

Abstract
Bartonella is a genetically diverse group of vector-borne bacteria. Over 40 species have been characterized to date, mainly from mammalian reservoirs and arthropod vectors. Rodent reservoirs harbor one of the largest Bartonella diversity described to date, and novel species and genetic variants are continuously identified from these hosts. Yet, it is still unknown if this significant genetic diversity stems from adaptation to different niches or from intrinsic high mutation rates. Here, we explored the vertical occurrence of spontaneous genomic alterations in 18 lines derived from two rodent-associated Bartonella elizabethae-like strains, evolved in nonselective agar plates under conditions mimicking their vector- and mammalian-associated temperatures, and the transmission cycles between them (i.e., 26 °C, 37 °C, and alterations between the two), using mutation accumulation experiments. After ∼1,000 generations, evolved genomes revealed few point mutations (average of one-point mutation per line), evidencing conserved single-nucleotide mutation rates. Interestingly, three large structural genomic changes (two large deletions and an inversion) were identified over all lines, associated with prophages and surface adhesin genes. Particularly, a prophage, deleted during constant propagation at 37 °C, was associated with an increased autonomous replication at 26 °C (the flea-associated temperature). Complementary molecular analyses of wild strains, isolated from desert rodents and their fleas, further supported the occurrence of structural genomic variations and prophage-associated deletions in nature. Our findings suggest that structural genomic changes represent an effective intrinsic mechanism to generate diversity in slow-growing bacteria and emphasize the role of prophages as promoters of diversity in nature.
Keywords: experimental evolution, vertical inheritance, structural variations, mutation accumulation, rodents, slow growing bacteria
Introduction
Bacterial genomes evolve through the accumulation of vertically (i.e., clonally) inherited genetic alterations and by the horizontal exchange of genetic material (Bryant et al. 2012). While the latter depends on several factors, such as the encounters and interactions with foreign DNA, the former depends mainly on the mutation rates of each bacterial species (Barrick and Lenski 2013). Correspondingly, vertically inherited genetic alterations include single-nucleotide variations (SNVs), such as base substitutions and/or short insertions and deletions (i.e., indels), and genomic structural variations (SVs) comprising larger deletions and insertions, and sequence rearrangements (e.g., inversions, translocations, duplications; Barrick and Lenski 2013; Bryant et al. 2012). Experimental evolutionary studies allow the screening and measurement of these genetic changes under controlled conditions, leading to the determination of the organism’s mutational profiles (Lind and Andersson 2008; Lee et al. 2012; Barrick and Lenski 2013). In particular, spontaneous vertically inherited changes can be explored by mutation accumulation assays and whole genome sequencing, through the evolution of lines derived from a common ancestor, without bias of fitness effects (Lind and Andersson 2008; Lee et al. 2012; Heilbron et al. 2014; Foster et al. 2015). Despite the current availability and accessibility of these methods, experimental evolutionary studies integrating both SNV and SV screenings in bacterial models, particularly using mutation accumulation experiments are scarce, and merely explored in only few laboratory-selected fast-growing bacteria, such as Escherichia coli and Pseudomonas aeruginosa (Barrick et al. 2014; Heilbron et al. 2014). Thus, the determination of the type and frequency of vertically inherited genetic changes experienced by bacteria, with different growth and ecological characteristics, are essential steps in understanding evolutionary potential of unexplored taxa.
The Bartonella genus (α2-Proteobacteria) comprises a remarkable genetic diverse group of slow growing and emerging pathogenic Gram-negative bacteria, adapted to a broad range of ecological niches. To date, >40 species have been characterized from multiple mammal and arthropod species, including pathogenic species of humans and domestic animals (Chomel et al. 2009; Harms and Dehio 2012; Kosoy et al. 2012; Kesnerova et al. 2016; Okaro et al. 2017). Remarkably, an extensive and increasing list of molecularly divergent variants of Bartonella spp. have been discovered in nature, especially from rodents and their flea hosts (Bai et al. 2009; Harrus et al. 2009; Inoue et al. 2009). Moreover, genomic comparative analyses of different Bartonella spp. have revealed large genomic variations, such as rearrangements and deletions (Alsmark et al. 2004; Lindroos et al. 2006; Berglund et al. 2009; Guy et al. 2013; Harms et al. 2017). These studies have suggested that phage-related elements (e.g., prophages and gene transfer agents (GTAs) associated with a run-off replication origin) have contributed largely to the variability and evolution of some Bartonella lineages (Alsmark et al. 2004; Lindroos et al. 2006; Berglund et al. 2009; Guy et al. 2013). On the other hand, single-nucleotide mutational divergence (nonrecombinational) has been disclosed as a major force of variation in other lineages, such as Bartonella bacilliformis and Bartonella apis (Paul et al. 2016; Segers et al. 2017). These in silico studies attest to the plasticity and capability of Bartonella to generate genetic diversity, and designated Bartonella as an attractive model for the study of molecular evolution and adaptability. However, no experimental study has evaluated the role of vertical-inherited mechanisms (either SNVs and/or SVs) generating genetic diversity in Bartonella genomes. Thus, to broaden our understanding of the emerging pathogenic potential and host-adaptability of Bartonella and other slow-growing bacteria, investigation of the evolutionary patterns and mechanisms of vertical inheritance in a whole-genome level are essential and were performed herein.
In the present study, two wild-type Bartonella spp. (i.e., Bartonella sp. OE 1-1 and Bartonella sp. Tel Aviv), closely related to the zoonotic rat-borne bacterium Bartonella elizabethae, were clonally evolved in mutation accumulation experiments under conditions mimicking their flea and mammalian associated temperatures. After >1,000 generations, vertical evolutionary alterations were investigated in the evolved genomes. Complementary genomic analyses of wild Bartonella sp. OE 1-1 strains, collected from sympatric wild rodents and fleas, were carried out to confirm that the genomic alterations observed in the experimentally evolved strains occur also in nature. This study sheds light on the drivers and underlying mechanisms that lead to the evolution of vector-borne slow-growing bacteria, emphasizing the role of prophages as relevant promoters of genome-diversity and their association with particular environmental niches.
Materials and Methods
Bacterial Strains and Media
For the evolutionary experimental assays, two wild-type Bartonella spp. maintained in the laboratory were chosen: 1) Bartonella sp. Tel Aviv strain was isolated from the blood of a Rattus rattus rat collected from Tel Aviv, Israel (Harrus et al. 2009); and 2) Bartonella sp. OE 1-1 was isolated from a Synosternus cleopatrae flea collected from a Gerbillus andersoni gerbil in the Negev Desert, Israel. To obtain genetically homogeneous founders, ancestral strains were passaged through at least four bottleneck-cultures before selecting the study ancestral lines. Nonselective chocolate agar plates (Novamed, Ltd., Jerusalem, ISR) were used for all agar cultures and passages included in this study. Agar plates were incubated at 26 °C or 37 °C with constant 5% CO2, to emulate the flea and the rodent associated temperatures. Additional details can be found in supplementary Methods, Supplementary Material online.
As a complementary evaluation, the genomic structure of wild Bartonella sp. OE 1-1 strains collected from sympatric wild animals were isolated and processed. Accordingly, wild Gerbillus spp. were caught in October 2016 from the Western Negev, Israel, and samples were collected following methods described elsewhere (Gutiérrez et al. 2014). Bartonella isolation was performed following previously described methods (Gutiérrez et al. 2017). The study was approved by the Hebrew University Institutional Animal Care and Use Committee (IACUC; approval number MD-14-14293-2), the Ben-Gurion University IACUC (approval number IL-28-04-2015), and by the Israel Nature and National Parks Protection Authority (approval number 2016/41428).
Mutation Accumulation Experiments
Mutation accumulation experiments were performed following methods described elsewhere (Lee et al. 2012). Briefly, nine parallel lines from each ancestral strain were repeatedly and independently passaged in a nonselective medium (chocolate agar) at three different temperature conditions, by a random picking of a single colony forming unit (CFU) and its subsequent spread into a new plate, for 50 times. Accordingly, 18 lines were evolved under the following conditions: group A) six lines were evolved constantly at 26 °C (i.e., all agar passages maintained at this temperature), mimicking the flea associated temperature (Shenbrot et al. 2002; Krasnov 2008); group B) six lines were evolved constantly at 37 °C (i.e., all agar passages maintained at this temperature), mimicking the mammalian body temperature; and group C) six lines were evolved by alternating the agar passages between 26 °C and 37 °C, mimicking the transmission cycles of Bartonella between fleas and rodents (fig. 1). The number of generations per colony-passage was estimated following previously described methods (Foster et al. 2015). For further details, see supplementary Methods, Supplementary Material online.
Fig. 1.

