

It is generally believed that the Pseudomonas aeruginosa biofilm matrix itself acts as a molecular sieve or sink that contributes to significant levels of drug resistance, but it is becoming more apparent that multidrug efflux pumps induced during biofilm growth significantly enhance resistance levels. We present here a novel transcriptional regulator, PA3898, which controls biofilm formation and multidrug efflux pumps in P. aeruginosa.
KEYWORDS: EmrAB, MexAB-OprM, PA2100, PA3898, Pseudomonas aeruginosa, phenazine
ABSTRACT
It is generally believed that the Pseudomonas aeruginosa biofilm matrix itself acts as a molecular sieve or sink that contributes to significant levels of drug resistance, but it is becoming more apparent that multidrug efflux pumps induced during biofilm growth significantly enhance resistance levels. We present here a novel transcriptional regulator, PA3898, which controls biofilm formation and multidrug efflux pumps in P. aeruginosa. A mutant of this regulator significantly reduced the ability of P. aeruginosa to produce biofilm in vitro and affected its in vivo fitness and pathogenesis in Drosophila melanogaster and BALB/c mouse lung infection models. Transcriptome analysis revealed that PA3898 modulates essential virulence genes/pathways, including multidrug efflux pumps and phenazine biosynthesis. Chromatin immunoprecipitation sequencing (ChIP-seq) identified its DNA binding sequences and confirmed that PA3898 directly interacts with promoter regions of four genes/operons, two of which are mexAB-oprM and phz2. Coimmunoprecipitation revealed a regulatory partner of PA3898 as PA2100, and both are required for binding to DNA in electrophoretic mobility shift assays. PA3898 and PA2100 were given the names MdrR1 and MdrR2, respectively, as novel repressors of the mexAB-oprM multidrug efflux operon and activators for another multidrug efflux pump, EmrAB. The interaction between MdrR1 and MdrR2 at the promoter regions of their regulons was further characterized via localized surface plasmon resonance and DNA footprinting. These regulators directly repress the mexAB-oprM operon, independent of its well-established MexR regulator. Mutants of mdrR1 and mdrR2 caused increased resistance to multiple antibiotics in P. aeruginosa, validating the significance of these newly discovered regulators.
INTRODUCTION
Bacterial biofilms are a source of recurrent problems in clinical and industrial settings. Mechanical blockages and microbial-induced corrosion result in billions of dollars of losses each year (1). In the medical fields, infections caused by biofilms are a significant socioeconomic burden (2, 3). The Centers for Disease Control and Prevention estimates that 2 million patients suffer from hospital-acquired infections annually, related mainly to the presence of biofilms, leading to nearly 100,000 mortalities (2, 3). The source of these problems lies in the particular physiology of the bacteria immobilized within biofilms, which are notoriously difficult to treat because of increased tolerance to antimicrobial agents (4–6). The biofilm development cycle includes (i) initial attachment (7), (ii) irreversible attachment, (iii) maturation, and (iv) dispersion (8–10). Once established, biofilms are extremely difficult to eradicate due to the physical barriers that hinder the penetration of antimicrobial compounds as well as the increased antimicrobial resistance of the bacteria in a biofilm. Therefore, having a comprehensive understanding of the biofilm formation process is fundamentally important for effective biofilm treatment strategies in both clinical and industrial settings.
Pseudomonas aeruginosa has been a model organism for the investigation of biofilms, pathogenesis, and drug resistance (11–17). It is an opportunistic pathogen responsible for both acute and chronic infections and one of the most frequent and severe causes of acute nosocomial infections (18). P. aeruginosa has the highest number of regulatory genes among sequenced bacterial genomes, and regulators and regulatory sensors make up almost 10% of all genes in the P. aeruginosa genome (19). This contributes to a complex regulatory network, controlling genes in response to different conditions, and facilitates its adaptation to constantly evolving environments. P. aeruginosa is naturally resistant to many antimicrobial compounds, escalated by its ability to form biofilms in various environments and produce many virulence factors, making it a dangerous human pathogen, particularly in immunocompromised individuals (17, 20). P. aeruginosa has outstanding antibiotic resistance machineries, and antibiotic therapies have been severely compromised by its high level of antibiotic resistance (15, 21). P. aeruginosa possesses several multidrug efflux pumps, which are capable of expelling a number of antimicrobials and enable its survival in harsh environments (15, 21). Many of these multidrug efflux pumps belong to two main families: the resistance-nodulation-division (RND) family and the major facilitator superfamily (MFS). The RND family efflux pumps consist of an inner membrane protein component belonging to the RND superfamily of secondary transporters, a channel-forming outer membrane factor (OMF), and a periplasmic membrane fusion protein (MFP). These three components were shown to form a tripartite protein complex that expands across both the inner and outer membranes, allowing the extrusion of drugs directly into the extracellular environment (22). Among all the multidrug efflux pumps, MexAB-OprM contributes most significantly to high levels of resistance to clinically relevant antibiotics, including most β-lactams and quinolones (15, 23). In this pump, the proteins MexA, MexB, and OprM act as the MFP, RND transporter, and OMF, respectively. The mexAB-oprM genes are organized in an operon, and early pioneering studies have identified two potential transcriptional start sites as well as four transcriptional regulators, MexR, NalD (24, 25), BrlR (26), and CpxR (27). The mexR gene encodes a transcriptional regulator of the MarR family, which acts as a repressor of the mexAB-oprM operon and controls one of the two identified transcriptional start sites (24). Thus far, the control of the second transcriptional start site has not been assigned to any regulator(s), although the TetR family regulator NalD, the MerR family regulator BrlR, and the response regulator CpxR also bind directly to the promoter of the operon (25–28). Other regulators have also been shown to affect the expression of mexAB-oprM indirectly, including NalC (29), ArmR (30), and PA3225 (31). It is currently unclear if an additional regulator(s) directly controls the expression of mexAB-oprM, via the second transcriptional start site downstream of the known binding sites of MexR and NalD. P. aeruginosa also possesses two operons that encode multidrug efflux pumps in the MFS family: PA3136-PA3137 and PA5157–PA5160. These genes have not been characterized in detail but are similar to the emrA and emrB genes of Escherichia coli (32, 33). MFS pumps have been shown to function with only the drug efflux transporter or as a three-component pump, coupling the transporter with a MFP and an outer membrane protein (OMP) (34). Both operons in P. aeruginosa contain genes coding for the membrane fusion protein (PA3136 and PA5159) and the drug efflux transporter (PA3137 and PA5160); however, the PA5157-PA5160 operon also contains genes coding for a putative regulator (PA5157) and an OMP (PA5158). A previous study demonstrated that both operons in P. aeruginosa are induced in the presence of pentachlorophenol stress (35).
In this study, we set out to investigate a novel transcriptional regulator, PA3898, which is shown here to be essential for biofilm formation and the pathogenesis of P. aeruginosa in two animal models. Through detailed analysis, the regulation network of PA3898 along with its regulatory partner PA2100 was revealed. These novel dual regulators play essential roles in regulating biofilm formation, phenazine production, and the multidrug efflux pumps MexAB-OprM and EmrAB.
RESULTS
A previously uncharacterized transcriptional regulator, PA3898, controls biofilm formation and pathogenesis.
Recent developments in single-prokaryotic-cell transcriptomic technology (36, 37) have allowed an in-depth global expression profile within the P. aeruginosa biofilm architecture in three distinct vertical locations (i.e., surface, middle, and interior of the biofilm structure) (38). Among genes that have spatially dependent expression patterns, many were predicted/putative regulators with unknown regulatory functions (38). One such putative transcriptional regulator, PA3898, has a distinct spatial expression pattern (Fig. 1A). We hypothesized that differential expression of such a regulator in biofilms is likely important for controlling unique aspects of biofilm formation in the different spatial regions. In two phenotypic screens, a mutant strain with an insertional mutation in the regulator PA3898 (PA3898::Tn) was shown to be defective in biofilm production under static biofilm growth conditions for 48 h at 30°C (Fig. 1B and C). This defect in biofilm formation does not result from a growth defect, as the PA3898::Tn mutant strain grows identically to the wild type in vitro (Fig. 1D).
FIG 1.

Identification of PA3898 as essential for biofilm formation. (A) Spatial expression pattern of PA3898 in the surface, middle, and interior of a biofilm from a previous study (38). Fold changes of spatially expressed genes are shown with a red-black-green double-color gradient. Green indicates upregulation and red indicates downregulation compared to levels under planktonic growth conditions, as shown in the color gradient bar at the top (log2 fold change, 2 to −2). For each location within the biofilm, the first three boxes represent the results from three biological replicates, and the fourth box represents the average fold changes of the replicates. (B) The PA3898::Tn mutant strain has significantly reduced biofilm formation via a crystal violet assay compared to wild-type (WT) PAO1 and the complemented strain (***, P < 0.0005 based on Student’s t test). The biofilm assay was performed in triplicate, and average amounts of biofilm and standard errors of the means are shown. (C) Defects in biofilm were observed and validated by confocal microscopy. Microscopic imaging was done in triplicate, and representative images are shown. (D) The PA3898::Tn mutant strain shows no in vitro growth defect compared to its complemented strain and wild-type PAO1.
