

Abstract
Different genes and proteins evolve at very different rates. To identify the factors that explain these differences is an important aspect of research in molecular evolution. One such factor is the role a protein plays in a large molecular network. Here, we analyze the evolutionary rates of enzyme-coding genes in the genome-scale metabolic network of Escherichia coli to find the evolutionary constraints imposed by the structure and function of this complex metabolic system. Central and highly connected enzymes appear to evolve more slowly than less connected enzymes, but we find that they do so as a by-product of their high abundance, and not because of their position in the metabolic network. In contrast, enzymes catalyzing reactions with high metabolic flux—high substrate to product conversion rates—evolve slowly even after we account for their abundance. Moreover, enzymes catalyzing reactions that are difficult to by-pass through alternative pathways, such that they are essential in many different genetic backgrounds, also evolve more slowly. Our analyses show that an enzyme’s role in the function of a metabolic network affects its evolution more than its place in the network’s structure. They highlight the value of a system-level perspective for studies of molecular evolution.
Keywords: evolutionary rates, molecular evolution, metabolism, flux balance analysis, Escherichia coli
Introduction
Different proteins evolve at very different rates (Zuckerkandl and Pauling 1965; Li et al. 1985; Alvarez-Ponce 2014). Half a century after this observation seeded the field of molecular evolution (Zuckerkandl and Pauling 1965), the reasons are still a subject of active research, and even more so since the genome-era made sequence and functional data about proteins abundantly available. Much of the variation in evolutionary rates stems from variation in selective constraints on proteins, and several factors influence these constraints (for recent reviews, see Alvarez-Ponce 2014; Zhang and Yang 2015). The most important is the amount of a protein that is expressed, and the breadth of its expression across cells or tissues in multicellular organisms (Duret and Mouchiroud 2000; Pál et al. 2001; Drummond et al. 2005). Highly and broadly expressed genes are under strong purifying selection, and therefore evolve slowly. Other factors influence evolutionary rates more weakly. They include protein length (Subramanian and Kumar 2004; Bloom et al. 2006; Ingvarsson 2006; Liao et al. 2006; Kryuchkova and Robinson-Rechavi 2014), essentiality (Hurst and Smith 1999; Jordan et al. 2002; Rocha and Danchin 2004), multifunctionality (Wilson et al. 1977; He and Zhang 2006; Salathé et al. 2006; Podder et al. 2009), subcellular localization (Liao et al. 2010), or being a chaperone client (Bogumil and Dagan 2010; Williams and Fares 2010; Aguilar-Rodríguez et al. 2016; Kadibalban et al. 2016). To gain deeper insights into the determinants of protein evolution, one must go beyond a gene-centered approach and embrace a systems-oriented view of protein evolution.
Inside a cell, proteins often form large and complex networks of interacting molecules. The position of a protein within such a network, as well as its role in the network’s function, can affect the protein’s evolution. In other words, the structure and function of a molecular network can impose selective constraints on its member proteins (Cork and Purugganan 2004). For example, proteins at the center of a protein–protein interaction network evolve more slowly (they are more constrained) than those at the periphery (Fraser et al. 2002; Jordan et al. 2003; Hahn and Kern 2005; Lemos et al. 2005; Alvarez-Ponce 2012; Alvarez-Ponce and Fares 2012). In contrast, in the yeast transcriptional regulation network, more central transcription factors evolve faster than less central ones (Jovelin and Phillips 2009). As these two types of cellular networks have similar topological properties (Barabasi et al. 2004), this difference in selective constraints over the network structure must ultimately be caused by different network functions. Nonetheless, despite being significant and consistent across many different organisms, the effects of network topology on protein evolution are weak. It could be caused by confounding factors such as expression level, and it can be affected by biased and low-quality data (Bloom and Adami 2003; Batada et al. 2006).
Metabolic networks constitute another important class of cellular network. They are well-studied in model organisms such as Escherichia coli (Feist et al. 2007), and comprise hundreds to thousands of chemical reactions, most of them catalyzed by enzymes encoded in genes. In a metabolic network, chemical reactions are organized in a highly reticulate manner to perform two main functions: Energy production and biosynthesis. Specifically, using energy and chemical elements from environmental nutrients, metabolic networks synthetize essential small molecules (i.e., amino acids, ribonucleotides, deoxynucleotides, lipids, and enzyme cofactors). The chemical reactions a metabolic network catalyzes are encoded in a metabolic genotype—a genome’s set of enzyme-encoding genes. The network’s phenotype can be defined as the set of molecules it can synthesize, and the rate at which it does so (Matias Rodrigues and Wagner 2009). Thanks to computational approaches such as flux balance analysis (FBA) (Orth et al. 2010; Bordbar et al. 2014), the relationship between metabolic genotypes and phenotypes can be studied computationally, which also allows us to study how selection for a given metabolic phenotype can constrain metabolic enzyme evolution. This type of analysis is currently not possible in other types of molecular networks, such as protein–protein interaction networks.
Previous work in eukaryotes has revealed that more central and more highly connected enzymes in metabolic networks, that is, those sharing metabolites with many other enzymes, evolve more slowly (Vitkup et al. 2006; Lu et al. 2007; Greenberg et al. 2008; Hudson and Conant 2011; Montanucci et al. 2011). Additionally, enzymes catalyzing reactions with a high metabolic flux—the rate at which a reaction transforms substrates into products—tend to evolve slowly (Vitkup et al. 2006; Colombo et al. 2014), and enzymatic domains with a greater influence on the dynamics of a metabolic pathway also tend to be more selectively constrained (Mannakee and Gutenkunst 2016). In this contribution, we study how the structure and function of a bacterial metabolic network affects the evolution of metabolic genes through point mutations. To our knowledge, this is the first time that such a study is performed using the whole-genome metabolic reconstruction of E. coli (Feist et al. 2007), which is arguably the best-known metabolic network of any living organism. Specifically, we study how quantities such as enzyme connectivity and metabolic flux affect evolutionary rate. To do so, we account for possible flux variation with Markov chain Monte Carlo (MCMC) sampling, a method that has not been used before in this type of evolutionary analysis. Additionally, we also study for the first time the influence of factors such as reaction superessentiality (Samal et al. 2010), which quantifies how easily a reaction can be bypassed in a metabolic network by other reactions or pathways, and the number of different chemical reactions that an enzyme catalyzes (enzyme multifunctionality). In performing these analyses, we comprehensively characterize metabolic determinants of enzyme evolution in E. coli.
Materials and Methods
Metabolic Network
To investigate how the topology of a metabolic network affects the evolution of metabolic genes, we constructed a reaction graph representation of the E. coli metabolic network model iAF1260 (Feist et al. 2007), which includes 2,382 reactions and 1,972 metabolites. In a reaction graph, nodes represent reactions, which are connected by an edge if they share at least one metabolite as either a substrate or a product (Montañez et al. 2010). When constructing this reaction graph, we did not consider the following currency metabolites, which are the most highly connected metabolites: H, H2O, ATP, orthophosphate, ADP, pyrophosphate, NAD, NADH, AMP, NADP, NADPH, CO2, and CoA (Vitkup et al. 2006). The inclusion of such metabolites, which participate in many different reactions, would create many reactions that are adjacent in the graph but not otherwise functionally related. Such reactions would come to dominate the structure of the network, and obscure patterns of connections between functionally related reactions. Our results are qualitatively insensitive to the exact number of metabolites removed. The reaction graph thus created comprises 2,382 nodes and 18,953 edges. Its diameter, that is, the longest of the shortest paths between any two nodes, is 15. It has a characteristic path length, that is, the average shortest distance between any pair of nodes, of 4.55. The clustering coefficient, that is, the fraction of a node’s neighbors that are also neighbors themselves, of this graph is 0.54, and its assortativity by degree, that is, the propensity for nodes with a similar number of neighbors to share an edge, is 0.17. In this graph, we computed the connectivity (or degree) of every reaction, which is its number of edges. In other words, the connectivity of a reaction is the number of other reactions that share at least one metabolite with the focal reaction. To determine the centrality of a reaction, we computed its betweenness centrality (Freeman 1977; Newman 2010), which is the number of shortest paths between any two nodes that pass through this reaction, using the Python package “igraph.” Mathematically, the betweenness centrality xi of node i is defined as , where is 1 if node i lies on the shortest path between nodes s and t, and 0 otherwise (or if s and t are in different components of the network) (Newman 2010).
