state for free drug matching doses of 200 mg, 400 mg, 600 mg, and 800 mg, respectively. The experiment with five arms was performed once.
HFIM 28-day studies using clinically relevant drug exposures for NRP-phase Mycobacterium tuberculosis.
As described above, the goals of this experiment were to examine bacterial cell killing and resistance suppression. The experimental arms were identical to those used in the acid-phase growth study, but strain ss-18b was used. The experiment with five arms was performed once.
Pharmacokinetic and protein binding data.
Human pharmacokinetic parameters and protein binding values for moxifloxacin were obtained from the Avelox package insert (18).
Detection of resistance amplification.
Serial bacterial specimens were collected from the hollow-fiber system arms over the course of the 28-day experiments. The samples were washed and then quantitatively plated onto antibiotic-free agar and antibiotic-supplemented agar, to characterize the effect of each treatment regimen on the total bacterial and less-susceptible bacterial populations. A volume of 200 µl was removed from the peripheral compartment and was streaked onto the zero-dilution plate. Another 100 µl was used to perform serial 10-fold dilutions to obtain accurate countable numbers. This procedure was performed for both antibiotic-free plates (total bacterial burden) and antibiotic-containing plates (moxifloxacin-resistant organisms).
For detection of mutants resistant to moxifloxacin, the agar (at pH 7.0 for ss-18b experiments) was supplemented with 10% OADC. For acid-phase experiments, the agar was adjusted to pH 6.0. The agar plates were read after 6 weeks of incubation at 37°C in a 5% CO2 atmosphere. Moxifloxacin concentrations incorporated into the agar were 3.0× the baseline MIC value.
Achievement of target exposure profiles.
Serial medium samples were collected from the hollow-fiber treatment arms for assay of drug content by liquid chromatography-tandem mass spectrometry (LC-MS/MS), as described below, to confirm that the targeted concentration-time profiles were achieved. Samples were stored at −80°C until assayed. Concentrations of moxifloxacin in7H9 samples were measured using a validated high-performance liquid chromatography assay with triple-quadrupole mass spectrometry; the equipment consisted of a Thermo Endura tandem mass spectrometer and a Dionex Ultimate 3000 ultra-high-performance liquid chromatography (UHPLC) system. The 7H9 standards ranged from 0.20 to 15.00 μg/ml. Accuracy was within 7% for all validation standards. The overall precision for the validation standards was 1.6 to 4.7% (coefficient of variation), and that for the quality control samples was 3.2 to 5.1%. The recovery of moxifloxacin from 7H9 was 100%.
Population pharmacokinetic/pharmacodynamic mathematical model.
We simultaneously modeled two or three system outputs for the analysis of the acid-phase and ss-18b M. tuberculosis data, in separate analyses. The system outputs were the measured concentrations of moxifloxacin, the total M. tuberculosis burden, and the less-moxifloxacin-susceptible burden. Population modeling was performed by employing the nonparametric adaptive grid (NPAG) program described by Leary et al. (19). Modeling choices (such as weighting) and goodness-of-fit evaluations were as published previously (20). Simulation was performed with the ADAPT V program described by D’Argenio et al. (21), using Bayesian posterior parameter estimates.
ACKNOWLEDGMENTS
This work was supported by NIAID grant P01AI123036. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
REFERENCES
- 1.Centers for Disease Control and Prevention. 2018. Tuberculosis: data and statistics. www.cdc.gov/tb/statistics/default.htm. Accessed 5 June 2018.
- 2.World Health Organization. 2016. What is multidrug-resistant tuberculosis (MDR-TB) and how do we control it? www.who.int/features/qa/79/en. Accessed June 2018.