Two well-established animal infection models for studying P. aeruginosa virulence, Drosophila melanogaster and the BALB/c mouse (39, 40), were used to assess PA3898. It was previously shown in the fruit fly feeding model that the ability of P. aeruginosa to colonize the fly crop correlates with its ability to form a biofilm (39). As expected, the PA3898::Tn mutant strain was outcompeted by its complemented strain in the in vivo fruit fly model (Fig. 2A and B), in agreement with its defect in biofilm formation in vitro (Fig. 1B). The PA3898::Tn mutant strain shows an increased ability to kill fruit flies (Fig. 2C). This is not entirely surprising, as a previous study found that a P. aeruginosa pelB::Tn mutant is defective in biofilm formation and significantly more virulent than PAO1 (39). The reduced in vivo fitness of the PA3898::Tn mutant strain in fruit flies was consistent with the mammalian BALB/c mouse lung infection model (Fig. 2D). PA3898 could regulate some aspect of virulence, possibly beyond biofilm formation, justifying further investigation into the genes controlled by the important putative regulator PA3898.
FIG 2.

Contribution of PA3898 to virulence in fruit fly and mouse lung infection models. (A) In vivo competition between the mutant strain (tetracycline resistant and red fluorescent protein [RFP] tagged) and its corresponding complemented strain (gentamicin resistant and green fluorescent protein [GFP] tagged). A control experiment was performed using a tetracycline-resistant and RFP-tagged PAO1 strain versus a gentamicin-resistant and GFP-tagged PAO1 strain. The competitive index of bacteria harvested from triplicate groups of five infected flies at 48 h postinfection shows that the PA3898::Tn mutant has a reduced ability to colonize fruit flies compared to its complemented strain. The solid red line represents the average CI in each competition group. A competitive index of <1 indicates that the mutant was outcompeted by its complemented strain in fruit flies (**, P < 0.005 by a one-sample t test). (B) Microscopic images of crops harvested from infected flies from panel A showing that the PA3898::Tn mutant was outcompeted by the PA3898::Tn complemented strain, which is displayed by a lack of red PA3898::Tn mutant bacteria. The control experiment showed equal amounts of RFP- and GFP-tagged bacteria. (C) Survivals of flies infected by different P. aeruginosa strains were monitored compared to the no-bacterium control. Survivals were tested in triplicate, and averages are shown. Mutation in PA3898 increased the level of virulence in fruit flies compared to WT PAO1. (D) The PA3898::Tn mutant exhibits a similar competitive disadvantage against the complemented strain in the mouse lung infection model (*, P < 0.05 by a one-sample t test).
PA3898 directly regulates two multidrug efflux pumps and the phenazine biosynthesis operon.
The AraC family transcriptional regulator PA3898 (41) is described above to be essential for biofilm production and pathogenesis of P. aeruginosa. We hypothesized that PA3898 could control virulence genes and pathways, beyond biofilm formation, that could contribute to the in vivo pathogenesis shown in Fig. 2. Therefore, microarray analysis was performed on wild-type PAO1 and PA3898::Tn mutant strains growing under static biofilm growth conditions for 48 h at 30°C to study the genes regulated by PA3898. Gene expression comparisons between wild-type PAO1 and PA3898::Tn mutant strains revealed many genes directly or indirectly regulated by PA3898 (see Tables S1 and S2 in the supplemental material), including two multidrug efflux pump systems, EmrAB (PA3136-PA3137) (32, 33) and MexAB-OprM (PA0425-PA0427) (42). Several virulence genes and pathways are also regulated by PA3898, among which are FtsY/PA0373 (43), Vfr/PA0652 (44), RsmA/PA0905 (45), the phenazine biosynthesis operon PA1899-PA1905 (46), and PilS/PA4546 (47) (Tables S1 and S2). FtsY, a docking protein involved in protein targeting and membrane biogenesis, was shown to be induced in vivo through in vivo expression technology (IVET) in both a rat lung model and a mouse model of infection (43). Vfr is a global regulator of virulence factor expression, including exotoxin A and protease production (44). RsmA, a posttranscriptional regulatory protein, controls the expression of several virulence genes and was shown to be involved in initial colonization and dissemination in a mouse lung model (45). The phenazine biosynthesis operon phz2 is produced during biofilm development and host infection (46). The sensor kinase PilS directly interacts with and regulates one of the major virulence determinants, the type VI pilus (47). Although it is too complicated to predict at this stage, the in vivo importance of PA3898 shown in Fig. 2 could be attributed to any combination of these virulence factors.
In order to differentiate between direct and indirect regulation, chromatin immunoprecipitation sequencing (ChIP-seq) analysis was performed, because transcriptome analysis does not delineate between genes that are directly and indirectly regulated by PA3898. We identified four operons that are directly regulated by PA3898, mexAB-oprM, phz2, emrAB, and a sensor/response regulator hybrid operon (Table 1 and Fig. 3A). Additionally, the binding sequence of PA3898 was predicted by the MEME-ChIP suite (48) to be an 18-bp palindromic binding motif consisting of two conserved 6-bp sequences forming an inverted repeat separated by a random 6-bp spacer (GCAGCGN6CGCTGC) (Fig. 3B). These binding sequences are located in the intergenic regions upstream of these four operons directly regulated by PA3898, and the two 6-bp inverted repeats are 100% conserved among all four sequences. Two of these four operons encode proteins that have known functions: mexA (PA0425) and phzA2 (PA1899). mexA (PA0425) is the first gene in the operon encoding the multidrug efflux pump MexAB-OprM, and phzA2 (PA1899) is the first gene in a seven-gene operon that encodes phenazine biosynthesis proteins. P. aeruginosa has two phenazine biosynthesis operons, phz1 and phz2 (46). A previous study has shown that the phz1 operon is mainly expressed under planktonic growth conditions, while phz2 is almost exclusively produced during biofilm development and host infection (46). Additionally, PA3898 directly regulates another predicted MFS multidrug efflux pump, comprising EmrA (PA3136) and EmrB (PA3137), as well as a probable sensor/response regulator hybrid, PA3462. All four directly regulated operons identified via ChIP-seq were present in the microarray data, with significant fold changes (≥2-fold; P < 0.05) (Tables S1 and S2). Collectively, the ChIP-seq and microarray results present a comprehensive overview of the PA3898 regulatory network and reveal several key genes/pathways potentially contributing to biofilm formation and pathogenesis.
TABLE 1.
P. aeruginosa genes/operons directly regulated by MdrR1/PA3898
| Operon | Gene name and/or function | Activated or repressed by PA3898 |
Avg fold change (fold change by real-time RT-PCR)a |
|---|---|---|---|
| PA0425-PA0427 | MexAB-OprM, multidrug efflux pump | Repressed | 2.2 (3.1) |
| PA1899-PA1905 | Phenazine biosynthesis | Activated | 2.4 (1.9) |
| PA3136-PA3137 | Multidrug efflux pump | Activated | 2.1 (2.9) |
| PA3462-PA3463 | Probable sensor/response regulator hybrid | Activated | 2.4 (2.2) |
Shown are average fold increases (repressed genes) or fold-decreases (activated genes) in expression levels of target genes in PA3898::Tn compared to the wild-type PAO1 strain based on microarray data. Numbers in parentheses are fold changes from real-time RT-PCR validation.
FIG 3.

Genes and operons directly regulated by PA3898 and discovery of the coregulator PA2100. (A) Mapped peaks from ChIP-seq results. Genes controlled directly by PA3898 are indicated. (B) DNA binding motifs were predicted using the online software MEME-ChIP. (C) EMSA with P. aeruginosa whole-cell lysates. Binding was detected only using whole-cell lysates with intact PA3898 (PAO1 and PA3898 complemented strains) and DNA fragments containing the binding motifs. (D) Coimmunoprecipitation using clarified lysates of the PA3898::Tn mutant strain containing TY1-tagged PA3898 obtained four bands on SDS-PAGE gels. Two of the bands were determined to be PA3898 (MdrR1) and PA2100 (MdrR2) via iron trap LC-MS/MS, and the other two were not determined (ND) due to insufficient amounts of peptides extracted. (E) EMSA with purified regulators MdrR1 and MdrR2. The recombinant regulatory proteins His6-MdrR1 and His6-MdrR2 together completely shifted the promoter region of mexAB-oprM.