To study how different properties of a metabolic reaction may affect the evolution of the enzyme-coding gene whose product catalyzes the reaction, it is preferable to work mostly with reactions that show a one-to-one relationship to enzyme-encoding genes. Therefore, we exclude from our evolutionary analyses reactions catalyzed by large macromolecular complexes that are encoded by multiple genes. Following Vitkup et al. (2006), for enzymes that catalyze more than one reaction, we use the reaction carrying the largest metabolic flux (the rate at which metabolites are converted into products) because it is the reaction imposing a higher evolutionary constraint. In addition, also following Vitkup et al. (2006), wherever different enzymes (isoenzymes) catalyze the same chemical reaction, we use the enzyme with the lowest rate of sequence evolution. The resulting data set comprises 659 enzyme-coding genes associated with the same number of metabolic reactions.
Metabolic Fluxes
We determined the distribution of fluxes that is allowable during growth on glucose for each reaction in the E. coli metabolic model iAF1260 (Feist et al. 2007) using MCMC sampling (Schellenberger and Palsson 2009). We used the artificially centered hit-and-run algorithm (ACHR) (Kaufman and Smith 1998) with minor modification as described by Bordbar et al. (2010) and Lewis, Schramm, et al. (2010). We implemented the ACHR algorithm with the ACHRSampler in COBRA Toolbox v.2.0.5 (Schellenberger, Que, et al. 2011), using the MATLAB (The MathWorks, Natick, MA) environment R2012b. We used a minimal (computational) medium in which glucose was the only carbon source, and set the uptake rate of glucose to the value of 8 millimoles per gram dry cell weight per hour. Following Nam et al. (2012), in order to restrict the sampling to the space of flux values relevant to in vivo E. coli growth on glucose, we established a lower bound to the biomass objective function of 90% of the optimal growth rate predicted by FBA (Orth et al. 2010). The mixed fraction is a metric introduced by Bordbar et al. (2010) to measure the uniformity of the sample from the space of allowed fluxes. We obtained a mixed fraction of 0.5096, which suggests that the space was nearly uniformly sampled (Bordbar et al. 2010). We removed reactions with a median flux value >15 millimoles per gram dry cell weight per hour from further analysis to ensure the exclusion of reactions involved in futile cycles (Beard et al. 2002; Schellenberger, Lewis, et al. 2011).
Reaction Superessentiality, Reaction’s Genome Occurrence, and Enzyme Multifunctionality
We obtained superessentiality indices of metabolic reactions for growth on glucose (SIglu) and for growth on 54 different sole carbon sources (SI54) from Barve et al. (2012). We obtained data about a reaction’s genome occurrence from the same study. A reaction’s genome occurrence is defined as the fraction of 1,093 prokaryotic species containing a gene encoding an enzyme known to catalyze the reaction.
We followed the classification of E. coli K-12 enzymes in generalists and specialists of Nam et al. (2012). Enzymes that only catalyze a specific chemical reaction were classified as specialists, while enzymes that catalyze more than one reaction were classified as generalists.
Evolutionary Rates
We obtained the values of dN/dS, dN, and dS in this analysis from the study by Alvarez-Ponce et al. (2016). In that study, orthologs in E. coli and S. enterica genomes were identified as reciprocal best hits (Tatusov et al. 1997) using the protein–protein Basic Local Alignment Search Tool (i.e., BLASTP with an E-value cut-off of 10−10). Each pair of orthologous proteins was aligned using ProbCons 1.2 (Do et al. 2005). The resulting alignments were back-translated into codon-based nucleotide alignments, and the ratio dN/dS was estimated using the program codeml from the package PAML 4.7 (one-ratio model M0) (Yang 2007). We removed dN/dS values >10 from our analyses.
Gene Expression and Protein Abundance
We obtained gene expression data for E. coli K-12 MG1655 grown in rich medium (LB) at 37°C from Chen and Zhang (2013), who quantified gene expression levels as numbers of RNA-seq reads per gene, normalized by gene length. We retrieved protein abundance data of E. coli K-12 MG1655 from the integrated data set of PaxDb 3.0 (Wang et al. 2012).
Statistical Analyses
We used R for all statistical analyses and plots. We performed the partial correlation analyses using the function “pcor.test” from the R package “ppcor.” We carried out the principal component regression analysis using the package “pls.” We performed a base-10 logarithmic transformation of continuous variables when such transformations lead to a higher percent of the variance in evolutionary rates explained by the model (R2). If data for a continuous variable included values equal to zero, we added a small constant of 0.001 to all values to allow its logarithmic transformation. We scaled the independent variables to zero mean and unit variance.
Results
The Effect of Metabolic Network Topology on Enzyme Evolution
To study how network structure affects enzyme evolution, we constructed a reaction graph representation of the whole-genome E. coli metabolic network, in which the nodes represent reactions. Two reactions are connected by an edge if they share at least one metabolite (Materials and Methods). In such a graph, the connectivity of a reaction corresponds to the number of other reactions that produce or consume the reaction’s substrates or products. The connectivity of an enzyme is equivalent to the connectivity of the reaction catalyzed by the enzyme. The centrality of an enzyme can be measured as the number of shortest pathways passing through the reaction node associated with the enzyme (betweenness centrality).
In a metabolic network, highly connected enzymes tend to occupy a central position in the network (as determined by their betweenness centrality, Materials and Methods), while less connected enzymes are more peripheral (fig. 1A; Spearman’s ρ = 0.524, P < 2.2 × 10−16, n = 635). In other words, enzymes in central metabolic processes, such as central carbon metabolism, tend to be highly connected, while enzymes in peripheral pathways tend to be less connected.
Fig. 1.

—Highly central and connected enzymes in a metabolic network do not evolve slowly. (A) The relationship between enzyme connectivity and centrality in the Escherichia coli metabolic network (Spearman’s ρ = 0.524, P < 2.2 × 10−16, n = 635). The centrality measure of a reaction is its betweenness centrality determined from the reaction graph (Materials and Methods). (B) The relationship between enzyme connectivity and evolutionary rate measured as dN/dS (Spearman’s ρ = −0.088, P = 0.028, n = 635). In both panels, a dashed line shows the best linear fit to the data and is provided as a visual guide. Note the double-logarithmic scale.
One might expect that more highly connected enzymes in a metabolic network are more constrained in their rate of evolution than less connected enzymes. The reason is that the reaction products of highly connected enzymes are substrates of many different reactions, such that any mutation disturbing product formation is bound to be more deleterious in a highly connected enzyme. However, a previous study on E. coli metabolism found no correlation between enzyme connectivity in core intermediary metabolism and evolutionary rate, determined as the rate of amino acid replacements, for 108 pairs of E. coli–Haemophilus influenzae orthologs (Hahn et al. 2004). In contrast, a later study found that highly connected enzymes in the metabolic network of Saccharomyces cerevisiae do evolve more slowly (Vitkup et al. 2006). We suspected that the original negative result in E. coli could be caused by small statistical power resulting from the many fewer enzymes analyzed by Hahn et al. (2004) (n = 108) than by Vitkup et al. (2006) (n = 671). We therefore repeated the E. coli analysis using the much larger whole-genome metabolic reconstruction. We estimated the evolutionary rate of an enzyme as the ratio of nonsynonymous substitutions to synonymous substitutions per nucleotide site (dN/dS) in the gene coding for the enzyme. We used values of dN/dS obtained by comparing genes in E. coli to orthologs in the closely related genome of Salmonella enterica (Alvarez-Ponce et al. 2016). A small value of dN/dS indicates a lower evolutionary rate due to higher constraints on enzyme evolution. Figure 1B shows the relationship between enzyme connectivity and the rate of evolution (Spearman’s ρ = −0.088, P = 0.028, n = 635; table 1). The negative correlation is very small but significant.