—Scheme of the experimental evolution assays. Mutation accumulation assays were performed to two Bartonella elizabethae-like strains. Nine bacterial line descendants from each ancestral strain were evolved independently in chocolate agar plates (represented by brown colored filled circles) at three different conditions: group A) three lines per strain were passaged and incubated constantly at 26 °C; group B) three lines per strain were passaged and incubated constantly at 37 °C; and group C) three lines per strain were passaged and incubated by alternating the two above temperatures (one passage at 26 °C, the next passage at 37 °C, and so on). Both ancestral and evolved lines were characterized by short-read sequencing (Illumina 250-bp paired-end reads) and PFGE assays. Ancestral strains and evolved lines with major PFGE variations were further sequenced with long-read sequencing (Nanopore Oxord MinION, with an average of 7,500 bp per read), and the genomes were assembled by a hybrid assembly protocol.
Estimation of the Expected Base-Pair Substitution Rates
Expected base-pair substitution (BPS) rates, relative to each Bartonella strain genome size, were estimated using the regression equation (i.e., log10µ = -8.663–1.096log10G) described earlier (Sung et al. 2012).
Classical Fluctuation Tests
Spontaneous mutation rates in the Bartonella ancestral strains were estimated by classical fluctuation tests using rifampicin, and following recommendations described earlier (Foster 2006). The mutation rate was calculated by the Ma–Sandri–Sarkar maximum likelihood method on FALCOR web tool (http://www.mitochondria.org/protocols/FALCOR.html, last accessed June 10, 2018; Hall et al. 2009). Mutation rates per nucleotide were calculated following Lynch (Lynch 2010). For further details, see supplementary Methods, Supplementary Material online.
Pulsed-Field Gel Electrophoresis Assays
SmaI- and EagI-pulsed-field gel electrophoresis (PFGE)-restriction fragment length polymorphism analyses were performed to the ancestors, evolved lines and wild strains following recommendations for Bartonella and Vibrio genomes, described elsewhere (Cooper et al. 2006; Xu et al. 2009). For further details, see supplementary Methods, Supplementary Material online.
DNA Extraction from Bacterial Cultures
DNA was extracted from all bacterial lines cultivated on fresh chocolate agar plates using Illustra Tissue & Cells GenomicPrep Mini Spin kit (GE Healthcare, Buckinghamshire, GBR).
Library Preparation and Short-Read-Based Sequencing
DNA obtained from ancestors (triplicates; to evaluate genetic homogeneity at time zero) and all evolved lines (after >1,000 generations) was processed for Illumina MiSeq sequencing (2 × 250 bp), as previously described (Blecher-Gonen et al. 2013; Kimmerling et al. 2018). Reads were trimmed for Illumina adapters using cutadapt (Martin 2011), and pairs in which either read was trimmed <40 bp were removed. For further details, see supplementary Methods, Supplementary Material online.
Library Preparation and Long-Read-Based Sequencing
DNA obtained from the ancestral strains and evolved lines that showed PFGE variation were end-repaired (NEBnext ultra II end repair kit, New England Biolabs, MA, USA) and cleaned up with 1× AmPure beads (BeckmannCoulter, USA). Native barcode ligation (BC02-05) was performed with NEB blunt/TA ligase (New England Biolabs, MA, USA) and cleaned with 0.5× AmPure beads. Qubit quantified barcode ligated DNA samples were pooled at equi-molar concentration to attain 1 µg pooled sample. Adapter ligation (BAM) was performed for 15 min using NEBNext Quick Ligation Module (New England Biolabs, MA, USA). Library mix was cleaned up using 0.4X AmPure beads (Beckmann Coulter, USA) and library was eluted in 15 µl of elution buffer and used for sequencing. Sequencing was performed on MinION Mk1b (Oxford Nanopore Technologies, Oxford, GBR) using SpotON flow cell (FLO-MIN107) in a 48 h sequencing protocol on MinKNOW 1.7.7. Base calling was performed using Albacore v.1.2.6. Reads were processed by albacore and poretools (Loman and Quinlan 2014) for converting fast5 files into fasta format.
Hybrid De Novo Genome Assembly
The base called reads were error corrected using VCF consensus reads and de novo assembled using “CANU” Overlap-Layout-Consensus assembler (Koren et al. 2017). Bowtie alignments for Illumina short reads, and BWA alignments for Nanopore reads, were run for all evolved and ancestral strains to error correct the assembled contig and derive a consensus whole genome sequence.
SNV Calling Analysis
The Illumina sequenced reads were aligned to their respective reference assemblies using BWA MEM v0.7.5a (Li and Durbin 2009). Mapped reads were processed using GATK best practices for single nucleotide polymorphisms (SNP) and short INDELs discovery. Variants were filtered using standard hard filtering parameters (McKenna et al. 2010; DePristo et al. 2011; Van der Auwera et al. 2013). In brief, we used the following thresholds for technical filtering: for SNPs, “QD < 2.0,” “FS > 60.0,” “MQ < 40.0,” “HaplotypeScore > 13.0,” “MQRankSum<-12.5,” and “ReadPosRankSum<-8.0”; and for INDELs, “QD < 2.0,” “FS > 200.0,” and “ReadPosRankSum<-20.0.” Then, we filtered the following variants that indicated low confidence positions or assembly artifacts: 1) multi-allelic SNPs/INDELs; and 2) positions for which all samples were homozygotes. We kept only the fixed variants, since it is expected that each sample represents a single clone.
SV Inference
SPEEDSEQ (Chiang et al. 2015) and LUMPY (Layer et al. 2014) were used to infer SVs, followed by in-house scripts for filtering and population analysis. All events that had at least five reads supporting split-reads and paired-end-reads (abnormal insert size or direction), were kept and the results were merged across all samples following the script mergeSVcallers (https://github.com/zeeev/mergeSVcallers, last accessed June 10, 2018) and using a window size of 200 base-pairs. Events in common to all samples were filtered and the results were further inspected and filtered manually using IGV genome browser (Robinson et al. 2011).
Validation of SV Events
Sequences involved in each detected SV event were identified. Then, primers were designed to validate each of the events (supplementary table S1, Supplementary Material online), using conventional PCR (Synthezza, Jerusalem, ISR) or Q5 (New England BioLabs, Massachusetts, USA) long PCR reactions. PCR products were visualized in TAE (0.4–1%) agarose gels stained with ethidium bromide. Products below 5,000 bp were characterized by Sanger sequencing, by BigDye Terminator cycle sequencing chemistry with the Applied Biosystems ABI 3700 DNA Analyzer and the ABI Data Collection and Sequence Analysis software (ABI, Carlsbad, CA, USA).
Bartonella Genome Comparisons and Annotations
The consensus sequence of each genome was obtained by mapping the short-read data to the hybrid assembly using Geneious software version 7.1.9 (Kearse et al. 2012). Then, entire or partial genome alignments were performed in MAUVE (Darling et al. 2004, 2010). In silico digestion of the genomes was performed by simulating SmaI digestion and the PFGE run in a virtual gel. Average nucleotide identity (ANI; Goris et al. 2007) was calculated between the Bartonella sp. genomes characterized in this study and other reference Bartonella spp. from GenBank database, using the ANI calculator online tool (http://enve-omics.ce.gatech.edu/ani/index, last accessed June 10, 2018).
Annotation of the genomes was assessed by RAST (Aziz et al. 2008), using classical, GLIMMER and RASTK pipelines. These results were compared and validated using GenBank database (nt and nr; Benson et al. 2004). Phage gene-related clusters were identified manually from the annotations, and defined as a region containing at least four phage-related genes, with a distance between phage related-genes of <20 kb. Prophages were predicted using PHASTER (Arndt et al. 2016) online server (http://phaster.ca, last accessed June 10, 2018). The genomes of Bartonella grahamii (RefSeq: NC_012846.1) and Bartonella tribocorum (NC_010161.1) were also annotated, using the same protocols described above, to compare their GenBank available annotations and our pipeline.
Prophage DNA Quantitative Real-Time PCR
An intercalating-dye based qPCR assay was developed to quantify relative and absolute copy numbers of prophage and phage sequences from the targeted samples. Primers employed for real-time qPCR reactions are detailed in supplementary table S1, Supplementary Material online. All reactions were performed following methods described earlier (Gutiérrez et al. 2015). For more details, see supplementary Methods, Supplementary Material online.
Bacteriophage Isolation
Bacteriophage particles were isolated from bacterial cultures by PEG-precipitation, following protocols previously described (Berglund et al. 2009; Guy et al. 2013).
Transmission Electron Microscopy
PEG-precipitates were diluted (1:2–1:20) in double-distilled water (DDW) and absorbed to Formvar coated copper grids for 30 s. Then, the grids were stained with 1% (w/v) uranyl acetate for 1 min and air-dried for 30 min. Transmission electron microscopy (TEM) visualization were performed in Tecnai 12 TEM 100 kV (Phillips, Eindhoven, NLD) equipped with MegaView II CCD camera and Analysis version 3.0 software (SoftImaging System GmbH, Münstar, DEU). The diameter of all visual bacteriophage capsids (with clear borders) were measured using ImageJ software (Schneider et al. 2012).