Since the predicted binding motif of PA3898 is highly conserved, we also searched for the existence of additional sequences throughout the whole PAO1 genome (48). Interestingly, eight additional sequences were found to have 100% identity in the two 6-bp inverted repeats compared to the predicted binding motif. Surrounding genes of these additional binding motifs include PA1566 (glutamylpolyamine synthetase), PA2086 (probable epoxide hydrolase), PA2513 (antB) (anthranilate dioxygenase small subunit), PA3018 (hypothetical protein), PA3358 (hypothetical protein), PA4322 (conserved hypothetical protein), PA4475 (conserved hypothetical protein), and PA4636 (hypothetical protein). All of these genes were cross-checked with microarray data, and surprisingly, none of these additional genes were regulated by PA3898 under these conditions. We cannot rule out that this is likely a result of the specific growth conditions under which the microarray and ChIP-seq analyses were performed, static biofilm. Therefore, it is possible that these additional genes are regulated by PA3898 under other growth conditions, such as planktonic culture, which would be an interesting subject for future investigation.
PA3898 binding to the DNA motif requires an interaction partner, PA2100.
After identification of the predicted DNA binding sequences of PA3898, the next logical step was to validate the binding of these sequences through an electrophoretic mobility shift assay (EMSA). Numerous attempts at EMSAs performed by multiple experimenters failed when the predicted binding regions upstream of operons regulated by PA3898 (mexA, phzA2, PA3136, and PA3462) were used (data not shown). However, shifting of the mexA promoter (PmexA) DNA fragments was achieved when clarified whole-cell lysates of PAO1 were used instead of purified His6-PA3898 (Fig. 3C). As expected, the clarified lysate of the PA3898 deletion mutant was unable to bind and shift the DNA fragments, verifying the importance of PA3898 in binding to the PmexA sequences (Fig. 3C). Additionally, deletion of the predicted 18-bp binding motif from the PmexA fragment completely abolished the shift (PmexA-Δmotif) (Fig. 3C). Taking these outcomes together, we suspected that other proteins or factors in P. aeruginosa are necessary for the interaction between PA3898 and the DNA binding motif. To further evaluate this possibility, we employed a protein coimmunoprecipitation screen, as previously described (49), to search for a potential protein partner(s) necessary for the interaction of PA3898 and the binding motif (Fig. 3D). Four protein bands were observed on an SDS-PAGE gel (Fig. 3D), and two bands were identified via ion trap liquid chromatography-tandem mass spectrometry (LC-MS/MS) to be PA3898 and PA2100. The other two protein bands were not successfully identified due to insufficient amounts of extracted peptide. These new data clearly suggested that PA2100 and PA3898 interact directly with each other and that both may be required to bind to the palindromic DNA motif. PA2100 was annotated a GntR family regulator (41), which is shown in Table S1 in the supplemental material to be activated by PA3898. Therefore, an EMSA was attempted again by using a combination of purified His6-PA3898 and His6-PA2100. As shown in Fig. 3E, PA3898 and PA2100 together shifted the PmexA DNA fragments completely, thereby confirming the interaction of the regulator-coregulator-DNA complex in vitro. Considering that these two regulators collaboratively control two multidrug efflux pump systems, we named PA3898 and PA2100 MdrR1 and MdrR2, respectively, and these names are used throughout this article. The interesting observation that MdrR1 (PA3898) activates MdrR2 (PA2100) (Table S1) also led us to further test the correlation between their gene expression levels through real-time reverse transcription-PCR (RT-PCR). It turns out that MdrR2 also activates the expression of MdrR1, where the expression level of MdrR1 is 2.1-fold lower in the MdrR2::Tn mutant strain than in wild-type PAO1. This connection between MdrR1 and MdrR2 expression is likely indirect through some feedback mechanism, since the specific DNA binding motif identified above was not found near mdrR1 or mdrR2.
Real-time imaging of MdrR1-MdrR2-DNA interaction and validation of the MdrR1 and MdrR2 binding motif via DNA footprinting.
Besides qualitative confirmation of regulator-coregulator-DNA binding by EMSAs, we sought to quantitatively measure the interaction via localized surface plasmon resonance (LSPR). His6-MdrR2 was coupled to a nitrilotriacetic acid (NTA)-Ni-gold chip, untagged MdrR1 and DNA containing the binding sequences were then run through the chip, and changes in absorbance were measured. As negative controls, untagged MdrR1 and DNA containing the binding motif were run through the MdrR2-coupled chip individually. Neither DNA nor MdrR1 alone showed any interaction with His6-MdrR2 (Fig. 4A). When a mixture of DNA and MdrR1 was injected through the chip, concentration-dependent interactions were observed (Fig. 4A). At all three concentrations (11, 33, and 100 nM each MdrR1 and DNA), DNA and MdrR1 completely dissociated from the chip within 10 min. The dissociation constant (Kd) for MdrR1 and DNA containing the binding motif was determined to be 0.17 μM, using the off-rate for the first 50 s (Fig. 4B) and the on-rate (Fig. 4C). An additional negative control was performed by using untagged MdrR1 and the DNA fragment with the binding motif deleted, at 100 nM each, showing no interaction with bound His6-MdrR2 (Fig. 4A). This indicates a specific interaction between MdrR1, MdrR2, and DNA containing the binding motif.
FIG 4.
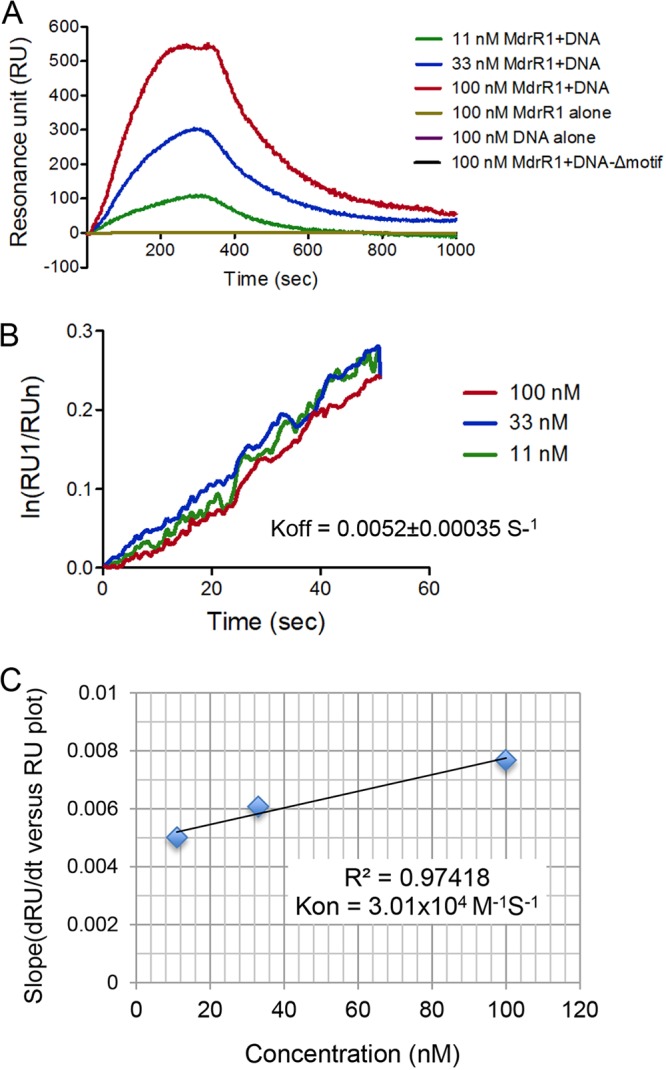
Determination of the kinetic parameters (on-rate and off-rate) of the interaction between MdrR1, MdrR2, and the DNA complex. His6-MdrR2 was covalently immobilized on an NTA sensor. Untagged MdrR1 and the DNA fragment containing the binding motif functioned as analytes. MdrR1 alone, DNA containing the binding motif alone, and MdrR1 and DNA with the binding motif deleted were included as negative controls. (A) Sensorgram data plotted on a dRU/dt-versus-RU plot. (B) Linearized data from sensorgrams for the determination of the off-rate (slope of the plot). (C) Linearized data from sensorgrams for the determination of the on-rate (slope of the plot).