Table 1.
Correlations of Various Quantities with dN/dS
| Quantity | Spearman’s ρ | P Value |
|---|---|---|
| Enzyme connectivity | −0.088 | 2.8 × 10−2 |
| Betweenness centrality | 0.074 | 6.1 × 10−2 |
| Metabolic flux | −0.299 | 1.1 × 10−13 |
| SIglu | −0.313 | 6.4 × 10−14 |
| SI54 | −0.274 | 6.7 × 10−11 |
| Gene expression | −0.340 | 9.4 × 10−15 |
| Protein abundance | −0.488 | < 2.2 × 10−16 |
One potentially important confounding factor in the association between enzyme connectivity and evolutionary constraint is enzyme expression. Highly connected enzymes tend to be highly abundant (Spearman’s ρ = 0.163, P = 9.5 × 10−5, n = 565), and in general, abundant proteins tend to evolve more slowly (Pál et al. 2001; Drummond et al. 2005). This association between expression level and evolutionary rate also holds for enzymes. Specifically, we observe that high enzyme expression is associated with slow evolution (low dN/dS) regardless of whether expression is measured on the mRNA level (Spearman’s ρ = −0.340, P = 9.4 × 10−15, n = 491; table 1) or on the protein level (Spearman’s ρ = −0.488, P < 2.2 × 10−16, n = 565; table 1). Since expression of enzyme-coding genes is correlated between the mRNA and protein level (Spearman’s ρ = 0.432, P < 2.2 × 10−16, n = 433), we focus our analysis below on the protein level (Wang et al. 2012), but note that all reported results also hold for the mRNA level. When controlling for enzyme abundance in a partial correlation analysis between enzyme connectivity and evolutionary rate, the correlation loses statistical significance (Spearman’s ρ = 0.009, P = 0.830, n = 565; table 2). In other words, while highly connected enzymes evolve at slightly lower rates than less connected enzymes, this association is a byproduct of the relationship between evolutionary rate and enzyme abundance.
Table 2.
Partial Correlations of Various Quantities with dN/dS
| Quantity | Controlled Quantity | Spearman’s ρ | P Value |
|---|---|---|
| Enzyme connectivity | Protein abundance | 0.009 | 8.3 × 10−1 |
| Betweenness centrality | Protein abundance | 0.082 | 5.1× 10−2 |
| Metabolic flux | Protein abundance | −0.164 | 1.5 × 10−4 |
| SIglu | Protein abundance | −0.198 | 6.9 × 10−6 |
| SIglu | Metabolic flux | −0.197 | 3.4 × 10−6 |
| SI54 | Protein abundance | −0.187 | 2.1 × 10−5 |
| SI54 | Metabolic flux | −0.190 | 7.4 × 10−6 |
Similarly to enzyme connectivity, one might expect that more central enzymes should be more constrained in their evolution, but this relationship is also not consistent across studies. Some studies in eukaryotic species have found a significant association (Lu et al. 2007; Hudson and Conant 2011), while others have not (Greenberg et al. 2008; Montanucci et al. 2011; Colombo et al. 2014). We find a very weak positive association that is not significant (Spearman’s ρ = 0.074, P = 0.061, n = 635; table 1), and that is also not significant after controlling for enzyme abundance in a partial correlation analysis (Spearman’s ρ = 0.082, P = 0.051, n = 565; table 2). Thus, there is no clear association between enzyme centrality and evolutionary rate.
Enzymes Catalyzing Reactions with High Metabolic Flux Evolve Slowly
A reaction’s metabolic flux refers to the rate at which the reaction converts substrates into products. One might expect that enzymes catalyzing high flux reactions may evolve more slowly. The reason is that such enzymes tend to supply products to a large number of reactions and pathways, such that the effects of flux-diminishing mutations may be more deleterious than in low-flux enzymes (Vitkup et al. 2006). To study the relationship between metabolic flux and the rate of enzyme evolution, we applied flux balance analysis (FBA) to the metabolism of E. coli (Feist et al. 2007). FBA is a linear programming method that maximizes the rate of biomass production in a given nutritional environment, simultaneously balancing all the metabolic fluxes under a steady state assumption and a set of flux constraints (Orth et al. 2010). FBA has been extensively used to predict the phenotype of a metabolism from its genotype, that is, to predict the ability of a metabolism to synthetize biomass in a given chemical environment from the genes encoding the metabolism’s enzymes (Matias Rodrigues and Wagner 2009; He et al. 2010; Barve et al. 2012; Barve and Wagner 2013; Harcombe et al. 2013; Bordbar et al. 2014; Hosseini et al. 2015; Plata et al. 2015). FBA predictions are in good agreement with experimental data for model organisms such as E. coli (Edwards and Palsson 2000b; Edwards et al. 2001; Ibarra et al. 2002; Segre et al. 2002; Fong and Palsson 2004; Feist et al. 2007; Lewis, Hixson, et al. 2010).
We applied FBA to the E. coli metabolic network iAF1260 (Feist et al. 2007), maximizing aerobic growth on glucose in an environment where glucose is the only carbon source. Analyzing the association between metabolic flux and evolutionary rate is complicated by the fact many distributions of fluxes through individual enzymes can produce the same maximal biomass synthesis rate. For example, if two different reactions can produce the same biomass molecule at the same maximal rate, one of the two reactions could carry the maximal flux, while the other carries no flux, or both reactions could be active, such that the sum of their individual fluxes produces the metabolite at the maximal rate. In other words, a metabolic network can solve the problem of synthesizing biomass in multiple equivalent ways. To account for this flux variation, we used MCMC sampling to uniformly sample the space of all possible flux values (Schellenberger and Palsson 2009). We then computed a distribution of flux values for each of the reactions in the E. coli metabolic network, and used the median of this distribution as the reaction flux. To our knowledge, this is the first time that the complete flux distribution, as determined by MCMC sampling, is taken into consideration in studying the relationship between metabolic flux and enzyme evolution.
Figure 2 shows that enzymes catalyzing high-flux reactions evolve more slowly (Spearman’s ρ = −0.299, P = 1.1 × 10−13, n = 592; table 1). Importantly, this association does not disappear if we account for enzyme abundance: While high-flux enzymes tend to be highly abundant (Spearman’s ρ = 0.370, P < 2.2 × 10−16, n = 529), they still evolve more slowly in a partial correlation analysis that controls for enzyme abundance (Spearman’s ρ = −0.164, P = 1.5 × 10−4, n = 529; table 2). This observation agrees with a previous finding that high-flux yeast metabolic enzymes are subject to more constrained evolution (Vitkup et al. 2006). A similar association has been found with experimental flux measurements in the human erythrocyte core metabolism (Colombo et al. 2014).
Fig. 2.

—Enzymes catalyzing reactions with high metabolic flux evolve more slowly. The relationship between metabolic flux and enzyme evolutionary rate measured as dN/dS (Spearman’s ρ = −0.299, P = 1.1 × 10−13, n = 592). The dashed line shows the best linear fit to the data and is provided as a visual guide. Flux values <10−5 are set to zero. Note the logarithmic scale on both axes.