Screening of SVs and P-BOE11-3 from Wild Strains
The genomic restriction profiles and the presence of prophage P-BOE11-3 were evaluated in DNA extracted from 33 Bartonella sp. OE 1-1 wild strains isolated from Gerbillus spp. rodents and their associated fleas. Accordingly, the same SmaI-PFGE assay used for the evolutionary experiment was applied to these wild type strains. Moreover, the wild strains were screened for the presence of the internal fragment of the prophage P-BOE11-3 (supplementary table S1, Supplementary Material online) by PCR and Sanger sequencing, following methods described above (see “Validation of SV events” section).
Statistical Analyses
One sided Exact Poisson tests were run to compare the obtained mutation rates to the lower bound of the expected mutation rates, using the R function “poisson.test” (R-Core-Team 2017). Two-tailed nonparametric statistical tests were run in IBM SPSS software version 20 (IBM, NY, USA) to compare coverage ratios and qPCR data. Statistical significance was defined as P < 0.05.
Results
Ancestral Bartonella Genomes Contained Several Different Phage-Related Gene Clusters
From each Bartonella sp. OE 1-1 and Bartonella sp. Tel Aviv ancestral strain, a large contig (>2.0 Mb, “chromosome”) and a small contig (<42 kb, “plasmid”) were obtained by de novo hybrid assembly (table 1). To evaluate the nucleotide homogeneity of the ancestors (i.e., founders at time zero), three independent colonies from each ancestral strain were sequenced by Illumina MiSeq assay, and SNVs were screened by variant calling analysis. Accordingly, no detectable point mutations were exclusively associated with one of these clones, ensuring the clonality of the ancestors.
Table 1.
Characteristics of the Bartonella spp. Ancestral Strains
| Characteristics | Bartonella sp. OE 1-1 | Bartonella sp. Tel Aviv |
|---|---|---|
| Genomics | ||
| Number of chromosomes | 1 | 1 |
| Chromosomal estimated length (bp) | 2,004,652 | 2,174,009 |
| Number of plasmids | 1 | 1 |
| Plasmid estimated length (bp) | 29,057 | 41,483 |
| Predicted protein coding genes | 1,985 | 2,330 |
| Predicted tRNA genes | 42 | 41 |
| Predicted rRNA operons | 2 | 2 |
| Predicted phage-related gene clusters | 7 | 9 |
| Genetic relatedness between spp. (ANI ± standard deviation)a | ||
| Between ancestral strains | 90.3 ± 4.2% | |
| Between ancestors and Bartonella elizabethae (NZ_AILW00000000.1)b | 91.2 ± 3.9% | 94.2 ± 4.3% |
| Between ancestors and Bartonella tribocorum (NC_010161.1) | 89.2 ± 4.2 | 93.0 ± 4.3% |
| Between ancestors and Bartonella grahamii (NC_012846.1) | 87.8 ± 4.1% | 88.4 ± 3.9% |
| Culture properties | ||
| Incubation time at 26 °Cc | 7 days | 10 days |
| Incubation time at 37 °Cc | 5 days | 6 days |
| Average number of generations/passage at 26 °C (±standard deviation) | 23.9 ± 0.8 | 20.1 ± 1.0 |
| Average number of generations/passage at 37 °C (± standard deviation) | 23.3 ± 1.1 | 20.4 ± 1.2 |
ANI, average nucleotide identity.
RefSeq, NCBI Reference Sequence Database accession number.
Time required to achieve a 1.0–2.0 mm colony.
The Bartonella relatedness of ancestral genomes was confirmed by calculating the ANI to closely related reference genomes (table 1). The annotations of the ancestral genomes revealed the presence of seven and nine phage gene-related clusters in the OE 1-1 and Tel-Aviv strains, respectively (supplementary table S2, Supplementary Material online). These clusters corresponded to complete prophages, incomplete prophages and prophage remnants. Moreover, the Bartonella associated GTA and run-off-replication (ROR) loci were identified in the ancestral genomes by manual inspection, following earlier descriptions (Quebatte et al. 2017; Tamarit et al. 2017).
Evolution of Bartonella Genomes Evidenced Large SVs and Conserved SNV Rates
Eighteen lines (nine per Bartonella strain) were evolved under three different temperature conditions as shown in figure 1. The number of generations per passage at the different temperatures ranged from 20.1 to 23.9 (table 1). After the 50 serial bottleneck passages on nonselective rich media (chocolate agar) at each of the three culture conditions, the Bartonella lines underwent between 1,005 and 1,195 generations. The experiment required eight to 17 months, depending on the Bartonella sp. and the incubation conditions. The 18 evolved genomes were characterized and SNVs and SVs were detected with respect to their ancestral genomes, as follows:
Single-Nucleotide Variations
The 250-bp paired-end MiSeq reads obtained from each evolved line mapped 93.0–97.5% to their corresponding ancestral genome, with a high coverage per nucleotide (∼100×). SNV analysis revealed few point mutations, ranging from 0 to 3 events per genome (average of 1 mutation/genome), found in coding and noncoding regions, in all treatment groups and in both chromosome and/plasmid replicons (table 2). The BPS rates averaged (95% CI range) 2.8 (0.27–5.3) × 10−10 and 4.5 (1.4–7.5) × 10−10 BPS/bp/generation (or 5.6 and 9.9 × 10−4 BPS/genome) for Bartonella sp. OE 1-1 and Bartonella sp. Tel Aviv strains, respectively. These rates were not higher than the expected mutation rate estimations: 9.2–9.6 × 10−10 BPS/bp/generation for organisms with 2.0–2.2 Mb genome size (One-side Poisson test, P = 0.604 and 0.946, respectively). To confirm these results, we ran classical fluctuation tests to determine the spontaneous mutation rates (i.e., using rifampicin) in the ancestral Bartonella strains. Accordingly, the ancestral Bartonella strains showed spontaneous mutation rates similar to those expected for their genome sizes (supplementary table S3, Supplementary Material online).
Table 2.
Single-Nucleotide Variation Events Detected in the Evolved Lines of Bartonella sp. Tel Aviv and Bartonella sp. OE 1-1 Strains
| ID | Evolutionary Condition | SNV | Nucleotide Change | Replicon | Region or Gene | Effect |
|---|---|---|---|---|---|---|
| Bartonella sp. Tel Aviv | ||||||
| TA-A1 | 26 °C | BS | G:C > T:A | Plasm. | NCR | NA |
| TA-A1 | 26 °C | BS | A:T > T:A | Chr. | NCR | NA |
| TA-A3 | 26 °C | BS | A:T > C:G | Chr. | NCR | NA |
| TA-A3 | 26 °C | BS | G:C > C:G | Plasm. | NCR | NA |
| TA-B1 | 37 °C | BS | G:C > T:A | Chr. | NCR | NA |
| TA-B3 | 37 °C | BS | A:T > G:C | Chr. | ibaE | SS |
| TA-B3 | 37 °C | BS | A:T > G:C | Chr. | NCR | NA |
| TA-B3 | 37 °C | BS | G:C > A:T | Chr. | sucA | AAS (P > L) |
| TA-C1 | 26 °C & 37 °C | D | G | Chr. | MFS | IFS |
| TA-C1 | 26 °C & 37 °C | BS | A:T > C:G | Chr. | ugpB | AAS (F > L) |
| TA-C2 | 26 °C & 37 °C | D | C | Chr. | NCR | NA |
| Bartonella sp. OE 1-1 | ||||||
| OE11-A1 | 26 °C | BS | A:T > C:G | Chr. | NCR | NA |
| OE11-A3 | 26 °C | BS | A:T > G:C | Chr. | NCR | NA |
| OE11-B1 | 37 °C | BS | G:C > A:T | Chr. | NCR | NA |
| OE11-B1 | 37 °C | BS | G:C > T:A | Chr. | rpmH | AAS (A > S) |
| OE11-B1 | 37 °C | I | G | Chr. | fhaB | IFS |
| OE11-B2 | 37 °C | BS | G:C > A:T | Chr. | ibpA2 | AAS (T > M) |
| OE11-C1 | 26 °C & 37 °C | BS | G:C > A:T | Chr. | mltB | AAS (R > C) |
Note.—ID, identifier; SNV, single nucleotide variation; BS, base substitution; D, deletion; I, insertion; Chr., chromosome; Plasm., plasmid; NCR, noncoding region; NA, not applicable; SS, synonymous substitution; AAS, amino acid substitution; IFS, in-frame shift; ibaE, inducible autotransporter E gene; sucA, 2-oxoglutarate dehydrogenase E1 component gene; MFS, major facilitator superfamily transporter gene; rpmH, 50S ribosomal protein L34 gene; fhaB, filamentous hemagglutinin gene; ibpA2, 16Kda heat shock protein A gene; mltB, membrane-bound lytic murein transglycolase B precursor gene; P, proline; L, leucine; F, phenylalanine; A, alanine; S, serine; T, threonine; M, methionine; R, arginine; C, cysteine.