Taken together, the ChIP-seq and EMSA results indicate that MdrR1 and MdrR2 bind to DNA sequences upstream of genes and operons that they directly regulate, and LSPR validates the specific interaction between MdrR1, MdrR2, and DNA. Furthermore, to verify the exact binding motif predicted by ChIP-seq, DNA footprinting was performed with an automated DNA analyzer as previously described (49, 50). A DNA fragment upstream of PmexA (from bases −189 to +224 relative to the start codon of mexA) was coincubated with PsrA (PA3006) (a regulator, as a negative control), MdrR1 alone, MdrR2 alone, and MdrR1 and MdrR2 together (Fig. 5). Only when both MdrR1 and MdrR2 were incubated with PmexA was a distinct region protected from DNase I digestion (Fig. 5, boxed region). As shown in Fig. 5, this protected region covers the entire 18-bp predicted binding sites (underlined and contained within the boxed region in Fig. 5). Since both regulators have a helix-turn-helix DNA binding domain, current data are insufficient to determine the binding sequences of MdrR1 and/or MdrR2. Future studies investigating the crystal structures of MdrR1 and MdrR2 could yield valuable information on the binding sequence specificity of these regulators.
FIG 5.

Region of binding of MdrR1 and MdrR2 to the mexA promoter (PmexA) identified via DNA footprinting. The MdrR1- and MdrR2-protected trace in the bottom panel shows the DNA region that is protected from DNase I digestion. In comparison, the transcriptional regulator PsrA, MdrR1, and MdrR2 individually as controls did not protect the DNA from DNase I digestion, as indicated by the distribution of DNA fragments across the whole region. The outlined region is the predicted binding sequence of MdrR1 and MdrR2.
MdrR1 and MdrR2 regulate the expression of the multidrug efflux pump mexAB-oprM independent of MexR.
Our data suggest that the mexAB-oprM operon (PA0425-PA0427) was modulated by this MdrR1-MdrR2 dual-regulation system (Fig. 3 and Table 1; see also Table S1 in the supplemental material). Since the mexAB-oprM operon and its regulation by MexR were extensively studied previously (24, 25, 51), we aimed to thoroughly validate the involvement of MdrR1-MdrR2 in the regulation of this multidrug efflux pump. ChIP-seq and DNA footprinting results indicate that MdrR1-MdrR2 binds to a different sequence of the promoter region of mexA (Fig. 3 and 5), which is downstream of both the MexR binding site and the second predicted transcriptional start site of mexA (Fig. 6A). Therefore, we sought to validate if MdrR1-MdrR2 regulates the expression of PmexA independently of MexR. Various combinations, including single, double, and triple mutants of the mdrR1, mdrR2, and mexR genes, were engineered to facilitate this study using an established method (52). All strains were grown under static growth conditions for 48 h at 30°C and harvested for β-galactosidase (β-Gal) activity quantification. We hypothesized that gene expression levels of mexAB-oprM, based on the promoter activities of mexA, would be affected by the mutations in the mdrR1 and mdrR2 genes. The mdrR1::Tn and mdrR2::Tn single mutants have mexAB-oprM expression levels comparable to that of the mdrR1::Tn ΔmdrR2 double mutant (Fig. 6B), in agreement with the prediction that they work collaboratively in controlling this efflux pump. There were additive effects of mdrR1, mdrR2, and mexR mutations on the expression level of mexAB-oprM, indicated by the highest level of activity of PmexA in the triple mutant background (mexR::Tn ΔmdrR1 ΔmdrR2) (Fig. 6B). These observations were further corroborated by MIC data for various regulator mutant strains (Table 2), correlating the expression levels of mexAB-oprM with the functional phenotypes of this important multidrug efflux pump in P. aeruginosa. Mutation in the regulators MdrR1 and MdrR2 resulted in increased levels of resistance of P. aeruginosa to multiple types of antibacterial agents, likely via deregulation of mexAB-oprM (Table 2). This increased resistance is consistent across all classes of antibiotic substrates of MexAB-OprM tested here, including carbenicillin, chloramphenicol, erythromycin, norfloxacin, novobiocin, and nalidixic acid (Table 2). In comparison, using an aminoglycoside that is not a substrate for this pump, such as amikacin, resistance levels were unchanged in all the mutant and complemented strains tested (Table 2). Since MICs are indicators of the susceptibility phenotypes of planktonic cells, our data demonstrated that MdrR1 and MdrR2 could affect the expression of mexAB-oprM under both biofilm and planktonic growth conditions.
FIG 6.

MdrR1 and MdrR2 interact with the mexA promoter region, independent of MexR. (A) Schematic showing the binding positions of the MdrR1-MdrR2 complex and MexR. (B) MdrR1 and MdrR2 significantly repress the expression of mexA, independent of MexR. An additive effect on the derepression of PmexA was observed in the triple mutant strain, as indicated by the PmexA-lacZ fusion activities in various strain backgrounds. A β-Gal assay was performed in triplicate, and average activities and standard errors of the means are shown (**, P < 0.005 based on Student’s t test).
TABLE 2.
MICs of various P. aeruginosa strains
| Strain | Antibiotic MIC (μg/ml)a |
||||||
|---|---|---|---|---|---|---|---|
| Cb | Cm | Ery | Norf | Nov | Nal | Amk | |
| PAO1 | 16 | 64 | 128 | 0.5 | 128 | 64 | 4 |
| mdrR1::Tn | 64 | 256 | 512 | 8 | 512 | 128 | 4 |
| mdrR1::Tn comp | 16 | 64 | 128 | 0.5 | 128 | 64 | 4 |
| mdrR2::Tn | 64 | 256 | 512 | 8 | 512 | 128 | 4 |
| mdrR2::Tn comp | 16 | 64 | 128 | 0.5 | 128 | 64 | 4 |
| mdrR1::Tn ΔmdrR2 | 64 | 256 | 512 | 8 | 512 | 128 | 4 |
| mdrR1::Tn ΔmdrR2 comp | 16 | 64 | 128 | 0.5 | 128 | 64 | 4 |
| mexR::Tn | 32 | 128 | 256 | 4 | 256 | 256 | 4 |
| mexR::Tn comp | 16 | 64 | 128 | 0.5 | 128 | 64 | 4 |
| mexR::Tn ΔmdrR1 ΔmdrR2 | 128 | 512 | 1,024 | 16 | 1,024 | 256 | 4 |
| Triple comp | 16 | 64 | 128 | 0.5 | 128 | 64 | 4 |
| emrA::Tn | 8 | 32 | 128 | 0.5 | 128 | 16 | 4 |
| emrA::Tn comp | 16 | 64 | 128 | 0.5 | 128 | 64 | 4 |
| emrB::Tn | 8 | 32 | 128 | 0.5 | 128 | 16 | 4 |
| emrB::Tn comp | 16 | 64 | 128 | 0.5 | 128 | 64 | 4 |
MexAB-OprM substrates were carbenicillin (Cb), chloramphenicol (Cm), erythromycin (Ery), norfloxacin (Norf), novobiocin (Nov), and nalidixic acid (Nal). EmrAB and MexAB-OprM pump overlapping substrates were carbenicillin, chloramphenicol, and nalidixic acid. The negative control for MexAB-OprM and EmrAB pumps was amikacin (Amk). comp, complemented.
MdrR1 and MdrR2 control EmrAB.
Another efflux pump, PA3136-PA3137, was also regulated by MdrR1-MdrR2. These genes have not been previously described in P. aeruginosa. However, PA3136 and PA3137 are similar to the emrA and emrB genes of Escherichia coli, respectively, which encode components of a MFS pump that is able to confer resistance to heavy metals, carbonylcyanide m-chlorophenylhydrazone, nalidixic acid, and a number of other toxic compounds (32). Therefore, we do not expect that the downregulation of emrAB in these mdrR mutants is the cause of the increased MICs of the tested antibiotics in Table 2. To validate this, we further performed MIC testing for the emrA::Tn and emrB::Tn mutants as well as their complemented strains. As presented in Table 2, the emrA::Tn and emrB::Tn mutants have slightly decreased levels of resistance to carbenicillin, chloramphenicol, and nalidixic acid, which were completely complemented back to wild-type levels in the complemented strains. Taken together, these data suggest that MdrR1 and MdrR2 regulate two different multidrug efflux pump systems, MexAB-OprM and EmrAB, which have unique and overlapping drug targets (Table 2). Among all the MexAB-OprM substrates tested here, including carbenicillin, chloramphenicol, erythromycin, norfloxacin, novobiocin, and nalidixic acid, the emrAB mutant strains also showed reduced levels of resistance to three of them: carbenicillin, chloramphenicol, and nalidixic acid.
MdrR1 and MdrR2 modulate the production of biofilm through phenazine biosynthesis.