Highly Superessential Enzymes Evolve Slowly
A central function of a metabolic network is to synthetize the small-molecule precursors of biomass (amino acids, nucleotides, and cofactors) that are indispensable for cell growth and survival. In a given chemical environment, a metabolic reaction is essential if its product is needed for viability, that is, for biomass synthesis, and if its removal (“knock-out”) eliminates this ability. Otherwise, the reaction is nonessential. Reaction essentiality depends not only on the environment but also on a network’s genotype, that is, on the genes encoding the enzymes of the network. For example, certain genes are only essential in some strains of Saccharomyces cerevisiae (Dowell et al. 2010). One reason for such variation in essentiality is that different organisms can synthesize the same biomass molecules via alternative metabolic pathways that comprise different biochemical reactions and enzymes, which are encoded by different genes (Edwards and Palsson 1999, 2000a, 2000b; Barve et al. 2012).
While it is easy to manipulate an organism’s environment experimentally to study how reaction essentiality depends on the environment, current technologies limit our ability to systematically alter metabolic genotypes to study how essentiality varies with metabolic genotypes, that is, with the presence or absence of genes encoding alternative metabolic pathways. This limitation calls for computational approaches. One such approach is suited to study comprehensively how the presence or absence of enzyme-coding genes affects the essentiality of other enzyme-coding genes (Barve et al. 2012). It builds on the ability of FBA to efficiently predict a metabolic network’s phenotype—whether the network can produce biomass in a given environment—from its genotype. Briefly, the approach samples the “universe” of >5,000 biochemical reactions known to occur in at least one species, to generate viable metabolic networks with a given phenotype, but an otherwise random complement of reactions (Matias Rodrigues and Wagner 2009). By analyzing large ensembles of such random viable networks, one can determine how difficult it is to bypass a reaction through an alternative metabolic pathway, by computing a reaction’s superessentiality index (SI) (Barve et al. 2012). The SI of a reaction, which ranges from zero to one, is the fraction of random viable networks in which the reaction is essential for viability. In any given environment, reactions with a SI close to zero are easily bypassed, and nonessential for viability in most metabolisms, whereas reactions with the highest SI of one are always essential and cannot be bypassed according to current biochemical knowledge.
It is possible that highly superessential reactions (large SI, not easily by-passed) evolve at lower rates, because they may be subject to stronger purifying selection caused by their greater importance for viability in different genetic backgrounds. This could be especially the case in bacteria, where gene content can evolve very fast via lateral gene transfer, so that a given enzyme may become part of many different metabolic networks during its evolutionary history. To find out whether this is the case, we used superessentiality indices of E.coli metabolic reactions computed for 1) an aerobic minimal environment with glucose as the only carbon source (SIglu) and 2) 54 minimal environments that contain different unique carbon sources (SI54) (Barve et al. 2012). Enzymes catalyzing highly superessential enzymes tend to be present in most prokaryotic genomes while enzymes catalyzing less superessential enzymes are less common (Barve et al. 2012). There is a positive association between the SIglu of a metabolic reaction and the fraction of prokaryotic genomes that carry a gene coding for an enzyme known to catalyze the reaction (Spearman’s ρ = 0.444, P < 2.2 × 10−16, n = 548). This association is also found for SI54 (Spearman’s ρ = 0.356, P < 2.2 × 10−16, n = 548).
Figure 3A shows that E. coli reactions with high SIglu evolve more slowly (Spearman’s ρ = −0.313, P = 6.4 × 10−14, n = 548; table 1). It is possible that this association could be explained by enzyme abundance, because superessential enzymes tend to be highly abundant (Spearman’s ρ = 0.287, P = 3.7 × 10−11, n = 510). However, the association between SIglu and dN/dS persists in a partial correlation analysis that controls for protein abundance (Spearman’s ρ = −0.198, P = 6.9 × 10−6, n = 510; table 2). In other words, enzymes that are difficult to bypass in a glucose minimal environment evolve slowly, and do so independently of their abundance.
Fig. 3.

—Enzymes with high superessentiality evolve more slowly. (A) Scatter-plot showing the negative association between enzyme superessentiality in glucose (SIglu) and evolutionary rate measured as dN/dS (Spearman’s ρ = −0.313, P = 6.4 × 10−14, n = 548). (B) Scatter-plot showing the association between enzyme superessentiality in 54 different carbon sources (SI54) and dN/dS (Spearman’s ρ = −0.274, P = 6.7 × 10−11, n = 548). In both panels, a dashed line shows the best linear fit to the data and is provided as a visual guide. Note the logarithmic scale of the y axes.
Like SIglu, SI54 quantifies how difficult it is to bypass a metabolic reaction, but does so for 54 different environments, each containing one of 54 nutrients as its sole carbon source. A reaction or enzyme has a high SI54 if its removal abolishes viability in at least one of the 54 different environments for a large fraction of random networks viable in these 54 environments. Enzymes with a high SI54 also evolve slowly (fig. 3B; Spearman’s ρ = −0.274, P = 6.7 × 10−11, n = 548; table 1). While these enzymes also tend to be highly abundant (Spearman’s ρ = 0.193, P = 1.2 × 10−5, n = 510), the association persists when we control for enzyme abundance in a partial correlation analysis (Spearman’s ρ = −0.187, P = 2.1 × 10−5, n = 510; table 2).
Reactions highly superessential in a glucose-minimal environment tend to carry a high metabolic flux in this environment (Spearman’s ρ = 0.500, P < 2.2 × 10−16, n = 548). Metabolic flux is thus an additional potentially confounding factor for the observed relationship between SIglu and evolutionary rate. However, a partial correlation analysis shows that enzymes with high SIglu still evolve more slowly after controlling for metabolic flux (Spearman’s ρ = −0.197, P = 3.4 × 10−6, n = 548; table 2). Similarly, the effect of SI54 on enzyme evolution still holds after controlling for metabolic flux (Spearman’s ρ = −0.190, P = 7.4 × 10−6, n = 548; table 2).
The Multifunctionality of an Enzyme Does Not Affect Its Rate of Evolution
Metabolic enzymes can be classified as either specialists or generalists (Nam et al. 2012). A specialist enzyme catalyzes one specific chemical reaction, while a generalist enzyme catalyzes more than one reaction. One might expect that generalist enzymes evolve more slowly than specialist enzymes, since mutations in the genes encoding them may affect more than one metabolic pathway or function. This would at least be predicted by existing work on mutations that are pleiotropic, that is, they affect multiple different phenotypes (Stern and Orgogozo 2008). For example, theoretical considerations (Baatz and Wagner 1997; Orr 2000; Otto 2004), and empirical evidence in yeast suggest that highly pleiotropic mutations tend to be more deleterious than less pleiotropic mutations (Cooper et al. 2007).
For metabolic enzymes in E. coli, we find that generalist enzymes have a lower average evolutionary rate (1.241; n = 216) than specialist enzymes (1.308; n = 424), but the difference between these two enzyme categories is not significant (Wilcoxon rank-sum test, P = 0.804). Thus, there is no connection between multifunctionality or pleiotropy on the one hand, and evolutionary rate on the other hand, at least for E. coli metabolic enzymes.
Principal Component Regression Analysis
Finally, we performed a principal component regression, which is an established method to study the relative contributions of different determinants of protein evolutionary rates (Drummond et al. 2006). Principal component regression computes new variables, called principal components, which are linear combinations of the original predictor variables, and then regresses the response variable against them. We performed principal component regression using protein abundance, enzyme connectivity, betweenness centrality, metabolic flux, SIglu, SI54, and enzyme multifunctionality as potential predictor variables. Table 3 shows the numerical data from the analysis, while figure 4 shows these data graphically.
Table 3.