Structural Variations
The genomic structure of ancestral and evolved lines was evaluated by PFGE and sequenced-based SV inference analyses. Accordingly, large structural genomic events in evolved lines, exposed either to constant 37 °C or alternating temperatures (26 °C and 37 °C), were revealed. Firstly, a Bartonella sp. OE 1-1 genome line, evolved at 37 °C (referred herein as line “OE11-B3”) evidenced a large deletion event. The SmaI-PFGE of this evolved genome revealed a shift on the electrophoretic run of a high molecular-weight band (i.e., difference of ∼30 kb, fig. 2A). In addition, short-read SV based analyses identified a deletion of 33,154 bp in this line (supplementary fig. S1A, Supplementary Material online). Moreover, the independent hybrid de novo assembly of this evolved genome, and the subsequent whole-genome alignment between evolved and ancestral genomes confirmed the ∼33 kb-deletion (fig. 2B). This event was validated using conventional PCR and Sanger sequencing, targeting an internal locus (1,500 bp) that was shown to be present in the ancestor but absent in line OE11-B3 (supplementary fig. S1B, Supplementary Material online). Finally, we tracked this deletion in stored OE11-B3 line from earlier passages, up to passage number 12 (∼283rd generation, supplementary fig. S1C, Supplementary Material online).
Fig. 2.

—Large structural variation events in experimentally evolved Bartonella genomes. (A) SmaI-PFGE run of Bartonella sp. OE 1-1 ancestral and evolved OE11-B3 genomes, showing a shift in the electrophoretic run of a high molecular band (red arrow), corresponding to a ∼30 kb molecular weight change in line OE11-B3. Lines corresponded to: 1. Salmonella enterica subsp. enterica serovar Braenderup (digested with XbaI, as a molecular marker); 2–3, Bartonella sp. OE 1-1 ancestral line (duplicates), and; 4–5, Bartonella sp. OE 1-1 line OE11-B3 (duplicates). (B) MAUVE alignments of whole-evolved and ancestral genomes evidencing a ∼33 kb region of the ancestral genome not covered in the evolved line (red-thick arrow). (C) SmaI-PFGE run of Bartonella sp. Tel Aviv ancestral and evolved genomes, showing a shift in the electrophoretic run of two high molecular-weight bands (∼320 and ∼130 kb) observed only in the evolved line TA-C2 (lines marked by pink arrows), and two bands (∼398 kb and ∼60 kb) which were not evident in this line, but present in the ancestors and other evolved lines. Lines corresponded to: 1&12: S. enterica subsp. enterica serovar Braenderup (digested with XbaI, as molecular marker); 2–3: Bartonella sp. Tel Aviv ancestral line; 4–5: Bartonella sp. Tel Aviv line TA-B2; 6–7: Bartonella sp. Tel Aviv line TA-C1; 8–9: Bartonella sp. Tel Aviv line TA-C2 (with a profile change: pink arrows); and, 10–11: Bartonella sp. Tel Aviv line TA-C3. (D) Whole genome comparison between ancestor and evolved lines, demonstrating a ∼800 kb inverted area in the evolved line. Figure was built using GenoplotR package in R software with the MAUVE alignment data. (E) Comparison of scaled coverage depth from Bartonella sp. Tel Aviv ancestor (triplicates) and evolved TA-B2 line in a 38,000 bp window. TA-B2 evolved line showed absence of reads covering a ∼13 kb region. (F) Conventional PCR amplification targeting the flanking sequences of the deletion detected in line TA-B2. A ∼7,000 bp product was obtained only in the TA-B2 line: L: GeneRuler High Range DNA Ladder; A: Bartonella sp. Tel Aviv ancestral strain; B1: Bartonella sp. Tel Aviv evolved TA-B1 line; B2: Bartonella sp. Tel Aviv evolved TA-B2 line; B3: Bartonella sp. Tel Aviv evolved TA-B3 line; EC: extraction control; NTC: nontemplate control. All gel pictures were color-inverted for clarity without any manipulation.
Secondly, one line of the Bartonella sp. Tel Aviv, evolved under alternating temperatures (referred herein as line “TA-C2”), revealed pronounced PFGE profile changes, after independent digestions with two different restriction enzymes (i.e., SmaI and EagI; fig. 2C and supplementary fig. S2A, Supplementary Material online). The short-read SV based inference was not conclusive with respect to a potential SV in this sample, however the hybrid de novo assembly (i.e., using large and short-read data) demonstrated a large inversion of ∼800 kb in comparison with the ancestral clone (fig. 2D). This event was confirmed and validated by in silico digestion of both ancestral and evolved genome assemblies (supplementary fig. S2B) and by a long-PCR assay targeting one end of the inversion (supplementary fig. S2C, Supplementary Material online). This inversion was detected as early as passage number 23 (∼467th generation, supplementary fig. S2D, Supplementary Material online).
Short-read based-SV analysis revealed an additional event, which was not apparent by the SmaI-PFGE assay (fig. 2C, lines 4–5). Specifically, a Bartonella sp. Tel Aviv clone evolved at constant 37 °C (referred herein as “TA-B2”), underwent a large deletion of ∼13 kb (fig. 2E). This event was validated by conventional PCR amplifying a 7,000 bp product of the flanking regions of the deletion in the evolved clone (fig. 2F).
Structural Genomic Changes Occurred in Prophage and Surface Protein-Gene-Related Regions
The annotation of the evolved and ancestral genomes showed that the 33-kb deletion identified in OE11-B3 line was localized within a cluster of phage-related genes (i.e., cluster IV; supplementary tables S2 and S4, Supplementary Material online). The independent prophage prediction analysis of the deleted sequence (extracted from the ancestral genome) identified this locus as an “incomplete prophage” (PHASTER score = 50, 30.9 kb length and 23 predicted genes). This locus was localized downstream an intact prophage (Mu-like) in the ancestral genome (PHASTER score = 140, 50 kb length, 52 predicted genes). The deleted locus was found to be closely related to prophages identified in B. grahamii as4aup (ANI = 86.6 ± 3.6%; positions 1,123,117–1,161,624), and Bartonella sp. Tel Aviv genomes (ANI of 86.5 ± 3.9%; fig. 3A). These homologous loci included integrase/recombinase, exonuclease, lysozyme, carboxypeptidase, repressor, methylase, portal, capsid, and structural phage-related genes. Moreover, all of them were delimited upstream by phage late control and tail protein-genes (proteins D, U, and X) and downstream by tRNA genes for Ser, Val and Asp. Accordingly, in the evolved OE11-B3 genome, the latter genes were not deleted (fig. 3A). Notably, in the ancestral genome, a phage attachment site (attL) was identified between the phage late control and tail protein-genes and a phage-related integrase/recombinase.
Fig. 3.