MdrR1 and MdrR2 were shown to directly regulate phenazine production, mexAB-oprM, emrAB, and a putative sensor/response regulator hybrid (Table 1), and somehow, this leads to a defect in biofilm formation (Fig. 1B and C). We sought to further investigate the interconnections between these genes/operons (Table 1) that could be directly linked to this biofilm formation defect. Mutant strains of multiple genes from each of the operons that MdrR1-MdrR2 directly regulated were tested for their ability to form biofilms (Fig. 7A). All mutant strains with an individual mutation in mexA, mexB, oprM, phz2, emrA, emrB, or PA3462 were obtained from the P. aeruginosa two-allele transposon mutant library (53). As shown in Fig. 7A, the majority of the mutant strains showed levels of biofilm production comparable to that of wild-type PAO1, indicating that the modulation of these genes through MdrR1-MdrR2 likely has no effect on biofilm formation. On the contrary, the phenazine synthesis genes were the only ones shown to be involved in biofilm production, as shown in Fig. 7A, as all three mutant strains that had the individual phz2 gene insertionally mutated had significantly decreased biofilm formation abilities in vitro. These effects of mutations in the phz2 genes on biofilm formation are not caused by defects in planktonic growth, as all mutant and wild-type strains have identical growth characteristics, as shown in Fig. S1A in the supplemental material. Additionally, the defects in biofilm formation are complemented by using a single-copy mini-Tn7 integration containing a copy of the wild-type phz2 gene. This new observation that phz2::Tn mutants are defective in biofilm formation is consistent with a previous study showing that phz2 is almost exclusively produced during biofilm development (46). Therefore, our data provided evidence that the phz2 operon is not only expressed exclusively in biofilm (46) but also functionally essential for biofilm development.
FIG 7.

MdrR1 and MdrR2 regulate biofilm formation through the phz2 operon. (A) Various mutant strains of phz2 genes regulated by MdrR1 and MdrR2 have decreased biofilm formation. A biofilm assay was performed in triplicate, and average amounts of biofilm and standard errors of the means are shown. (B) Model of the MdrR1 and MdrR2 regulatory network summarizing the genes that they control. Heat maps display the spatial expression patterns of these genes summarized by using data from a previous study (38). Fold changes of spatially expressed genes are shown with a red-black-green double-color gradient. Green indicates upregulation and red indicates downregulation compared to levels under planktonic growth conditions, as shown in the color gradient bar (log2 fold change, 2 to −2). For each location within the biofilm, the box represents the average fold change of data from three biological replicates. MdrR1 and MdrR2 were shown to repress one multidrug efflux pump (MexAB-OprM) while activating another (EmrAB). Additionally, MdrR1 and MdrR2 act together as an activator for a phenazine biosynthesis operon, phz2, and PA3462, a probable sensor/response regulator hybrid.
DISCUSSION
Through these studies, we have identified the intricate connection between biofilm production and multidrug efflux pumps by the novel dual regulators MdrR1 and MdrR2. Figure 7B summarizes the model of the MdrR1-MdrR2 regulatory network that our data suggested. MdrR1 and MdrR2 were shown to modulate biofilm formation through activating the expression of the phenazine biosynthesis operon phz2. Reduced biofilm formation in the mdrR1::Tn mutant, through reduced expression of phz2 genes, could likely explain the defect of the mdrR1::Tn mutant in colonizing fruit flies and BALB/c mouse lungs. Although not tested here, we suspect that the mdrR2::Tn mutant has a similar defect in in vivo models, since expressions of mdrR1 and mdrR2 were shown by real-time RT-PCR to be correlated through an unidentified mechanism. Our microarray data show that these regulators activate one multidrug efflux pump, EmrAB, while repressing MexAB-OprM (validated by real-time RT-PCR [Table 1] and gene fusion [Fig. 6B]). Additionally, MdrR1 and MdrR2 act as activators for PA3462, a probable sensor/response regulator hybrid (Table 1; see also Table S1 in the supplemental material). We have previously shown global gene expression of genes within the different spatial regions of biofilms (38); hence, it is curious to see what the spatial expression pattern of MdrR1 and MdrR2 is and the genes that they regulate. Heat maps showing the spatial expression patterns of mdrR1, mdrR2, and all the genes that they directly regulate were reanalyzed from our recent publication (38) (Fig. 7B). MdrR1 and MdrR2 have low to moderate expression levels on the surface and the middle of biofilms and are downregulated in the interior of the biofilm compared to planktonic growth (Fig. 7B). Overall, the spatial expression patterns of MdrR1 and MdrR2 agree mostly, because the genes that they activate, including the phz2 operon, the sensor/response regulator hybrid PA3462, and the emrAB genes, all show similar trends of low to moderate levels of expression on the surface and middle of biofilms and downregulation in the interior of biofilms (Fig. 7B). However, the MexAB-OprM operon, on the other hand, shows a unique spatial expression pattern with relatively high expression levels on the surface of the biofilm and downregulated expression in the middle, and the expression level in the interior of the biofilm is then comparable to that of planktonic growth. This unique spatial expression pattern of mexAB-oprM is likely the result of its complex regulatory network, involving multiple regulators working together to fine-tune the spatial expression of this important efflux system, as previous studies have revealed (24–26, 29–31).
The data presented here provide valuable insight into the regulation mechanisms of P. aeruginosa biofilms and multidrug efflux pumps. The regulation of P. aeruginosa biofilms and antibiotic resistance is a very complex and sophisticated system that our data revealed to be connected through the novel dual regulators MdrR1 and MdrR2. These dual regulators regulate two multidrug efflux pump systems, a phenazine biosynthesis operon, as well as another putative transcriptional regulator, PA3462. Future study on this additional transcriptional regulator could lead to the expansion of the regulation network controlled by MdrR1 and MdrR2. It is particularly interesting that MdrR1 and MdrR2 act as repressors for one multidrug efflux pump (MexAB-OprM) and as activators for another multidrug efflux pump (EmrAB). Since these efflux pumps have mostly different ranges of substrate specificities, MdrR1 and MdrR2 could act as a master modulator controlling the activities of various pumps under unique growth conditions as well as in different microenvironments within the stratified biofilm structure. Our data showed that emrAB mutations caused decreased MICs of a few antibiotics that MexAB-OprM also targets (i.e., carbenicillin, nalidixic acid, and chloramphenicol). This observation is somewhat surprising because emr::Tn mutants still harbor intact MexAB-OprM, which is efficient at expelling these drugs. There are two possible explanations. One is that EmrAB is very effective in effluxing these antibiotics, and therefore, the deletion of emrAB resulted in observable reductions in MICs. Another possibility is that this effect of the emrAB mutation on MICs is due to feedback inhibition of MexAB-OprM expression through MdrR1 and MdrR2. The first hypothesis is more simple and direct; however, the second hypothesis is supported by the fact that the MdrR12 mutant showed increased resistance to carbenicillin, nalidixic acid, and chloramphenicol through upregulation of MexAB-OprM, masking the effect of EmrAB downregulation. This suggests that MexAB-OprM is more efficient at expelling these antibiotics than EmrAB.
Our data directly link the spatial gene expression profiles of MdrR1 and MdrR2 to the stratified expression patterns of MexAB-OprM and EmrAB. Therefore, these new findings challenge the role of the biofilm matrix passively acting as a molecular sieve or sink that leads to drug resistance and support the idea that multidrug efflux pumps induced during biofilm growth under various conditions contribute to drug resistance. Additionally, MdrR1 and MdrR2 act as an activator for the phz2 operon; this results in phenazine production genes coregulated with emrAB and reciprocally regulated with mexAB-oprM. The ability of MdrR1 and MdrR2 to act as activators or repressors for different genes allows, to some extent, the independent modulation of different pathways (phenazine production and efflux pumps) and phenotypes (biofilm formation and drug resistance). These regulation mechanisms suggest the existence of an environmental or internal signal(s) that modulates biofilm formation and drug resistance through MdrR1 and MdrR2. Future studies could focus on this potential environmental or intracellular signal(s) that affects the regulation mechanism of MdrR1 and MdrR2 and their biological relevance in vivo. In conclusion, the present study identified a regulation bridge between diverse virulence processes of P. aeruginosa that furthers the understanding of this important opportunistic pathogen.
MATERIALS AND METHODS
Bacterial strains, media, and culture conditions.
E. coli EPMax10B-lacIq-pir was routinely used as a cloning strain. The P. aeruginosa wild-type strain PAO1 and its derivatives were cultured in Luria-Bertani (LB) medium with appropriate antibiotics, as previously described (38, 49).
Molecular reagents.
All reagents and molecular methods used are as previously described (54).
Microarray data analysis and real-time RT-PCR validation.
Growth of the wild-type PAO1 strain and the PA3898::Tn mutant strain, total RNA purification, and microarray analysis were performed as previously described (38). Strains were grown overnight in LB medium, diluted 200-fold into fresh LB medium, and grown under static biofilm growth conditions (no shaking) for 48 h at 30°C. Bacteria were harvested, and total RNA was isolated by using a Qiagen RNeasy minikit (Qiagen), with on-column DNase I treatment. Final RNA concentrations and purities were determined on a Thermo Scientific NanoDrop spectrophotometer (A260/280 of between 1.8 and 2.0). cDNA was synthesized and used for microarray analysis in triplicate as previously described (55).