Results from the Principal Component Regression Analysis
| Principal Components |
||||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | All | |
| Percentage of explained variance in | ||||||||
| dN/dS | 12.55*** | 3.52*** | 1.77*** | 8.51*** | 0.21 | 0.01 | 0.28 | 26.85 |
| Percent contributions of each variable | ||||||||
| Protein abundance | 7.3 | 2.3 | 5.9 | 71.9 | 3.5 | 8.8 | 0.1 | |
| Enzyme connectivity | 3.0 | 42.2 | 1.9 | 10.9 | 0.0 | 41.8 | 0.2 | |
| Betweenness centrality | 6.4 | 36.2 | 8.8 | 2.2 | 2.9 | 42.1 | 1.4 | |
| Superessentiality index, SIglu | 32.4 | 5.0 | 0.0 | 2.7 | 2.5 | 4.6 | 52.7 | |
| Superessentiality index, SI54 | 29.6 | 3.6 | 0.8 | 6.2 | 15.1 | 1.3 | 43.3 | |
| Metabolic flux | 21.2 | 0.2 | 5.0 | 0.4 | 69.7 | 1.3 | 2.3 | |
| Enzyme multifunctionality | 0.1 | 10.4 | 77.5 | 5.7 | 6.2 | 0.0 | 0.0 | |
Note.—We indicate in bold the contributions of a variable to a principal component when >20%.
Significance levels:
P < 0.05, **P < 0.001, ***P < 10−5.
Fig. 4.
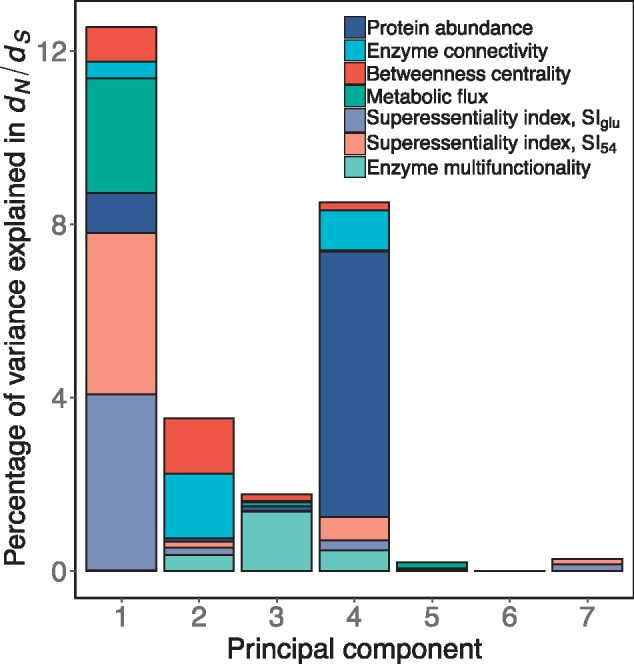
—Principal component regression on the rate of enzyme evolution (dN/dS). For each principal component, the height of the bar represents the percent of variance in dN/dS explained by the component. The relative contribution of each variable to a principal component is represented with different colors. This analysis was performed with 485 genes for which information for all variables is available. Table 3 contains the numerical data used to draw this figure.
We found four significant principal components. The component explaining the largest fraction of the variance in dN/dS (∼13%) was mostly determined (>60%) by roughly equal contributions from both superessentiality indices (SIglu and SI54). Metabolic flux determined ∼20% of the variance in dN/dS explained by this component. Protein abundance contributed ∼70% of the variance explained by a principal component explaining ∼9% of the variance in the rate of enzyme evolution. Network structure (enzyme connectivity and betweenness centrality) was the main contributor (∼78%) to a principal component explaining ∼4% of the variance in dN/dS. Finally, enzyme multifunctionality mostly determined (78%) a component explaining just ∼2% of the variance in evolutionary rate.
While protein abundance explained the largest fraction of the total variance in the rate of evolution (7.2%), the two superessentiality indices together explained ∼9% of the variance (table 4). Network topology explained ∼5%, metabolic flux explained ∼3%, and enzyme multifunctionality explained ∼2% of the total variance in dN/dS. In summary the main determinant of the rate of enzyme evolution is superessentiality in combination with protein abundance, followed by metabolic network structure. Enzyme multifunctionality has a very minor effect on enzyme evolution.
Table 4.
Percentage of total Variance in dN/dS Explained by Each Variable in the Principal Component Regression Analysis
| Protein abundance | 7.238 |
| Enzyme connectivity | 2.825 |
| Betweenness centrality | 2.432 |
| Superessentiality index, SIglu | 4.623 |
| Superessentiality index, SI54 | 4.540 |
| Metabolic flux | 2.934 |
| Enzyme multifunctionality | 2.253 |
Discussion
Natural selection on the function of a molecular network constrains how the network’s genes evolve. Conversely, changes in network genes affect the function of the whole network. In other words, the evolution of a network’s parts affects the evolution of the whole network, and vice versa. These two types of influence are entangled, because changes in network function that result from changes in network genes can themselves impose new evolutionary constraints on network genes. Here, we study how the structure and function of a large metabolic network (the whole) influences the evolution of its constituent enzymes (the parts). In doing so, we perform a comprehensive exploration of the metabolic determinants of enzyme evolution. Our analysis is part of a research tradition aiming to understand the molecular evolution of living systems by relating the evolutionary rates of genes with their function and position in a biological network (Fraser et al. 2002; Hahn et al. 2004; Vitkup et al. 2006; Alvarez-Ponce 2012, 2014; Alvarez-Ponce and Fares 2012; Zhang and Yang 2015). An advantage of using metabolic systems in such studies is that the relationship between the functions of the enzymes and the network is especially well understood (Papp et al. 2011; Wagner 2012; Bordbar et al. 2014).
First, we show that the position of an enzyme in the E. coli metabolic network does not affect its rate of evolution. Previous studies have found significant but very modest correlations between some topological network parameters and evolutionary rates in other metabolic networks and pathways (Vitkup et al. 2006; Lu et al. 2007; Greenberg et al. 2008; Hudson and Conant 2011). However, in the E. coli metabolic network, central and highly connected enzymes do not evolve at different rates when we control for their abundance. This corroborates previous findings in small-scale metabolic systems of mammals (Hudson and Conant 2011; Colombo et al. 2014) and E. coli (Hahn et al. 2004). Other studies in yeast (Vitkup et al. 2006) and Drosophila (Greenberg et al. 2008) have found that the connectivity of an enzyme influences its rate of evolution. However, even where significant, this association is very weak. Such a weak or absent association is not unreasonable, considering the “bow-tie” architecture of a metabolic network (Csete and Doyle 2004; Friedlander et al. 2015), where numerous input pathways of nutrient conversion feed into a highly interconnected central core metabolism, which feeds many output biosynthetic pathways. Some of these biosynthetic pathways are linear sequences of reactions that produce essential and complex biomass molecules, such as amino acids or enzyme cofactors. A loss-of-function mutation of an enzyme in one such linear and peripheral pathway would be lethal (Wagner 2005), even though the enzyme is not highly connected. In other words, mutations in both central and peripheral enzymes can be deleterious, albeit for different reasons.
Metabolic flux control measures how perturbations to an enzyme’s activity affect the steady-state global flux of a pathway (Kacser and Burns 1973), and it can also explain some variation on enzyme evolutionary rates. Flux control is not uniformly distributed in metabolic networks. Upstream enzymes in linear metabolic pathways and enzymes in bifurcation points of branched pathways tend to have higher flux control (Flowers et al. 2007; Wright and Rausher 2010; Rausher 2013). Over short evolutionary time scales, these enzymes are subjected to higher selective constraints, as well as positive selection (Eanes 2011; Dallolio et al. 2012; Olson-Manning et al. 2013; Hermansen et al. 2015). These observations show how selection on pathway function can constrain the evolution of individual enzymes. However, these evolutionary pressures are not stable because flux control can change considerably over longer evolutionary periods (Orlenko, Hermansen, et al. 2016; Orlenko, Teufel, et al. 2016; Orlenko et al. 2017). Unfortunately, the absence of metabolic-flux control measures for genome-scale metabolic networks prevented us to explore their impact on enzyme evolution in this study.
In agreement with previous studies in other organisms (Vitkup et al. 2006; Colombo et al. 2014), we find that enzyme-specific metabolic flux—the rate at which a reaction converts substrates into products—affects enzyme evolution by itself. We find that enzymes catalyzing reactions with high flux tolerate fewer amino acid substitutions than enzymes catalyzing reactions with lower fluxes. In other words, the function of a metabolic network, that is, biomass production, constrains the evolution of network genes through amino acid substitutions in a nonuniform way: Enzymes with high flux experience greater constraints than enzymes with low flux, since they are more important for network function.