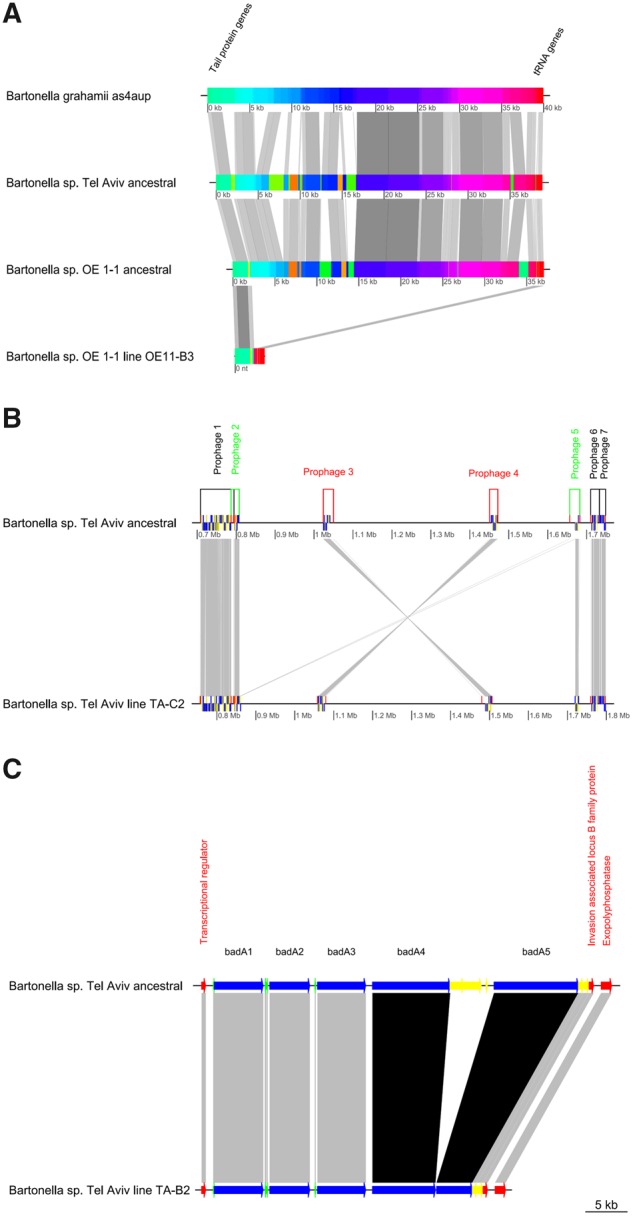
—Structural variation events occurred in prophage and surface protein-gene related regions. (A) MAUVE alignment of homologous prophages from B. grahamii as4aup strain, Bartonella sp. Tel Aviv ancestral genome, Bartonella sp. OE 1-1 ancestral genome (P-BOE11-3) and Bartonella sp. OE 1-1 evolved line OE11-B3 (with deleted prophage). Common genes flaking the loci are indicated in the figure. (B) Comparison of the location of the prophages in Bartonella sp. Tel Aviv ancestral and evolved TA-C2 genomes predicted by PHASTER. Colors indicate: black: prophage in homologous position and orientation; green: prophages that underwent recombination; red: homologous prophages in opposite orientation as a result of an inversion event. (C) Short-read sequencing analysis of Bartonella sp. Tel Aviv ancestor and evolved TA-B2 line (deletion), revealed a ∼13-kb deletion occurred in the evolved line, involving the badA4, badA5 and the intercalated badA remnant genes. All figures were constructed with GenoplotR package in R software.
The annotation analyses of the Bartonella sp. Tel Aviv ancestral and the evolved TA-C2 genomes revealed that the flanking regions of the 800 kb-inversion corresponded to two prophages (fig. 3B;supplementary table S5, Supplementary Material online). Within these prophages, a 15 kb-identical sequence was identified as a potential recombination site. This region included filamentous hemagglutinin gene fha, transcriptional regulator genes, phage related genes (such as integrase, portal, terminase genes), replicon stabilization toxin and antitoxin genes (copG-like) and other hypothetical protein genes. Consequently, the SV event resulted in the formation of chimeric prophage clusters (fusion of prophages 2 and 5 in TA-C2, fig. 3B).
The 13-kb deletion detected in Bartonella sp. Tel Aviv line TA-B2 was identified within the Bartonella adhesin gene cluster (fig. 3C;supplementary table S6, Supplementary Material online). The annotation of the Bartonella sp. Tel Aviv ancestral genome, predicted 5 badA gene copies, with sequences >5,000 bp (encoding 1,667–3,452 amino acids), intercalated by at least 5 gene remnants (lengths ranging 194–2,735 bp). The deletion detected in line TA-B2 involved the downstream part of the BadA cluster, including part of the 4th and 5th badA genes and the intercalating badA gene remnants (fig. 3C).
A Prophage, Deleted during Constant Evolution at 37 °C, Was Associated with an Increased Autonomous Replication at 26 °C
Short-read based coverage depth analysis revealed a highly covered 33-kb locus in all and exclusively the Bartonella sp. OE 1-1 lines evolved at 26 °C (i.e., “OE11-A1-A3”; fig. 4A). This locus corresponded exactly to the deleted prophage identified in OE11-B3 line (referred herein as prophage “P-BOE11-3”). Accordingly, the ratio between the average coverage at this prophage locus and that of four nearby loci was 3-fold higher in the lines evolved at 26 °C, and significantly different between all the evolved groups (Kruskal–Wallis test, χ2 = 6.3, df = 2, P = 0.044; fig. 4B). To test if these events corresponded to an evolutionary outcome, such as copy-number variation (CNV), or due to autonomous amplification of the prophage, the relative quantification of this locus in evolved and ancestral lines was performed by qPCR. Results showed a 3-fold higher amplification of this locus in the lines evolved at 26 °C in comparison with the other evolved lines, but this difference was absent when the former were recultured at 37 °C (supplementary fig. S3, Supplementary Material online). Although the latter comparison was marginally nonsignificant (Kruskal–Wallis test, χ2 = 7.5, df = 3, P = 0.058), probably due to the low number of evolved lines, it suggests an amplification associated with the 26 °C treatment rather than an integrated-CNV event. To confirm this, we analyzed ancestral cultures exposed at both study temperatures (N = 5 each group). Results confirmed the significantly higher prophage amplification of this locus in ancestral cultures at 26 °C compared with 37 °C (Mann–Whitney U test, U = 0, P = 0.009; fig. 4C). In Bartonella sp. Tel Aviv, the P-BOE11-3 homolog showed no differences in the relative quantification on ancestors cultured at 26 °C and 37 °C (Mann–Whitney U test, N = 5 each group, P = 0.361).
Fig. 4.

—Autonomous replication of Bartonella sp. OE 1-1 prophage is promoted at the flea-associated temperature (26 °C). (A) Coverage depth of Illumina 250 bp paired-end reads mapped to the reference ancestral genome. Read counts were normalized by subtracting the mean for each sample. Bartonella sp. OE 1-1 evolved lines at 26 °C showed higher coverage depth of the prophage region (positions 137–140 kb) in comparison with the ancestral and other evolved lines. OE11-A1-A3: lines evolved at 26 °C; OE11-B1-B3: lines evolved at 37 °C; OE11-C1-C3: lines evolved at 26–37 °C; OE11-O1-O3: ancestral lines. (B) Bartonella sp. OE 1-1 lines evolved at 26 °C showed a higher coverage ratio of the prophage locus in comparison with the other evolved groups. Dashed line indicates the coverage ratio of the ancestral lines (ratio = 1.17). Line OE11-B3 was excluded due to lack of coverage at this locus. (C) The relative quantity of the prophage locus was significantly higher in the ancestral lines cultured at 26 °C rather than 37 °C. Figure (A) was constructed using ggplot2 function in R software. Figures (B) and (C), were constructed using IBM SPSS Statistics software. *Kruskal–Wallis test, P < 0.05; and, **Mann–Whitney test, P < 0.05. Error bars represent ±2 standard error (SE).
Packed P-BOE11-3-DNA Levels and Overall Bacteriophage Production Vary According to Temperature
Bacteriophage production of Bartonella sp. OE 1-1 was evaluated by qPCR and TEM assays. Firstly, the absolute quantification of P-BOE11-3-DNA was demonstrated to be significantly higher in bacteriophage preparations obtained from ancestral 26 °C-cultures compared with 37 °C-cultures (Mann–Whitney test, N = 5, U = 0, P = 0.009; fig. 5A). Secondly, TEM visualizations showed that at 37 °C, the bacteriophage polyethylene glycol (PEG) preparations from the ancestor were dominated by small capsids (i.e., 87.1% of the measured phages had an average diameter of 41.3 ± 2.4 nm; fig. 5B). In contrast, at 26 °C, a diverse range of capsid diameters was observed, with two peak diameters (i.e., modes) at 42.5 and 54.5 nm (fig. 5C). Notably, at this condition many medium capsid diameters were recorded (i.e., 50–75 nm), which were not evident at 37 °C (fig. 5C). Large bacteriophage particles (with capsids diameter >86 nm) were identified at both temperatures, but represented <2% of the observed bacteriophages (supplementary fig. S4A, Supplementary Material online). Additionally, the examination of the bacteriophage production from OE11-B3 evolved line (without the P-BOE11-3), showed a marked dominance of small capsids at both temperatures (≤47 nm, supplementary fig. S4B, Supplementary Material online). Nevertheless, at 26 °C, a wide range of medium capsids was still observed in the evolved line (supplementary fig. S4C, Supplementary Material online).
Fig. 5.

—Bacteriophage production from the Bartonella sp. OE 1-1 ancestral strain at 26 °C and 37 °C. (A) Absolute copy number of phage P-BOE11-3 DNA in poly-ethylene glycol (PEG) precipitations from cultures of Bartonella sp. OE 1-1 ancestor at 26 °C and 37 °C. qPCR was run using primers that amplified a 131-bp internal fragment of the P-BOE11-3. Error bars represent ±2 standard error (SE). (B) Transmission electron microscopy (TEM) visualizations of bacteriophage particles (right panel; showing the typical bacteriophage particles observed) and the obtained histogram of capsid sizes (left panel; n = 70) from Bartonella sp. OE 1-1 ancestral strain at 37 °C. The scale is indicated in the image. (C) TEM visualizations of bacteriophage particles (right panel; showing the typical bacteriophage particles observed) and the obtained histogram of capsid sizes (left panel; n = 106) from Bartonella sp. OE 1-1 ancestral strain at 26 °C. The scale is indicated in the image. *Mann–Whitney U test, P < 0.01.