The same RNA samples as the ones described above were processed with additional off-column DNase I digestion and then used for real-time RT-PCR validation. Primers for each gene were designed by using Integrated DNA Technologies Primer Quest software. Supermixes for all reactions were made and aliquoted into subsupermixes for each gene assayed. Each real-time PCR mixture contained 10 μl of cDNA, 12.5 μl of iQ SYBR green supermix (Bio-Rad), and 120 nM each forward and reverse primers, in a final volume of 25 μl. Real-time PCR was performed on the iCycler iQ instrument (Bio-Rad) with the following protocol: a denaturation step (95°C for 2 min) and 55 cycles of amplification and quantification (95°C for 20 s, 60°C for 30 s, and 72°C for 20 s). Melt curve analysis was performed for all reactions, and single peaks for all amplicons confirmed specific PCR amplification. Real-time RT-PCR values were averaged over 8 replicates for each gene/condition, and fold changes were calculated by using data analysis for real-time PCR (DART-PCR) as previously described (56).
Gene fusion and mutant strain construction.
All standard genetic manipulations were performed as previously described (38, 49, 52, 54, 55, 57, 58). All single mutant strains used throughout this study, including mdrR1::Tn, mdrR2::Tn, mexR::Tn, emrA::Tn, emrB::Tn, and all the mutant strains shown in Fig. 7A, were obtained from the two-allele P. aeruginosa transposon library (53). The double and triple mutant strains in Table 2, mdrR1::Tn ΔmdrR2 and mexR::Tn ΔmdrR1 ΔmdrR2, were constructed by using an established allelic-exchange method in the transposon mutant background (52). Complementation was done via single-copy mini-Tn7 integration as previously described (38).
Phenotypic studies.
A crystal violet biofilm assay and confocal microscopy for static biofilms were carried out in triplicate as previously described (38, 49). Briefly, wild-type PAO1 and PA3898::Tn mutant strains were first grown overnight in LB medium with shaking. Cultures grown overnight were then harvested, washed twice, and subcultured (200× dilution) into fresh LB medium. Biofilm was grown in triplicate in a 96-well plate with a non-tissue-culture-treated surface for 48 h at 30°C. Planktonic cells were then washed off, and biofilm formed in the wells was stained with crystal violet and quantitated according to an established protocol (59). Average amounts of biofilm and standard errors of the means are shown. Statistical analysis (Student’s t test) was done by using Prism software. Biofilms were grown identically on non-tissue-culture-treated glass-bottom plates for confocal microscopy. After incubation at 48 h at 30°C, biofilms were washed as described above, fixed using 4% paraformaldehyde, and stained using Thermo Fisher FilmTracer SYPRO ruby biofilm matrix stain. Images were scanned using an Olympus FV-1000 confocal microscope and processed with ImageJ software.
D. melanogaster and BALB/c mouse infection studies were performed similarly to methods previously described (38, 49). Wild-type PAO1, the PA3898::Tn mutant, and its complemented strains were used to infect D. melanogaster Canton-S strain flies to investigate their in vivo pathogenesis. For easy visualization, pUCP20-rfp and pUCP20-gfp were introduced into the PA3898::Tn mutant and complemented strains, respectively. In vivo competitive index determinations, crop dissection for imaging, and survival studies were performed in triplicate as previously described (38, 49). All BALB/c mouse experiments were approved by the University of Hawaii Institutional Animal Care and Use Committee (protocol no. 06–023) and performed in compliance with guidelines of the National Institutes of Health Guide for the Care and Use of Laboratory Animals (60). Six- to eight-week-old male BALB/c mice were purchased from Charles River Laboratories and used for the study. An in vivo competition study was performed by using 30 μl of a mixture of the mutant strain and the complemented strain (3 × 107 CFU each) resuspended in purified bacterium-free alginate as previously described (38, 49). Statistical analysis for competitive index studies (one-sample t test) was done using Prism software.
Molecular and biochemical characterization of the interaction between the dual regulator and DNA.
ChIP-seq, protein purification, coimmunoprecipitation, electrophoretic mobility shift assays (EMSAs), and DNA footprinting were performed as previously described (49).
Kinetics measurements using localized surface plasmon resonance.
LSPR was performed by using an NTA-gold sensor chip on a Nicoya OpenSPR instrument, according to the manufacturer’s instructions. For studying the binding interaction of the complex, purified His6-MdrR2 was first covalently immobilized on an NTA sensor in a nonrandom orientation using the capture-coupling method (61). Untagged MdrR1 was purified by using expression vector pTXB1 as previously described (62). Binding of untagged MdrR1 and/or DNA containing the predicted binding motif to the immobilized ligand on the biosensor was performed under the following conditions. Association measurements were obtained by injecting solutions of purified untagged MdrR1 and DNA containing the binding motif at various concentrations (11 nM, 33 nM, and 300 nM each MdrR1 and DNA) in HBS buffer (20 mM HEPES, 150 mM NaCl [pH 7.0]) and measuring the increase in signals for 300 s. Dissociation measurements were obtained by injecting a fresh solution of HBS buffer and measuring the decrease in signals for 700 s. Three negative controls were tested, including MdrR1 alone, DNA containing the binding motif alone, as well as MdrR1 and DNA with the binding motif deleted, at 100 nM each.
Antibiotic susceptibility test.
The MIC of each antibiotic was determined on Mueller-Hinton agar by a previously described 2-fold dilution method (27, 63). Mueller-Hinton agar plates containing serial 2-fold dilutions of each antibiotic were prepared. The range of antibiotic concentrations were as follows: 2 to 256 μg/ml carbenicillin, 8 to 1,024 μg/ml chloramphenicol, 16 to 2,048 μg/ml erythromycin and novobiocin, 0.125 to 16 μg/ml norfloxacin, 4 to 512 μg/ml nalidixic acid, and 0.25 to 32 μg/ml amikacin. P. aeruginosa cultures grown overnight were diluted 100-fold in fresh Mueller-Hinton broth, grown to the mid-log phase (optical density at 600 nm [OD600] of approximately 0.6), harvested, and washed in 1× phosphate-buffered saline (PBS). The Mueller-Hinton agar plates were spotted with 3 μl of the diluted bacterial suspensions (approximately 1 × 104 CFU). The MIC was defined as the concentration of antibiotic at which bacterial growth was absent after incubation at 37°C for 20 h. The MIC tests were performed in triplicate.
Data availability.
Microarray and ChIP-seq data sets are available in the NCBI Gene Expression Omnibus (GEO) (64) and accessible through GEO series accession no. GSE120009 and GSE120082, respectively.
Supplementary Material
ACKNOWLEDGMENTS
This project was supported by U.S. National Institutes of Health (NIH)/National Institute of General Medical Sciences (NIGMS) grant no. R01GM103580 and in part by U.S. NIH/National Institute of Allergy and Infectious Diseases (NIAID) grant no. R21AI123913. P. aeruginosa DNA arrays were obtained through the NIAID Pathogen Functional Genomics Resource Center, managed and funded by the Division of Microbiology and Infectious Diseases, NIAID, NIH, DHHS, and operated by the J. Craig Venter Institute.
We acknowledge Dawson Fogenn and Allexa Dow for assisting in the LSPR experimental setup and Sladjana Prisic for the use of the plasmon resonance instrument. We thank Darlene Cabanas for language editing and proofreading of the manuscript.
We declare no competing financial interests.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.01459-18.