In any one metabolic network, a loss of function mutation in a given enzyme may be lethal (in a specific environment), because it abolishes the network’s ability to produce biomass. In other metabolic networks with the same phenotype but a different metabolic genotype—a different complement of enzyme-coding genes—the enzyme may not be essential, because alternative reactions or pathways can assume its role. The extent to which an enzyme or reaction is easy or difficult to bypass is a function of metabolic biochemistry, and can be quantified through a reaction’s superessentiality index (Barve et al. 2012). Highly superessential reactions (enzymes) are difficult to bypass and their loss would be lethal in many different genetic backgrounds, while the loss of lowly superessential enzymes would be lethal in only a few backgrounds.
We find that highly superessential enzymes evolve more slowly. Relevant for this observation is that the metabolic genotypes of bacteria can evolve very rapidly. That is, bacterial enzymes can rapidly get lost via gene deletion or loss-of-function mutations, and new enzymes may be acquired via horizontal gene transfer (Ochman et al. 2000). For example, closely related E. coli strains may differ in >20% of their genomes, and in hundred or more metabolic genes, a difference that is partly due to horizontal gene transfer and gene deletions (Ochman and Jones 2000; Wagner 2009). On evolutionary time scales, bacterial metabolic enzymes can thus find themselves operating in different genotypic backgrounds, such that differences in superessentiality matter for their rate of evolution, as our data show. Superessentiality might influence the rate of evolution less in organisms whose metabolic genotypes change more slowly.
Finally, we also tested if generalist enzymes, which catalyze many reactions, are subjected to higher selective constraints than enzymes just catalyzing a single chemical reaction, as theoretical expectations would predict (Baatz and Wagner 1997; Orr 2000; Otto 2004). Previous studies have found that multifunctional genes in yeast evolve slowly (He and Zhang 2006; Salathé et al. 2006), corroborating theoretical expectations (Waxman 1998), although the magnitude of this effect is very modest. In mammals, multifunctional proteins also tend to be constrained, and the more functions a protein is involved in, the lower is its rate of evolution (Podder et al. 2009). However, generalist (multifunctional) enzymes do not evolve more slowly, indicating that pleiotropy is not constraining enzyme evolution, at least in E. coli.
We note that myriad other, nonmetabolic factors may influence the evolution of enzyme-coding genes. These include protein structure (Plotkin et al. 2012), chaperone targeting (Bogumil and Dagan 2010; Williams and Fares 2010; Bogumil et al. 2012; Pechmann and Frydman 2014; Aguilar-Rodríguez et al. 2016; Kadibalban et al. 2016), and many others, but the dominant factor is usually gene expression level (Alvarez-Ponce 2014; Zhang and Yang 2015). It is thus remarkable that the associations between evolutionary rate and metabolic flux or superessentiality are moderately high, comparable in strength to that between evolutionary rate and mRNA expression level, and only below the association between evolutionary rate and protein abundance.
In conclusion, our analysis of the rates of evolution of enzyme-coding genes in the E. coli metabolic network shows how a gene’s role in the function of a larger network can affect its evolution. In doing so, we show how a systems-level perspective can help understand the factors that contribute to protein evolution.
Acknowledgments
J.A.-R. acknowledges support by Swiss National Science Foundation grant P2ZHP3_174735. A.W. acknowledges support by ERC Advanced Grant 739874, by Swiss National Science Foundation grant 31003A_172887, as well as by the University Priority Research Program in Evolutionary Biology at the University of Zurich. We thank David Alvarez-Ponce for critical reading of the manuscript. We also thank Aditya Barve and Magdalena San Román for discussions.
Literature Cited
- Aguilar-Rodríguez J, et al. 2016. The molecular chaperone DnaK is a source of mutational robustness. Genome Biol Evol. 8(9):2979–2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Ponce D. 2012. The relationship between the hierarchical position of proteins in the human signal transduction network and their rate of evolution. BMC Evol Biol. 12(1):192.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Ponce D. 2014. Why proteins evolve at different rates: the determinants of proteins’ rates of evolution In: Fares MA, editor. Natural selection: methods and applications. Taylor & Francis: CRC Press; p. 126–178. [Google Scholar]
- Alvarez-Ponce D, Fares MA.. 2012. Evolutionary rate and duplicability in the Arabidopsis thaliana protein-protein interaction network. Genome Biol Evol. 4(12):1263–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Ponce D, Sabater-Muñoz B, Toft C, Ruiz-González MX, Fares MA.. 2016. Essentiality is a strong determinant of protein rates of evolution during mutation accumulation experiments in Escherichia coli. Genome Biol Evol. 8(9):2914–2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baatz M, Wagner GP.. 1997. Adaptive inertia caused by hidden pleiotropic effects. Theor Popul Biol. 51(1):49–66. [Google Scholar]
- Barabasi A-L, Oltvai ZNZN, Barabási A-L.. 2004. Network biology: understanding the cell’s functional organization. Nat Rev Genet. 5(2):101–113. [DOI] [PubMed] [Google Scholar]
- Barve A, Rodrigues JFM, Wagner A.. 2012. Superessential reactions in metabolic networks. Proc Natl Acad Sci U S A. 109(18):E1121–E1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barve A, Wagner A.. 2013. A latent capacity for evolutionary innovation through exaptation in metabolic systems. Nature 500(7461):203–206. [DOI] [PubMed] [Google Scholar]
- Batada NN, Hurst LD, Tyers M.. 2006. Evolutionary and physiological importance of hub proteins. PLoS Comput Biol. 2:0748–0756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beard DA, Liang S, Qian H.. 2002. Energy balance for analysis of complex metabolic networks. Biophys J. 83(1):79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom JD, Adami C.. 2003. Apparent dependence of protein evolutionary rate on number of interactions is linked to biases in protein-protein interactions data sets. BMC Evol Biol. 3:21.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom JD, Drummond DA, Arnold FH, Wilke CO.. 2006. Structural determinants of the rate of protein evolution in yeast. Mol Biol Evol. 23(9):1751–1761. [DOI] [PubMed] [Google Scholar]
- Bogumil D, Dagan T.. 2010. Chaperonin-dependent accelerated substitution rates in prokaryotes. Genome Biol Evol. 2:602–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogumil D, Landan G, Ilhan J, Dagan T.. 2012. Chaperones divide yeast proteins into classes of expression level and evolutionary rate. Genome Biol Evol. 4(5):618–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordbar A, Lewis NE, Schellenberger J, Palsson B, Jamshidi N.. 2010. Insight into human alveolar macrophage and M. tuberculosis interactions via metabolic reconstructions. Mol Syst Biol. 6:422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordbar A, Monk JM, King ZA, Palsson BO.. 2014. Constraint-based models predict metabolic and associated cellular functions. Nat Rev Genet. 15(2):107–120. [DOI] [PubMed] [Google Scholar]
- Chen X, Zhang J.. 2013. No gene-specific optimization of mutation rate in Escherichia coli. Mol Biol Evol. 30(7):1559–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo M, Laayouni H, Invergo BM, Bertranpetit J, Montanucci L.. 2014. Metabolic flux is a determinant of the evolutionary rates of enzyme-encoding genes. Evolution 68(2):605–613. [DOI] [PubMed] [Google Scholar]
- Cooper TF, Ostrowski EA, Travisano M.. 2007. A negative relationship between mutation pleiotropy and fitness effect in yeast. Evolution 61(6):1495–1499. [DOI] [PubMed] [Google Scholar]