The bacteriophage production of Bartonella sp. Tel Aviv ancestral strain cultured at 26 °C or 37 °C was characterized by the presence of small particles at both temperatures, with average capsid diameter of 39.2 ± 5.4 nm and 40.1 ± 0.8 nm, respectively (supplementary fig. S5, Supplementary Material online). Medium size capsids were also observed in cultures at 26 °C (supplementary fig. S5A, Supplementary Material online), but it represented <10% of the measured capsids.
Bartonella sp. OE 1-1 Wild Strains Demonstrated Large Genomic SVs and Frequent Deletions of the P-BOE11-3 Prophage
The extent of SVs and the association of P-BOE11-3 deletions as SV hotspot in nature was explored in 33 Bartonella sp. OE 1-1 wild type strains isolated from desert rodents and their associated fleas, collected from a restricted location in the Negev desert, Israel, during the same period. Accordingly, wild type Bartonella sp. OE 1-1 strains exhibited remarkable differences in their restriction genomic profiles, with up to 16 different PFGE profiles identified among the strains (supplementary fig. S6, Supplementary Material online). Furthermore, 45% (15/33) of tested Bartonella sp. OE 1-1 wild strains were negative for the presence of the P-BOE11-3 (supplementary fig. S7, Supplementary Material online), demonstrating their deletion or the occurrence of genomic alterations that could prevent their amplification.
Discussion
We present here evolutionary experimental evidence of the intrinsic occurrence of SVs, driving the genome dynamics of the slow growing vector-borne bacteria, Bartonella. Accordingly, rodent-associated Bartonella spp. exhibited major genomic structural changes, associated with prophages and surface membrane protein genes. These experimental findings support previous in silico evidence of extensive rearrangements in Bartonella genomes from mammalian origin (Alsmark et al. 2004; Lindroos et al. 2006; Guy et al. 2013). Our results further demonstrated that these events occur spontaneously, in the absence of foreign DNA input. Importantly, we provided evidence that such structural events occur in nature. Finally, the evolution of the strains under different conditions led to the discovery of a Bartonella-prophage highly induced at the arthropod-associated temperature.
Bartonella is characterized by a remarkable genetic diversity in certain mammalian orders, especially rodents (Kosoy et al. 2012). This great nucleotide diversity is reflected in “housekeeping” genes, posing a taxonomical challenge and resulting in the description of dozens of novel and Candidatus spp. from rodent origin. Here, we demonstrated that two different rodent-associated Bartonella spp. underwent large genomic SVs, under vertical evolution, while their genomes remained highly similar to their ancestors at the nucleotide level. Moreover, we further demonstrated that such structural genomic changes are common and co-occur in wild Bartonella sp. OE 1-1 strains isolated from desert rodent and flea communities. Interestingly, Bartonella spp. are often exposed to population bottlenecks through the transmission between their mammalian hosts and hematophagous arthropod vectors, highlighting the role of genetic drift in this system, as simulated in the mutation accumulation experiments. Similarly, B. grahamii isolated from woodland rodents from Sweden displayed low sequence diversity, but evidenced variation in gene presence/absence patterns and PFGE profiles (Berglund, Ehrenborg, et al. 2010). When comparing the genomes of B. grahamii from different parts of the world, it was suggested that European populations may have suffered recent population bottlenecks, decreasing the B. grahamii genetic diversity (i.e., at the nucleotide level; Berglund, Ellegaard, et al. 2010). Coupling our laboratory experimental findings and the evidence from wild Bartonella strains, it becomes evident that large genomic structural events represent an effective way of generating diversity in the wild, while the accumulation of point mutations is maintained in a conserved pace. Several authors proposed horizontal gene transfer (HGT) mechanisms, particularly those associated with GTA elements and a ROR phenomenon, as the major genetic variability drivers in this bacterial genus (Berglund et al. 2009; Guy et al. 2013; Quebatte et al. 2017). HGTs are potentially promoted by the frequent encounter of different Bartonella spp. within their co-infected hosts (Abbot et al. 2007; Brinkerhoff et al. 2010; Gutiérrez et al. 2014). Thus, it will be interesting to evaluate how vertical acquired SVs and HGT mechanisms interplay and promote the evolution of these bacteria, simultaneously. Regardless of the mechanism, the constant emergence of novel structural genomic variants is expected to promote coexistence within hosts through nontransitive interactions (Stubbendieck et al. 2016), which might ultimately result in the large genetic diversity of Bartonella detected at the nucleotide level in nature (Bai et al. 2009; Harrus et al. 2009; Inoue et al. 2009).
The characterization of closely related bacterial genomes from the wild have shown that prophages represent a great degree of their genetic differences (Canchaya et al. 2003; Nakagawa 2003). In this study, both evolved Bartonella spp. underwent large SV events associated with prophages. In agreement with our results, the role of prophages was evident in the evolution of the fast-growing bacteria E. coli, including large inversions and deletions, under both selective and nonselective conditions (Iguchi et al. 2006; Barrick et al. 2014; Raeside et al. 2014). Here, we complement these findings by providing evidence for potential mechanisms underlying the promotion of diversity by prophage-elements, under no selection, within a short period. Accordingly, these loci served as anchoring sites for homologous recombination, as occurred with the large inversion (∼800 kb) detected in Bartonella sp. Tel Aviv line TA-C2 between identical sequences of two prophages, and as a hotspot for deletion events, as determined with the complete excision of P-BOE11-3 prophage from Bartonella sp. OE 1-1 line OE11-B3 (evolved at constant 37 °C). Since bacteria commonly contain multiple prophages (Bobay et al. 2014), as also observed in our studied Bartonella genomes, it is suggested that these loci represent hotspots for SVs in nature. As a support, we provided evidence that P-BOE11-3 is a SV hotspot in nature, as 45% of wild type Bartonella sp. OE 1-1 strains isolated from desert rodents and their fleas had evidence of losing this prophage. As discussed below, the relative lower induction of this prophage at 37 °C may promote its deletion during the constant mammalian infection transits. Lastly, due to the unselective nature of the mutation accumulation experiments, a large amount of SV related events must have been undetected, thus, the fact that we were able to detect prophage-associated SVs in both strains supports an intrinsic ongoing mechanism rather than sporadic events.
Bacterial surface associated proteins are key elements in the host–bacteria interactions (Jedrzejas 2007). In this study, we detected a large deletion in the surface protein gene locus (i.e., badA adhesin genes) of a Bartonella sp. Tel Aviv evolved line (TA-B2). The badA genes and homologs (e.g., variable-expressed-membrane-proteins “VOMPs” in B. quintana) encode for several copies of trimeric autotransporter adhesins (Riess et al. 2004; Harms and Dehio 2012). These proteins have been associated with several roles in Bartonella spp. including adhesion (e.g., to other bacteria, to host cells and to extracellular matrix), phagocytosis inhibition and induction of proangiogenic response in host cells (Riess et al. 2004; Zhang et al. 2004; Harms and Dehio 2012). In general, sequence analysis of surface protein encoding genes has revealed a molecular evolution enhanced by expansion and reduction events (Goward et al. 1993; Wang et al. 1997). Alike, CNV (from 1 to 5) and length variation (from 2.8 to 15.1 kb) of badA loci have been recorded in different Bartonella genomes (Riess et al. 2004; Harms and Dehio 2012). Moreover, deletions at this locus have been detected in B. quintana during in vivo experiments (Zhang et al. 2004), and via in silico comparisons of Bartonella henselae genomes (Riess et al. 2004; Lindroos et al. 2006). Our results confirm the dynamic nature of these genes readily undergoing spontaneous deletions.
Environmental stressors may trigger spontaneous induction of prophages (Nanda et al. 2015). Here, we demonstrated the induction of a Bartonella-prophage (i.e., P-BOE11-3) at 26 °C, a temperature associated with the arthropod. This phenomenon was discovered by CNV analysis and was later confirmed as an intrinsic mechanism rather than an experimental outcome (i.e., present already in the ancestral strain). Complementarily, we demonstrated that a more diverse profile of bacteriophage particles is produced at 26 °C in comparison to the mammalian body temperature (37 °C), evidencing further differences between these conditions. Diverse sizes of bacteriophage capsids (40–80 nm) have been recorded in other Bartonella spp. (Anderson et al. 1994; Barbian and Minnick 2000; Maggi and Breitschwerdt 2005; Berglund et al. 2009) and were associated with the induction of GTA (i.e., phage-elements that packed randomly 14-kb-DNA segments) and complete prophages (Berglund et al. 2009). Thus, our results suggest that in Bartonella sp. OE 1-1, capsids from multiple prophage origins are being produced at the vector-associated temperature. In the case of the P-BOE11-3, molecular and microscopic evaluations suggested its capacity to be packed, potentially in medium-size capsids (i.e., 50–70 nm). Notably, the extracellular encounters within the arthropod’s gut might have promoted the selection of these prophage-inductions, possibly to promote HGT events. The fact that all the SV events observed in this study occurred in lines that were exposed to 37 °C (either constantly or alternatively), including the loss of this P-BOE11-3, deserves special attention and further investigation. A potential temperature-response phenomenon promoting these SVs cannot be ruled out (i.e., faster growth rate of these Bartonella spp. or their reduced prophage induction at the warmer temperature).