REFERENCES
- 1.Mittelman MW. 1998. Structure and functional characteristics of bacterial biofilms in fluid processing operations. J Dairy Sci 81:2760–2764. doi: 10.3168/jds.S0022-0302(98)75833-3. [DOI] [PubMed] [Google Scholar]
- 2.Reed D, Kemmerly SA. 2009. Infection control and prevention: a review of hospital-acquired infections and the economic implications. Ochsner J 9:27–31. [PMC free article] [PubMed] [Google Scholar]
- 3.Leaper D, McBain AJ, Kramer A, Assadian O, Sanchez JL, Lumio J, Kiernan M. 2010. Healthcare associated infection: novel strategies and antimicrobial implants to prevent surgical site infection. Ann R Coll Surg Engl 92:453–458. doi: 10.1308/003588410X12699663905276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van Acker H, Van Dijck P, Coenye T. 2014. Molecular mechanisms of antimicrobial tolerance and resistance in bacterial and fungal biofilms. Trends Microbiol 22:326–333. doi: 10.1016/j.tim.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 5.Davies D. 2003. Understanding biofilm resistance to antibacterial agents. Nat Rev Drug Discov 2:114–122. doi: 10.1038/nrd1008. [DOI] [PubMed] [Google Scholar]
- 6.Mah T-FC, O’Toole GA. 2001. Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol 9:34–39. doi: 10.1016/S0966-842X(00)01913-2. [DOI] [PubMed] [Google Scholar]
- 7.O’Toole GA, Kolter R. 1998. Initiation of biofilm formation in Pseudomonas fluorescens WCS365 proceeds via multiple, convergent signalling pathways: a genetic analysis. Mol Microbiol 28:449–461. doi: 10.1046/j.1365-2958.1998.00797.x. [DOI] [PubMed] [Google Scholar]
- 8.Sauer K, Camper AK, Ehrlich GD, Costerton JW, Davies DG. 2002. Pseudomonas aeruginosa displays multiple phenotypes during development as a biofilm. J Bacteriol 184:1140–1154. doi: 10.1128/jb.184.4.1140-1154.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stoodley P, Sauer K, Davies DG, Costerton JW. 2002. Biofilms as complex differentiated communities. Annu Rev Microbiol 56:187–209. doi: 10.1146/annurev.micro.56.012302.160705. [DOI] [PubMed] [Google Scholar]
- 10.Kostakioti M, Hadjifrangiskou M, Hultgren SJ. 2013. Bacterial biofilms: development, dispersal, and therapeutic strategies in the dawn of the postantibiotic era. Cold Spring Harb Perspect Med 3:a010306. doi: 10.1101/cshperspect.a010306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fazli M, Almblad H, Rybtke ML, Givskov M, Eberl L, Tolker-Nielsen T. 2014. Regulation of biofilm formation in Pseudomonas and Burkholderia species. Environ Microbiol 16:1961–1981. doi: 10.1111/1462-2920.12448. [DOI] [PubMed] [Google Scholar]
- 12.Balasubramanian D, Schneper L, Kumari H, Mathee K. 2013. A dynamic and intricate regulatory network determines Pseudomonas aeruginosa virulence. Nucleic Acids Res 41:1–20. doi: 10.1093/nar/gks1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mann EE, Wozniak DJ. 2012. Pseudomonas biofilm matrix composition and niche biology. FEMS Microbiol Rev 36:893–916. doi: 10.1111/j.1574-6976.2011.00322.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jimenez PN, Koch G, Thompson JA, Xavier KB, Cool RH, Quax WJ. 2012. The multiple signaling systems regulating virulence in Pseudomonas aeruginosa. Microbiol Mol Biol Rev 76:46–65. doi: 10.1128/MMBR.05007-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Breidenstein EBM, de la Fuente-Núñez C, Hancock REW. 2011. Pseudomonas aeruginosa: all roads lead to resistance. Trends Microbiol 19:419–426. doi: 10.1016/j.tim.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 16.Schuster M, Greenberg EP. 2006. A network of networks: quorum-sensing gene regulation in Pseudomonas aeruginosa. Int J Med Microbiol 296:73–81. doi: 10.1016/j.ijmm.2006.01.036. [DOI] [PubMed] [Google Scholar]
- 17.Kipnis E, Sawa T, Wiener-Kronish J. 2006. Targeting mechanisms of Pseudomonas aeruginosa pathogenesis. Med Mal Infect 36:78–91. doi: 10.1016/j.medmal.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 18.Vincent JL. 2003. Nosocomial infections in adult intensive-care units. Lancet 361:2068–2077. doi: 10.1016/S0140-6736(03)13644-6. [DOI] [PubMed] [Google Scholar]
- 19.Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, Brinkman FS, Hufnagle WO, Kowalik DJ, Lagrou M, Garber RL, Goltry L, Tolentino E, Westbrock-Wadman S, Yuan Y, Brody LL, Coulter SN, Folger KR, Kas A, Larbig K, Lim R, Smith K, Spencer D, Wong GK, Wu Z, Paulsen IT, Reizer J, Saier MH, Hancock RE, Lory S, Olson MV. 2000. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406:959–964. doi: 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- 20.Doring G. 1997. Cystic fibrosis respiratory infections: interactions between bacteria and host defense. Monaldi Arch Chest Dis 52:363–366. [PubMed] [Google Scholar]
- 21.Poole K. 2011. Pseudomonas aeruginosa: resistance to the max. Front Microbiol 2:65. doi: 10.3389/fmicb.2011.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Daury L, Orange F, Taveau JC, Verchere A, Monlezun L, Gounou C, Marreddy RK, Picard M, Broutin I, Pos KM, Lambert O. 2016. Tripartite assembly of RND multidrug efflux pumps. Nat Commun 7:10731. doi: 10.1038/ncomms10731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Terzi HA, Kulah C, Ciftci IH. 2014. The effects of active efflux pumps on antibiotic resistance in Pseudomonas aeruginosa. World J Microbiol Biotechnol 30:2681–2687. doi: 10.1007/s11274-014-1692-2. [DOI] [PubMed] [Google Scholar]
- 24.Evans K, Adewoye L, Poole K. 2001. MexR repressor of the mexAB-oprM multidrug efflux operon of Pseudomonas aeruginosa: identification of MexR binding sites in the mexA-mexR intergenic region. J Bacteriol 183:807–812. doi: 10.1128/JB.183.3.807-812.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morita Y, Cao L, Gould VC, Avison MB, Poole K. 2006. nalD encodes a second repressor of the mexAB-oprM multidrug efflux operon of Pseudomonas aeruginosa. J Bacteriol 188:8649–8654. doi: 10.1128/JB.01342-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liao J, Schurr MJ, Sauer K. 2013. The MerR-like regulator BrlR confers biofilm tolerance by activating multidrug efflux pumps in Pseudomonas aeruginosa biofilms. J Bacteriol 195:3352–3363. doi: 10.1128/JB.00318-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tian Z-X, Yi X-X, Cho A, O’Gara F, Wang Y-P. 2016. CpxR activates MexAB-OprM efflux pump expression and enhances antibiotic resistance in both laboratory and clinical nalB-type isolates of Pseudomonas aeruginosa. PLoS Pathog 12:e1005932. doi: 10.1371/journal.ppat.1005932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang F, He Q, Yin J, Xu S, Hu W, Gu L. 2018. BrlR from Pseudomonas aeruginosa is a receptor for both cyclic di-GMP and pyocyanin. Nat Commun 9:2563. doi: 10.1038/s41467-018-05004-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cao L, Srikumar R, Poole K. 2004. MexAB-OprM hyperexpression in NalC-type multidrug-resistant Pseudomonas aeruginosa: identification and characterization of the nalC gene encoding a repressor of PA3720-PA3719. Mol Microbiol 53:1423–1436. doi: 10.1111/j.1365-2958.2004.04210.x. [DOI] [PubMed] [Google Scholar]
- 30.Wilke MS, Heller M, Creagh AL, Haynes CA, McIntosh LP, Poole K, Strynadka NC. 2008. The crystal structure of MexR from Pseudomonas aeruginosa in complex with its antirepressor ArmR. Proc Natl Acad Sci U S A 105:14832–14837. doi: 10.1073/pnas.0805489105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hall CW, Zhang L, Mah TF. 2017. PA3225 is a transcriptional repressor of antibiotic resistance mechanisms in Pseudomonas aeruginosa. Antimicrob Agents Chemother 61:e02114-16. doi: 10.1128/AAC.02114-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lomovskaya O, Lewis K. 1992. Emr, an Escherichia coli locus for multidrug resistance. Proc Natl Acad Sci U S A 89:8938–8942. doi: 10.1073/pnas.89.19.8938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Poole K. 2004. Efflux-mediated multiresistance in Gram-negative bacteria. Clin Microbiol Infect 10:12–26. doi: 10.1111/j.1469-0691.2004.00763.x. [DOI] [PubMed] [Google Scholar]