- Cork JM, Purugganan MD.. 2004. The evolution of molecular genetic pathways and networks. BioEssays 26(5):479–484. [DOI] [PubMed] [Google Scholar]
- Csete M, Doyle J.. 2004. Bow ties, metabolism and disease. Trends Biotechnol. 22(9):446–450. [DOI] [PubMed] [Google Scholar]
- Dallolio GM, et al. 2012. Distribution of events of positive selection and population differentiation in a metabolic pathway: the case of asparagine N-glycosylation. BMC Evol Biol. 12:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do CB, Mahabhashyam MSP, Brudno M, Batzoglou S.. 2005. ProbCons: probabilistic consistency-based multiple sequence alignment. Genome Res. 15(2):330–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowell R, et al. 2010. Genotype to phenotype: a complex problem. Science 328(5977):469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond DA, Bloom JD, Adami C, Wilke CO, Arnold FH.. 2005. Why highly expressed proteins evolve slowly. Proc Natl Acad Sci U S A. 102(40):14338–14343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond DA, Raval A, Wilke CO.. 2006. A single determinant dominates the rate of yeast protein evolution. Mol Biol Evol. 23(2):327–337. [DOI] [PubMed] [Google Scholar]
- Duret L, Mouchiroud D.. 2000. Determinants of substitution rates in mammalian genes: expression pattern affects selection intensity but not mutation rate. Mol Biol Evol. 17(1):68–70. [DOI] [PubMed] [Google Scholar]
- Eanes WF. 2011. Molecular population genetics and selection in the glycolytic pathway. J Exp Biol. 214(2):165–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards JS, Ibarra RU, Palsson BO.. 2001. In silico predictions of Escherichia coli metabolic capabilities are consistent with experimental data. Nat Biotechnol. 19(2):125–130. [DOI] [PubMed] [Google Scholar]
- Edwards JS, Palsson BO.. 1999. Systems properties of the Haemophilus influenzae Rd metabolic genotype. Cell Biol Metab. 274(25):17410–17416. [DOI] [PubMed] [Google Scholar]
- Edwards JS, Palsson BO.. 2000a. Robustness analysis of the Escherichia coli metabolic network. Biotechnol Prog. 16(6):927–939. [DOI] [PubMed] [Google Scholar]
- Edwards JS, Palsson BO.. 2000b. The Escherichia coli MG1655 in silico metabolic genotype: its definition, characteristics, and capabilities. Proc Natl Acad Sci U S A. 97(10):5528–5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feist AM, et al. 2007. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol Syst Biol. 3:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flowers JM, et al. 2007. Adaptive evolution of metabolic pathways in Drosophila. Mol Biol Evol. 24(6):1347–1354. [DOI] [PubMed] [Google Scholar]
- Fong SS, Palsson BØ.. 2004. Metabolic gene-deletion strains of Escherichia coli evolve to computationally predicted growth phenotypes. Nat Genet. 36(10):1056–1058. [DOI] [PubMed] [Google Scholar]
- Fraser HB, Hirsh AE, Steinmetz LM, Scharfe C, Feldman MW.. 2002. Evolutionary rate in the protein interaction network. Science 296(5568):750–752. [DOI] [PubMed] [Google Scholar]
- Freeman LC. 1977. A set of measures of centrality based on betweenness. Sociometry 40(1):35. [Google Scholar]
- Friedlander T, Mayo AE, Tlusty T, Alon U.. 2015. Evolution of Bow-Tie architectures in biology. PLoS Comput Biol. 11:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg AJ, Stockwell SR, Clark AG.. 2008. Evolutionary constraint and adaptation in the metabolic network of Drosophila. Mol Biol Evol. 25(12):2537–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn MW, Conant GC, Wagner A.. 2004. Molecular evolution in large genetic networks: does connectivity equal constraint? J Mol Evol. 58(2):203–211. [DOI] [PubMed] [Google Scholar]
- Hahn MW, Kern AD.. 2005. Comparative genomics of centrality and essentiality in three eukaryotic protein-interaction networks. Mol Biol Evol. 22(4):803–806. [DOI] [PubMed] [Google Scholar]
- Harcombe WR, Delaney NF, Leiby N, Klitgord N, Marx CJ.. 2013. The ability of flux balance analysis to predict evolution of central metabolism scales with the initial distance to the optimum. PLoS Comput Biol. 9: e1003091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Qian W, Wang Z, Li Y, Zhang J.. 2010. Prevalent positive epistasis in Escherichia coli and Saccharomyces cerevisiae metabolic networks. Nat Genet. 42(3):272–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Zhang J.. 2006. Toward a molecular understanding of pleiotropy. Genetics 173(4):1885–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermansen RA, Mannakee BK, Knecht W, Liberles DA, Gutenkunst RN.. 2015. Characterizing selective pressures on the pathway for de novo biosynthesis of pyrimidines in yeast. BMC Evol Biol. 15:232.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosseini S-R, Barve A, Wagner A.. 2015. Exhaustive analysis of a genotype space comprising 1015 central carbon metabolisms reveals an organization conducive to metabolic innovation. PLoS Comput Biol. 11(8):e1004329.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson C, Conant G.. 2011. Expression level, cellular compartment and metabolic network position all influence the average selective constraint on mammalian enzymes. BMC Evol Biol. 11:89.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst LD, Smith NGCC.. 1999. Do essential genes evolve slowly? Curr Biol. 9(14):747–750. [DOI] [PubMed] [Google Scholar]
- Ibarra RU, Edwards JS, Palsson BO.. 2002. Escherichia coli K-12 undergoes adaptive evolution to achieve in silico predicted optimal growth. Nature 420(6912):186–189. [DOI] [PubMed] [Google Scholar]
- Ingvarsson PK. 2006. Gene expression and protein length influence codon usage and rates of sequence evolution in Populus tremula. Mol Biol Evol. 24(3):836–844. [DOI] [PubMed] [Google Scholar]
- Jordan IK, Rogozin IB, Wolf YI, Koonin EV.. 2002. Essential genes are more evolutionarily conserved than are nonessential genes in bacteria. Genome Res. 12(6):962–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan IK, Wolf YI, Koonin EV.. 2003. No simple dependence between protein evolution rate and the number of protein-protein interactions: only the most prolific interactors tend to evolve slowly. BMC Evol Biol. 3:1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovelin R, Phillips PC.. 2009. Evolutionary rates and centrality in the yeast gene regulatory network. Genome Biol. 10(4):R35.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kacser H, Burns JA.. 1973. The control of flux. Symp Soc Exp Biol. 27:65–104. [PubMed] [Google Scholar]
- Kadibalban AS, Bogumil D, Landan G, Dagan T.. 2016. DnaK-dependent accelerated evolutionary rate in prokaryotes. Genome Biol Evol. 8(5):1590–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman DE, Smith RL.. 1998. Direction choice for accelerated convergence in hit-and-run sampling. Oper Res. 46(1):84–95. [Google Scholar]
- Kryuchkova N, Robinson-Rechavi M.. 2014. Determinants of protein evolutionary rates in light of ENCODE functional genomics. BMC Bioinformatics 15(Suppl 3):A9. [Google Scholar]
- Lemos B, Bettencourt BR, Meiklejohn CD, Hartl DL.. 2005. Evolution of proteins and gene expression levels are coupled in Drosophila and are independently associated with mRNA abundance, protein length, and number of protein-protein interactions. Mol Biol Evol. 22(5):1345–1354. [DOI] [PubMed] [Google Scholar]
- Lewis NE, Hixson KK, et al. 2010. Omic data from evolved E. coli are consistent with computed optimal growth from genome-scale models. Mol Syst Biol. 6:390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis NE, Schramm G, et al. 2010. Large-scale in silico modeling of metabolic interactions between cell types in the human brain. Nat Biotechnol. 28(12):1279–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WH, Wu CI, Luo CC.. 1985. A new method for estimating synonymous and nonsynonymous rates of nucleotide substitution considering the relative likelihood of nucleotide and codon changes. Mol Biol Evol. 2:150–174. [DOI] [PubMed] [Google Scholar]