- 3.Chung-Delgado K, Guillen-Bravo S, Revilla-Montag A, Bernabe-Ortiz A. 2015. Mortality among MDR-TB cases: comparison with drug-susceptible tuberculosis and associated factors. PLoS One 10:e0119332. doi: 10.1371/journal.pone.0119332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Srivastava S, Pasipanodya JG, Meek C, Leff R, Gumbo T. 2011. Multidrug-resistant tuberculosis not due to noncompliance but to between-patient pharmacokinetic variability. J Infect Dis 204:1951–1959. doi: 10.1093/infdis/jir658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gomez JE, McKinney JD. 2004. M. tuberculosis persistence, latency, and drug tolerance. Tuberculosis (Edinb) 84:29–44. doi: 10.1016/j.tube.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 6.Wang X, Kang Y, Luo C, Zhao T, Liu L, Jiang X, Fu R, An S, Chen J, Jiang N, Ren L, Wang Q, Baillie JK, Gao Z, Yu J. 2014. Heteroresistance at the single-cell level: adapting to antibiotic stress through a population-based strategy and growth-controlled interphenotypic coordination. mBio 5:e00942-13. doi: 10.1128/mBio.00942-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gumbo T, Louie A, Deziel MR, Parsons LM, Salfinger M, Drusano GL. 2004. Selection of a moxifloxacin dose that suppresses Mycobacterium tuberculosis resistance using an in vitro pharmacodynamic infection model and mathematical modeling. J Infect Dis 190:1642–1651. doi: 10.1086/424849. [DOI] [PubMed] [Google Scholar]
- 8.Drusano GL, Sgambati N, Eichas A, Brown DL, Kulawy R, Louie A. 2010. The combination of rifampin plus moxifloxacin is synergistic for resistance suppression but antagonistic for cell kill of Mycobacterium tuberculosis as determined in a hollow fiber infection model. mBio 1:e00139-10. doi: 10.1128/mBio.00139-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drusano GL, Sgambati N, Eichas A, Brown D, Kulawy R, Louie A. 2011. Effect of administering moxifloxacin plus rifampin against Mycobacterium tuberculosis 7 of 7 days versus 5 of 7 days in an in vitro pharmacodynamic system. mBio 2:e00108-11. doi: 10.1128/mBio.00108-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gillespie SH, Crook AM, McHugh TD, Mendel CM, Meredith SK, Murray SR, Pappas F, Phillips PP, Nunn AJ, REMoxTB Consortium . 2014. Four-month moxifloxacin-based regimens for drug-sensitive tuberculosis. N Engl J Med 371:1577–1587. doi: 10.1056/NEJMoa1407426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Honoré N, Marchal G, Cole ST. 1995. Novel mutation in 16S rRNA associated with streptomycin dependence in Mycobacterium tuberculosis. Antimicrob Agents Chemother 39:769–770. doi: 10.1128/AAC.39.3.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drusano GL, Myrick J, Maynard M, Nole J, Duncanson B, Brown D, Schmidt S, Neely M, Scanga CA, Peloquin C, Louie A. 2018. Linezolid kills acid-phase and nonreplicative-persister-phase Mycobacterium tuberculosis in a hollow-fiber infection model. Antimicrob Agents Chemother 62:e00221-18. doi: 10.1128/AAC.00221-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sala C, Dhar N, Hartkoorn RC, Zhang M, Ha YH, Schneider P, Cole ST. 2010. Simple model for testing drugs against nonreplicating Mycobacterium tuberculosis. Antimicrob Agents Chemother 54:4150–4158. doi: 10.1128/AAC.00821-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Sullivan DM, Hinds J, Butcher PD, Gillespie SH, McHugh TD. 2008. Mycobacterium tuberculosis DNA repair in response to subinhibitory concentrations of ciprofloxacin. J Antimicrob Chemother 62:1199–1202. doi: 10.1093/jac/dkn387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lemaire S, Tulkens PM, Van Bambeke F. 2011. Contrasting effects of acidic pH on the extracellular and intracellular activities of the anti-Gram-positive fluoroquinolones moxifloxacin and delafloxacin against Staphylococcus aureus. Antimicrob Agents Chemother 55:649–658. doi: 10.1128/AAC.01201-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blaser J, Lüthy R. 1988. Comparative study on the antagonistic effects of low pH and cation supplementation on in vitro activity of quinolones and aminoglycosides against Pseudomonas aeruginosa. J Antimicrob Chemother 22:15–22. doi: 10.1093/jac/22.1.15. [DOI] [PubMed] [Google Scholar]
- 17.de Miranda Silva C, Hajihosseini A, Myrick J, Nole J, Louie A, Schmidt S, Drusano GL. 2018. Effect of linezolid plus bedaquiline against Mycobacterium tuberculosis in log phase, acid phase and nonreplicating persister phase in an in vitro assay. Antimicrob Agents Chemother 62:e00856-18. doi: 10.1128/AAC.00856-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bayer. 1999. Avelox™ (moxifloxacin hydrochloride) tablets, package insert. Bayer, Whippany, NJ: https://www.accessdata.fda.gov/drugsatfda_docs/label/1999/21085lbl.pdf. [Google Scholar]
- 19.Leary R, Jelliffe R, Schumitzky A, Van Guilder M. 2001. An adaptive grid non-parametric approach to pharmacokinetic and dynamic (PK/PD) models. Conf Proc 14th IEEE Symp Comput Based Med Syst 2001:389–394. doi: 10.1109/CBMS.2001.941750. [DOI] [Google Scholar]
- 20.Brown AN, Drusano GL, Adams JR, Rodriquez JL, Jambunathan K, Baluya DL, Brown DL, Kwara A, Mirsalis JC, Hafner R, Louie A. 2015. Preclinical evaluations to identify optimal linezolid regimens for tuberculosis therapy. mBio 6:e01741-15. doi: 10.1128/mBio.01741-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.D’Argenio DZ, Schumitzky A, Wang X. 2009. ADAPT 5 user’s guide: pharmacokinetic/pharmacodynamic systems analysis software. Biomedical Simulations Resource, Los Angeles, CA. [Google Scholar]