- 34.Kumar A, Schweizer HP. 2005. Bacterial resistance to antibiotics: active efflux and reduced uptake. Adv Drug Deliv Rev 57:1486–1513. doi: 10.1016/j.addr.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 35.Muller JF, Stevens AM, Craig J, Love NG. 2007. Transcriptome analysis reveals that multidrug efflux genes are upregulated to protect Pseudomonas aeruginosa from pentachlorophenol stress. Appl Environ Microbiol 73:4550–4558. doi: 10.1128/AEM.00169-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang Y, Norris MH, Zarzycki-Siek J, Nierman WC, Donachie SP, Hoang TT. 2011. Transcript amplification from single bacterium for transcriptome analysis. Genome Res 21:925–935. doi: 10.1101/gr.116103.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kang Y, McMillan I, Norris MH, Hoang TT. 2015. Single prokaryotic cell isolation and total transcript amplification protocol for transcriptomic analysis. Nat Protoc 10:974–984. doi: 10.1038/nprot.2015.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heacock-Kang Y, Sun Z, Zarzycki-Siek J, McMillan IA, Norris MH, Bluhm AP, Cabanas D, Fogen D, Vo H, Donachie SP, Borlee BR, Sibley CD, Lewenza S, Schurr MJ, Schweizer HP, Hoang TT. 2017. Spatial transcriptomes within the Pseudomonas aeruginosa biofilm architecture. Mol Microbiol 106:976–985. doi: 10.1111/mmi.13863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mulcahy H, Sibley CD, Surette MG, Lewenza S. 2011. Drosophila melanogaster as an animal model for the study of Pseudomonas aeruginosa biofilm infections in vivo. PLoS Pathog 7:e1002299. doi: 10.1371/journal.ppat.1002299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hoffmann N, Rasmussen TB, Jensen PO, Stub C, Hentzer M, Molin S, Ciofu O, Givskov M, Johansen HK, Hoiby N. 2005. Novel mouse model of chronic Pseudomonas aeruginosa lung infection mimicking cystic fibrosis. Infect Immun 73:2504–2514. doi: 10.1128/IAI.73.4.2504-2514.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Finn RD, Attwood TK, Babbitt PC, Bateman A, Bork P, Bridge AJ, Chang HY, Dosztanyi Z, El-Gebali S, Fraser M, Gough J, Haft D, Holliday GL, Huang H, Huang X, Letunic I, Lopez R, Lu S, Marchler-Bauer A, Mi H, Mistry J, Natale DA, Necci M, Nuka G, Orengo CA, Park Y, Pesseat S, Piovesan D, Potter SC, Rawlings ND, Redaschi N, Richardson L, Rivoire C, Sangrador-Vegas A, Sigrist C, Sillitoe I, Smithers B, Squizzato S, Sutton G, Thanki N, Thomas PD, Tosatto SC, Wu CH, Xenarios I, Yeh LS, Young SY, Mitchell AL. 2017. InterPro in 2017—beyond protein family and domain annotations. Nucleic Acids Res 45:D190–D199. doi: 10.1093/nar/gkw1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li XZ, Nikaido H, Poole K. 1995. Role of mexA-mexB-oprM in antibiotic efflux in Pseudomonas aeruginosa. Antimicrob Agents Chemother 39:1948–1953. doi: 10.1128/AAC.39.9.1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Handfield M, Lehoux DE, Sanschagrin F, Mahan MJ, Woods DE, Levesque RC. 2000. In vivo-induced genes in Pseudomonas aeruginosa. Infect Immun 68:2359–2362. doi: 10.1128/IAI.68.4.2359-2362.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.West SE, Sample AK, Runyen-Janecky LJ. 1994. The vfr gene product, required for Pseudomonas aeruginosa exotoxin A and protease production, belongs to the cyclic AMP receptor protein family. J Bacteriol 176:7532–7542. doi: 10.1128/jb.176.24.7532-7542.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mulcahy H, O’Callaghan J, O’Grady EP, Macia MD, Borrell N, Gomez C, Casey PG, Hill C, Adams C, Gahan CG, Oliver A, O’Gara F. 2008. Pseudomonas aeruginosa RsmA plays an important role during murine infection by influencing colonization, virulence, persistence, and pulmonary inflammation. Infect Immun 76:632–638. doi: 10.1128/IAI.01132-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Recinos DA, Sekedat MD, Hernandez A, Cohen TS, Sakhtah H, Prince AS, Price-Whelan A, Dietrich LE. 2012. Redundant phenazine operons in Pseudomonas aeruginosa exhibit environment-dependent expression and differential roles in pathogenicity. Proc Natl Acad Sci U S A 109:19420–19425. doi: 10.1073/pnas.1213901109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kilmury SL, Burrows LL. 2016. Type IV pilins regulate their own expression via direct intramembrane interactions with the sensor kinase PilS. Proc Natl Acad Sci U S A 113:6017–6022. doi: 10.1073/pnas.1512947113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. 2009. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37:W202–W208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heacock-Kang Y, Zarzycki-Siek J, Sun Z, Poonsuk K, Bluhm AP, Cabanas D, Fogen D, McMillan IA, Chuanchuen R, Hoang TT. 31 July 2018. Novel dual regulators of Pseudomonas aeruginosa essential for productive biofilms and virulence. Mol Microbiol doi: 10.1111/mmi.14063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zianni M, Tessanne K, Merighi M, Laguna R, Tabita FR. 2006. Identification of the DNA bases of a DNase I footprint by the use of dye primer sequencing on an automated capillary DNA analysis instrument. J Biomol Tech 17:103–113. [PMC free article] [PubMed] [Google Scholar]
- 51.Poole K, Tetro K, Zhao Q, Neshat S, Heinrichs D, Bianco N. 1996. Expression of the multidrug resistance operon mexA-mexB-oprM in Pseudomonas aeruginosa: mexR encodes a regulator of operon expression. Antimicrob Agents Chemother 40:2021–2028. doi: 10.1128/AAC.40.9.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77–86. doi: 10.1016/S0378-1119(98)00130-9. [DOI] [PubMed] [Google Scholar]
- 53.Jacobs MA, Alwood A, Thaipisuttikul I, Spencer D, Haugen E, Ernst S, Will O, Kaul R, Raymond C, Levy R, Chun-Rong L, Guenthner D, Bovee D, Olson MV, Manoil C. 2003. Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc Natl Acad Sci 100:14339–14344. doi: 10.1073/pnas.2036282100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kang Y, Lunin VV, Skarina T, Savchenko A, Schurr MJ, Hoang TT. 2009. The long-chain fatty acid sensor, PsrA, modulates the expression of rpoS and the type III secretion exsCEBA operon in Pseudomonas aeruginosa. Mol Microbiol 73:120–136. doi: 10.1111/j.1365-2958.2009.06757.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kang Y, Nguyen DT, Son MS, Hoang TT. 2008. The Pseudomonas aeruginosa PsrA responds to long-chain fatty acid signals to regulate the fadBA5 b-oxidation operon. Microbiology 154:1584–1598. doi: 10.1099/mic.0.2008/018135-0. [DOI] [PubMed] [Google Scholar]
- 56.Peirson SN, Butler JN, Foster RG. 2003. Experimental validation of novel and conventional approaches to quantitative real-time PCR data analysis. Nucleic Acids Res 31:e73. doi: 10.1093/nar/gng073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Choi KH, Schweizer HP. 2006. Mini-Tn7 insertion in bacteria with single attTn7 sites: example Pseudomonas aeruginosa. Nat Protoc 1:153–161. doi: 10.1038/nprot.2006.24. [DOI] [PubMed] [Google Scholar]
- 58.Choi K-H, Gaynor JB, White KG, Lopez C, Bosio CM, Karkhoff-Schweizer RR, Schweizer HP. 2005. A Tn7-based broad-range bacterial cloning and expression system. Nat Methods 2:443–448. doi: 10.1038/nmeth765. [DOI] [PubMed] [Google Scholar]
- 59.Merritt JH, Kadouri DE, O’Toole GA. 2005. Growing and analyzing static biofilms. Curr Protoc Microbiol Chapter 1:Unit 1B.1. doi: 10.1002/9780471729259.mc01b01s00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
- 61.Kimple AJ, Muller RE, Siderovski DP, Willard FS. 2010. A capture coupling method for the covalent immobilization of hexahistidine tagged proteins for surface plasmon resonance. Methods Mol Biol 627:91–100. doi: 10.1007/978-1-60761-670-2_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chong S, Mersha FB, Comb DG, Scott ME, Landry D, Vence LM, Perler FB, Benner J, Kucera RB, Hirvonen CA, Pelletier JJ, Paulus H, Xu MQ. 1997. Single-column purification of free recombinant proteins using a self-cleavable affinity tag derived from a protein splicing element. Gene 192:271–281. doi: 10.1016/S0378-1119(97)00105-4. [DOI] [PubMed] [Google Scholar]
- 63.European Committee for Antimicrobial Susceptibility Testing of the European Society of Clinical Microbiology and Infectious Diseases. 2000. EUCAST definitive document E.DEF 3.1, June 2000: determination of minimum inhibitory concentrations (MICs) of antibacterial agents by agar dilution. Clin Microbiol Infect 6:509–515. doi: 10.1046/j.1469-0691.2000.00142.x. [DOI] [PubMed] [Google Scholar]
- 64.Edgar R, Domrachev M, Lash AE. 2002. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Microarray and ChIP-seq data sets are available in the NCBI Gene Expression Omnibus (GEO) (64) and accessible through GEO series accession no. GSE120009 and GSE120082, respectively.