- Liao BY, Scott NM, Zhang J.. 2006. Impacts of gene essentiality, expression pattern, and gene compactness on the evolutionary rate of mammalian proteins. Mol Biol Evol. 23(11):2072–2080. [DOI] [PubMed] [Google Scholar]
- Liao BY, Weng MP, Zhang J.. 2010. Impact of extracellularity on the evolutionary rate of mammalian proteins. Genome Biol Evol. 2:39–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Zhang Z, Leach L, Kearsey M, Luo Z.. 2007. Impacts of yeast metabolic network structure on enzyme evolution. Genome Biol. 8(8):407.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannakee BK, Gutenkunst RN.. 2016. Selection on network dynamics drives differential rates of protein domain evolution. PLoS Genet. 12:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matias Rodrigues JF, Wagner A.. 2009. Evolutionary plasticity and innovations in complex metabolic reaction networks. PLoS Comput Biol. 5(12):e1000613.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montañez R, Medina MA, Solé RV, Rodríguez-Caso C.. 2010. When metabolism meets topology: reconciling metabolite and reaction networks. BioEssays 32(3):246–256. [DOI] [PubMed] [Google Scholar]
- Montanucci L, Laayouni H, Dall’Olio GM, Bertranpetit J.. 2011. Molecular evolution and network-level analysis of the N-glycosylation metabolic pathway across primates. Mol Biol Evol. 28(1):813–823. [DOI] [PubMed] [Google Scholar]
- Nam H, et al. 2012. Network context and selection in the evolution to enzyme specificity. Science 337:1101–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman M. 2010. Networks: an introduction. Oxford: Oxford University Press. [Google Scholar]
- Ochman H, Jones IB.. 2000. Evolutionary genomics of full genome content in Escherichia coli. EMBO J. 19:6637–6643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman H, Lawrence JG, Groisman EA.. 2000. Lateral gene transfer and the nature of bacterial innovation. Nature 405(6784):299–304. [DOI] [PubMed] [Google Scholar]
- Olson-Manning CF, Lee CR, Rausher MD, Mitchell-Olds T.. 2013. Evolution of flux control in the glucosinolate pathway in Arabidopsis thaliana. Mol Biol Evol. 30(1):14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlenko A, Chi PB, Liberles DA.. 2017. Characterizing the roles of changing population size and selection on the evolution of flux control in metabolic pathways. BMC Evol Biol. 17:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlenko A, Hermansen RA, Liberles DA.. 2016. Flux control in glycolysis varies across the tree of life. J Mol Evol. 82(2–3):146–161. [DOI] [PubMed] [Google Scholar]
- Orlenko A, Teufel AI, Chi PB, Liberles DA.. 2016. Selection on metabolic pathway function in the presence of mutation-selection-drift balance leads to rate-limiting steps that are not evolutionarily stable. Biol Direct. 11:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr HA. 2000. Adaptation and the cost of complexity. Evolution 54(1):13–20. [DOI] [PubMed] [Google Scholar]
- Orth JD, Thiele I, Palsson BOØ.. 2010. What is flux balance analysis? Nat Biotechnol. 28(3):245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto SP. 2004. Two steps forward, one step back: the pleiotropic effects of favoured alleles. Proc R Soc B Biol Sci. 271(1540):705–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pál C, Papp B, Hurst LD.. 2001. Highly expressed genes in yeast evolve slowly. Genetics 158:927–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp B, Notebaart RA, Pál C.. 2011. Systems-biology approaches for predicting genomic evolution. Nat Rev Genet. 12(9):591–602. [DOI] [PubMed] [Google Scholar]
- Pechmann S, Frydman J.. 2014. Interplay between chaperones and protein disorder promotes the evolution of protein networks. PLoS Comput Biol. 10(6):e1003674.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plata G, Henry CS, Vitkup D.. 2015. Long-term phenotypic evolution of bacteria. Nature 517(7534):369–372. [DOI] [PubMed] [Google Scholar]
- Plotkin JB, et al. 2012. Structure and age jointly influence rates of protein evolution. PLoS Comput Biol. 8: e1002542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podder S, Mukhopadhyay P, Ghosh TC.. 2009. Multifunctionality dominantly determines the rate of human housekeeping and tissue specific interacting protein evolution. Gene 439(1–2):11–16. [DOI] [PubMed] [Google Scholar]
- Rausher MD. 2013. The evolution of genes in branched metabolic pathways. Evolution 67(1):34–48. [DOI] [PubMed] [Google Scholar]
- Rocha EPC, Danchin A.. 2004. An analysis of determinants of amino acids substitution rates in bacterial proteins. Mol Biol Evol. 21(1):108–116. [DOI] [PubMed] [Google Scholar]
- Salathé M, Ackermann M, Bonhoeffer S.. 2006. The effect of multifunctionality on the rate of evolution in yeast. Mol Biol Evol. 23:721–722. [DOI] [PubMed] [Google Scholar]
- Samal A, Matias Rodrigues JF, Jost J, Martin OC, Wagner A.. 2010. Genotype networks in metabolic reaction spaces. BMC Syst Biol. 4(1):30.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schellenberger J, Lewis NE, Palsson BØ.. 2011. Elimination of thermodynamically infeasible loops in steady-state metabolic models. Biophys J. 100(3):544–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schellenberger J, Palsson B.. 2009. Use of randomized sampling for analysis of metabolic networks. J Biol Chem. 284(9):5457–5461. [DOI] [PubMed] [Google Scholar]
- Schellenberger J, Que R, et al. 2011. Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox v2.0. Nat Protoc. 6(9):1290–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segre D, et al. 2002. Analysis of optimality in natural and perturbed metabolic networks. Proc Natl Acad Sci U S A. 99(23):15112–15117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern DL, Orgogozo V.. 2008. The loci of evolution: how predictable is genetic evolution? Evolution 62(9):2155–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian S, Kumar S.. 2004. Gene expression intensity shapes evolutionary rates of the proteins encoded by the vertebrate genome. Genetics 168(1):373–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Koonin EVE, Lipman DJ.. 1997. A genomic perspective on protein families. Science 278(5338):631–637. [DOI] [PubMed] [Google Scholar]
- Vitkup D, Kharchenko P, Wagner A.. 2006. Influence of metabolic network structure and function on enzyme evolution. Genome Biol. 7(5):R39.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner A. 2005. Robustness and evolvability in living systems. Princeton, NJ: Princeton University Press. [Google Scholar]
- Wagner A. 2009. Evolutionary constraints permeate large metabolic networks. BMC Evol Biol. 9:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner A. 2012. Metabolic networks and their evolution In: Soyer OS, editor. Evolutionary systems biology. Vol. 751 NY: Springer; p. 29–52. [Google Scholar]
- Wang M, et al. 2012. PaxDb, a database of protein abundance averages across all three domains of life. Mol Cell Proteomics. 11(8):492–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman P. 1998. Pleiotropy and the preservation of perfection. Science 279(5354):1210–1213. [PubMed] [Google Scholar]
- Williams TA, Fares MA.. 2010. The effect of chaperonin buffering on protein evolution. Genome Biol Evol. 2:609–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson AC, Carlson SS, White TJ.. 1977. Biochemical evolution. Annu Rev Biochem. 46(1):573–639. [DOI] [PubMed] [Google Scholar]
- Wright KM, Rausher MD.. 2010. The evolution of control and distribution of adaptive mutations in a metabolic pathway. Genetics 184(2):483–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 24(8):1586–1591. [DOI] [PubMed] [Google Scholar]
- Zhang J, Yang J-R.. 2015. Determinants of the rate of protein sequence evolution. Nat Rev Genet. 16(7):409–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuckerkandl E, Pauling L.. 1965. Evolutionary divergence and convergence in proteins In: Bryson V, Vogel HJ, editors. Evolving genes and proteins. New York: Academic Press; p. 97–166. [Google Scholar]