Bartonella sp. Tel Aviv and B. grahamii, harbor P-BOE11-3-homologs, which exhibit common integration origins. In contrast to the P-BOE11-3, the Bartonella sp. Tel Aviv prophage homolog did not exhibit a temperature-related induction. In B. grahamii, it was recently demonstrated that its P-OE11-3 homolog (i.e., BGL-1) encodes for GTA-like and ROR-like elements (Tamarit et al. 2017). The authors suggested that the BGL-1 represents an example of a bacteriophage in the process of becoming a GTA (Tamarit et al. 2017). Although this prophage exhibits certain degree of evolution among wild B. grahamii strains, no autonomous amplification was demonstrated from bacteriophage preparations (Berglund, Ellegaard, et al. 2010). Thus, the P-BOE11-3 offers evidence of a prestage of the GTA transition, conserving its capacity to be induced at 26 °C.
The detection of SV events at the sequence level in bacterial genomes is a challenging task. Here, we demonstrated that the combination of short (Illumina MiSeq) and long read (Oxford Nanopore MinION) based sequencing, together with PFGE was valuable in enabling the discovery and validation of major SVs. SV-inference based only on short sequencing data may miss large SV events, as observed with the 800-kb inversion detected on a Bartonella sp. Tel Aviv line. In that case, short reads were unable to identify the event, due to long duplicated prophage regions flanking the inversion.
In conclusion, a remarkable plasticity of Bartonella genomes was displayed in this study, by vertical inherited SV events, in a short period in evolution. Additionally, the induction of different prophages at an arthropod-associated temperature (26 °C) in one of the Bartonella strains reflected an induction-mechanism adapted to this ecological niche. The latter findings, coupled with the constant encounter of different Bartonella spp. with other co-infecting bacterial species within the arthropod gut and/or the host, and their potential contribution via HGT (such as GTA elements) elucidate mechanisms involved in the generation of the great genetic diversity observed in the slow-growing bacteria, Bartonella.
Supplementary Material
Acknowledgments
This research was supported by the ISRAEL SCIENCE FOUNDATION (grant No. 688/17 to S.H.). We thank Dr Carmit Cohen, Dr Mario Garrido, Ron Flatau, and Snir Halle from the Ben Gurion University, Israel, for their assistance in collecting the wild animals for Bartonella isolation; Dr Ohad Gal-Mor from The Infectious Diseases Research Laboratory, Sheba Medical Center, Tel-Hashomer, Israel, for kindly providing us the Salmonella ser. Braenderup H9812 strain; Dr Jonathan Barlev from the INCPM, Weizmann Institute, Israel, for his bioinformatics assistance; Soumyalipa Das, Dr Harpreet Kukreja, Sushmita Maitra, and Dr Manjunatha B L, from Genotypic Technology PVT LTD, India, for their sequencing assistance; Dr Saar Tal from The egg and poultry board, Israel, for his assistance in the phages preparations; Dr Yael Friedmann and Minna Angenitsky from the Bio-Imaging Unit, The Alexander Silberman Institute of Life Science, Edmond Safra Campus, and Dr Einat Zellinger, from the Centre for Scientific Imaging, The Robert H. Smith faculty of Agriculture, The Hebrew University of Jerusalem, Israel, for their contribution to the electron microscopy.
Author Contributions
R.G., B.M., S.C., and S.H. designed the research. H.H. was responsible for the field sampling collection. R.G., B.M., K.C.M.S., Y.N.B., E.M.H., S.C., and S.H. performed the research. B.M., S.C., E.M.H., and R.C.M. contributed with the introduction of new analytic tools.R.G., B.M., Y.N.B., R.C.M., S.C., and S.H. analyzed the data. R.G. and S.H. wrote the paper.
Literature Cited
- Abbot P, Aviles AE, Eller L, Durden LA.. 2007. Mixed infections, cryptic diversity, and vector-borne pathogens: evidence from Polygenis fleas and Bartonella species. Appl Environ Microbiol. 73(19):6045–6052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsmark CM, et al. 2004. The louse-borne human pathogen Bartonella quintana is a genomic derivative of the zoonotic agent Bartonella henselae. Proc Natl Acad Sci U S A. 101(26):9716–9721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson B, Goldsmith C, Johnson A, Padmalayam I, Baumstark B.. 1994. Bacteriophage-like particle of Rochalimaea henselae. Mol Microbiol. 13(1):67–73. [DOI] [PubMed] [Google Scholar]
- Arndt D, et al. 2016. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44(W1):W16–W21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aziz RK, et al. 2008. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 9(1):75.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y, Kosoy MY, Lerdthusnee K, Peruski LF, Richardson JH.. 2009. Prevalence and genetic heterogeneity of Bartonella strains cultured from rodents from 17 provinces in Thailand. Am J Trop Med Hyg. 81(5):811–816. [DOI] [PubMed] [Google Scholar]
- Barbian KD, Minnick MF.. 2000. A bacteriophage-like particle from Bartonella bacilliformis. Microbiology 146(3):599–609. [DOI] [PubMed] [Google Scholar]
- Barrick JE, et al. 2014. Identifying structural variation in haploid microbial genomes from short-read resequencing data using breseq. BMC Genomics. 15(1):1039.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrick JE, Lenski RE.. 2013. Genome dynamics during experimental evolution. Nat Rev Genet. 14(12):827–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL.. 2004. GenBank. Nucleic Acids Res. 33(Database issue):D34–D38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglund EC, Ehrenborg C, et al. 2010. Genome dynamics of Bartonella grahamii in micro-populations of woodland rodents. BMC Genomics. 11(1):152.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglund EC, Ellegaard K, et al. 2010. Rapid diversification by recombination in Bartonella grahamii from wild rodents in Asia contrasts with low levels of genomic divergence in Northern Europe and America. Mol Ecol. 19(11):2241–2255. [DOI] [PubMed] [Google Scholar]
- Berglund EC, et al. 2009. Run-off replication of host-adaptability genes is associated with gene transfer agents in the genome of mouse-infecting Bartonella grahamii. PLoS Genet. 5(7):e1000546.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blecher-Gonen R, et al. 2013. High-throughput chromatin immunoprecipitation for genome-wide mapping of in vivo protein–DNA interactions and epigenomic states. Nat Protoc. 8(3):539–554. [DOI] [PubMed] [Google Scholar]
- Bobay LM, Touchon M, Rocha EP.. 2014. Pervasive domestication of defective prophages by bacteria. Proc Natl Acad Sci U S A. 111(33):12127–12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkerhoff RJ, Kabeya H, Inoue K, Bai Y, Maruyama S.. 2010. Detection of multiple Bartonella species in digestive and reproductive tissues of fleas collected from sympatric mammals. ISME J. 4(7):955–958. [DOI] [PubMed] [Google Scholar]
- Bryant J, Chewapreecha C, Bentley SD.. 2012. Developing insights into the mechanisms of evolution of bacterial pathogens from whole-genome sequences. Future Microbiol. 7(11)1283–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canchaya C, Proux C, Fournous G, Bruttin A, Brussow H.. 2003. Prophage genomics. Microbiol Molec Biol Rev. 67(2):238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C, et al. 2015. SpeedSeq: ultra-fast personal genome analysis and interpretation. Nat Methods. 12(10):966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomel BB, et al. 2009. Ecological fitness and strategies of adaptation of Bartonella species to their hosts and vectors. Vet Res. 40(2):29.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper KLF, et al. 2006. Development and validation of a PulseNet standardized pulsed-field gel electrophoresis protocol for subtyping of Vibrio cholerae. Foodborne Pathog Dis. 3(1):51–58. [DOI] [PubMed] [Google Scholar]
- Darling AC, Mau B, Blattner FR, Perna NT.. 2004. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14(7):1394–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling AE, Mau B, Perna NT.. 2010. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE. 5(6):e11147.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, et al. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 43(5):491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL. 2006. Methods for determining spontaneous mutation rates. Methods Enzymol. 409:195–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL, Lee H, Popodi E, Townes JP, Tang H.. 2015. Determinants of spontaneous mutation in the bacterium Escherichia coli as revealed by whole-genome sequencing. Proc Natl Acad Sci U S A. 112(44):E5990–E5999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goris J, et al. 2007. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol. 57(1):81–91. [DOI] [PubMed] [Google Scholar]
- Gutiérrez R, Nachum-Biala Y, Harrus S.. 2015. Relationship between the presence of Bartonella species and bacterial loads in cats and cat fleas (Ctenocephalides felis) under natural conditions. Appl Environ Microbiol. 81(16):5613–5621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutiérrez R, Morick D, Cohen C, Hawlena H, Harrus S.. 2014. The effect of ecological and temporal factors on the composition of Bartonella infection in rodents and their fleas. ISME J. 8(8):1598–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutiérrez R, Vayssier-Taussat M, Buffet JP, Harrus S.. 2017. Guidelines for the isolation, molecular detection, and characterization of Bartonella species. Vector Borne Zoonotic Dis. 17(1):42–50. [DOI] [PubMed] [Google Scholar]
- Goward CR, Scawen MD, Murphy JP, Atkinson T.. 1993. Molecular evolution of bacterial cell-surface proteins. Trends Biochem Sci. 18:136–140. [DOI] [PubMed] [Google Scholar]
- Guy L, et al. 2013. A gene transfer agent and a dynamic repertoire of secretion systems hold the keys to the explosive radiation of the emerging pathogen Bartonella. PLoS Genet. 9(3):e1003393.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall BM, Ma CX, Liang P, Singh KK.. 2009. Fluctuation analysis CalculatOR: a web tool for the determination of mutation rate using Luria–Delbruck fluctuation analysis. Bioinformatics 25(12):1564–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms A, Dehio C.. 2012. Intruders below the radar: molecular pathogenesis of Bartonella spp. Clin Microbiol Rev. 25(1):42–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms A, et al. 2017. Evolutionary dynamics of pathoadaptation revealed by three independent acquisitions of the VirB/D4 Type IV secretion system in Bartonella. Genome Biol Evol. 9(3):761–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrus S, et al. 2009. Isolation and genetic characterization of a Bartonella strain closely related to Bartonella tribocorum and Bartonella elizabethae in Israeli commensal rats. Am J Trop Med Hyg. 81:55–58. [PubMed] [Google Scholar]
- Heilbron K, Toll-Riera M, Kojadinovic M, MacLean RC.. 2014. Fitness is strongly influenced by rare mutations of large effect in a microbial mutation accumulation experiment. Genetics 197(3):981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iguchi A, Iyoda S, Terajima J, Watanabe H, Osawa R.. 2006. Spontaneous recombination between homologous prophage regions causes large-scale inversions within the Escherichia coli O157:H7 chromosome. Gene 372:199–207. [DOI] [PubMed] [Google Scholar]
- Inoue K, et al. 2009. Exotic small mammals as potential reservoirs of zoonotic Bartonella spp. Emerg Infect Dis. 15(4):526–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jedrzejas MJ. 2007. Unveiling molecular mechanisms of bacterial surface proteins: Streptococcus pneumoniae as a model organism for structural studies. Cell Mol Life Sci. 64(21):2799–2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28(12):1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesnerova L, Moritz R, Engel P.. 2016. Bartonella apis sp. nov., a honey bee gut symbiont of the class Alphaproteobacteria. Int J Syst Evol Microbiol. 66(1):414–421. [DOI] [PubMed] [Google Scholar]
- Kimmerling N, et al. 2018. Quantitative species-level ecology of reef fish larvae via metabarcoding. Nat Ecol Evol. 2(2):306–316. [DOI] [PubMed] [Google Scholar]
- Koren S, et al. 2017. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27(5):722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosoy M, Hayman DTS, Chan K-S.. 2012. Bartonella bacteria in nature: where does population variability end and a species start? Infect Genet Evol. 12(5):894–904. [DOI] [PubMed] [Google Scholar]
- Krasnov BR. 2008. Functional and evolutionary ecology of fleas: a model for ecological parasitology. Cambridge/New York: Cambridge University Press. [Google Scholar]
- Layer RM, Chiang C, Quinlan AR, Hall IM.. 2014. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol. 15(6):R84.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Popodi E, Tang H, Foster PL.. 2012. Rate and molecular spectrum of spontaneous mutations in the bacterium Escherichia coli as determined by whole-genome sequencing. Proc Natl Acad Sci U S A. 109(41):E2774–E2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R.. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25(14):1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind PA, Andersson DI.. 2008. Whole-genome mutational biases in bacteria. Proc Natl Acad Sci U S A. 105(46):17878–17883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindroos H, et al. 2006. Genome rearrangements, deletions, and amplifications in the natural population of Bartonella henselae. J Bacteriol. 188(21):7426–7439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loman NJ, Quinlan AR.. 2014. Poretools: a toolkit for analyzing nanopore sequence data. Bioinformatics 30(23):3399–3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. 2010. Evolution of the mutation rate. Trends Genet. 26(8):345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggi RG, Breitschwerdt EB.. 2005. Isolation of bacteriophages from Bartonella vinsonii subsp. berkhoffii and the characterization of Pap31 gene sequences from bacterial and phage DNA. J Mol Microbiol Biotechnol. 9(1):44–51. [DOI] [PubMed] [Google Scholar]
- Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17(1):10. [Google Scholar]
- McKenna A, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20(9):1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa I. 2003. Genome sequence of an M3 strain of Streptococcus pyogenes reveals a large-scale genomic rearrangement in invasive strains and new insights into phage evolution. Genome Res. 13(6):1042–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanda AM, Thormann K, Frunzke J.. 2015. Impact of spontaneous prophage induction on the fitness of bacterial populations and host–microbe interactions. J Bacteriol. 197(3):410–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okaro U, Addisu A, Casanas B, Anderson B.. 2017. Bartonella species, an emerging cause of blood-culture-negative endocarditis. Clin Microbiol Rev. 30(3):709–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul S, Minnick MF, Chattopadhyay S.. 2016. Mutation-driven divergence and convergence indicate adaptive evolution of the intracellular human-restricted pathogen, Bartonella bacilliformis. PLoS Negl Trop Dis. 10(5):e0004712.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quebatte M, et al. 2017. Gene transfer agent promotes evolvability within the fittest subpopulation of a bacterial pathogen. Cell Syst. 4:611–621.e616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raeside C, et al. 2014. Large chromosomal rearrangements during a long-term evolution experiment with Escherichia coli. MBio 5:e01377–e01314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R-Core-Team 2017. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- Riess T, et al. 2004. Bartonella adhesin a mediates a proangiogenic host cell response. J Exp Med. 200(10):1267–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, et al. 2011. Integrative genomics viewer. Nat Biotechnol. 29(1):24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW.. 2012. NIH Image to ImageJ:25 years of image analysis. Nat Methods. 9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segers FH, Kesnerova L, Kosoy M, Engel P.. 2017. Genomic changes associated with the evolutionary transition of an insect gut symbiont into a blood-borne pathogen. ISME J. 11(5):1232–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenbrot G, Krasnov B, Khokhlova I, Demidova T, Fielden L.. 2002. Habitat-dependent differences in architecture and microclimate of the burrows of Sundevall's jird (Meriones crassus) (Rodentia: gerbillinae) in the Negev Desert, Israel. J Arid Environ. 51(2):265–279. [Google Scholar]
- Stubbendieck RM, Vargas-Bautista C, Straight PD.. 2016. Bacterial communities: interactions to scale. Front Microbiol. 7:1234.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung W, Ackerman MS, Miller SF, Doak TG, Lynch M.. 2012. Drift-barrier hypothesis and mutation-rate evolution. Proc Natl Acad Sci U S A. 109(45):18488–18492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamarit D, Neuvonen MM, Engel P, Guy L, Andersson SGE.. 2017. Origin and evolution of the Bartonella gene transfer agent. Mol Biol Evol.35(2):451–464. [DOI] [PubMed] [Google Scholar]
- Van der Auwera GA, et al. 2013. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 43:11.10.1–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Masuzawa T, Li M, Yanagihara Y.. 1997. Deletion in the genes encoding outer surface proteins OspA and OspB of Borrelia garinii isolated from patients in Japan. Microbiol Immunol. 41:673–679. [DOI] [PubMed] [Google Scholar]
- Xu C, et al. 2009. Optimization of pulse-field gel electrophoresis for Bartonella subtyping. J Microbiol Methods. 76(1):6–11. [DOI] [PubMed] [Google Scholar]
- Zhang P, et al. 2004. A family of variably expressed outer-membrane proteins (Vomp) mediates adhesion and autoaggregation in Bartonella quintana. Proc Natl Acad Sci U S A. 101(37):13630–13635. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.