

Abstract
With the aim of developing new molecular devices having higher photo-hydrogen-evolving activity, Pt(ppy)ClX units (ppy = 2-phenylpyridinate, X = Cl- or DMSO; DMSO = dimethylsulfoxide) have been employed as an H2-evolving site, as the catalytic activity of [Pt(ppy)Cl2]- was confirmed to be higher than those of other mononuclear platinum(II) complexes. In the present study, two new heterodinuclear Ru(II)Pt(II) complexes, produced by condensation of [Ru(bpy)2(5-amino-phen)]2+ (bpy = 2,2'-bipyridine, phen = 1,10-phenanthroline) with [Pt(cppy)Cl2]- and Pt(cppy)(DMSO)Cl (cppy = 9-carboxy-phenylpyridinate), respectively, have been prepared and their photo-hydrogen-evolving activities have been evaluated in detail. The ineffectiveness of these systems as photo-hydrogen-evolving molecular devices are interpreted in terms of their negative driving forces for the photoinduced electron transfer from the triplet MLCT excited state of the Ru chromophore to the π*(ppy) orbital of the catalyst moiety.
Keywords: solar energy conversion and storage, hydrogen energy, photocatalysis, platinum(II) catalysts, ruthenium complexes, molecular devices
1. Introduction
Visible light-induced water splitting into dihydrogen and dioxygen (2H2O + 4hν → 2H2 + O2) has attracted considerable attention towards the development of artificial photosynthesis generating renewable energy from sunlight. Around the beginning of 1980s, tris(2,2'-bipyridine)ruthenium(II) ([Ru(bpy)3]2+) was extensively investigated due to its potential application as a photosensitizer driving these water splitting reactions [1,2,3,4,5,6]. In extended studies in this area, our group has made continuous efforts in the last two decades to better understand the hydrogen evolution reaction (HER) from water catalyzed by Pt(II)-based molecular catalysts by employing the so-called “three-component system”, which consists of EDTA (ethylenediaminetetraacetic acid disodium salt) as a sacrificial electron donor, [Ru(bpy)3]2+ as a photosensitizer, and methyl viologen (N,N’-dimethyl-4,4’-bipyridinium, abbreviated as MV2+); as an electron relay (Scheme 1) [7,8,9,10,11,12].
Scheme 1.

Three-component system for H2-evolving cycle.
Important findings so far may be summarized as follows: (i) amidate-bridged cis-diammineplatinum(II) dimers [Pt(II)2(NH3)4(μ-amidato)2]2+ (amidate = α-pyridonate, α-pyrrolidinonate, acetamidate, 2-fluoroacetamidate) [7,8,9] with a shorter Pt-Pt distance display higher H2-evolving activity in comparison with those having a longer Pt-Pt distance; (ii) the mononuclear Pt(II) complexes having negatively charged chloride ligands, e.g., cis-PtCl2(NH3)2, PtCl2(ethylenediamine), and PtCl2(4,4’-dicarboxy-bpy), show substantially higher activity than those ligated with only neutral ligands, e.g., [Pt(NH3)4]2+ and [Pt(bpy)2]2+, even though their activities are still lower in comparison with those of the dinuclear Pt(II) complexes [10]; (iii) the Pt(II) compounds having electron acceptor ligands, e.g., those tethered to viologen type moieties, exhibit higher activity than those without such ligands, regardless of the Pt nuclearity [11]; (iv) the catalytic activity is lowered when the axial sites of the Pt(II) coordination plane are sterically hindered with regard to the access of a hydrogen atom [10,11]; (v) the destabilization of the HOMO which often corresponds to the filled Pt(II) dz2 orbital results in the catalytic enhancement [7,8,9,12].
During the above studies, we were also successful in inventing the first active model of a ‘photo-hydrogen-evolving’ molecular device (A; R = COOH; see Scheme 2), which is capable of driving photoreduction of water by EDTA into H2 as a single-molecular photocatalyst [13]. Some other examples of such hybrid molecules serving both as a photosensitizer and an H2-evolving catalyst were also reported by several other researchers [14,15,16,17,18,19,20,21]. We further undertook extended studies on the Ru(II)Pt(II)-based photo-hydrogen-evolving molecular devices [22,23,24], in which several important concepts have been developed as follows: (vi) the photoinduced HER driven by A was ascertained to proceed via a bimolecular pathway, which was suggested to involve the formation of a diplatinum intermediate [13,22]; (vii) the HER driven by A was shown to obey saturation kinetics as a function of the EDTA concentration, revealing that an ion-pair adduct of the dicationic A and the dianionic form of EDTA (dideprotonated form YH22-, where EDTA = YH4) is a key intermediate [22]; (viii) it was also realized that the driving force for the photoinduced electron transfer from the 3MLCT excited state of the [Ru(bpy)2(phen)]2+ moiety to the π*(bpy) orbital attached to the Pt(II) ion, leading to the HER, can be rationally controlled by changing the substituent group R (see Scheme 2; A-C) [23,24].
Scheme 2.

Covalently linked single-component system.
In order to improve the photocatalytic efficiency of these photo-hydrogen-evolving molecular devices, it is important for us to develop new systems by replacing the Pt(II)-based H2-evolving unit of A-C with a more highly active catalyst. In this context, we previously examined the H2-evolving activity of (2-phenylpyridinato)platinum(II), [Pt(ppy)Cl2]- (Hppy = 2-phenylpyridine), in the hope of destabilizing the Pt(II) dz2 orbital by the ligation of a strong σ-donating carbanion ligand. This has a relevance to the above-mentioned structure-activity relationship (v) [12]. In the study, we ascertained that [Pt(ppy)Cl2]- indeed exhibits higher H2-evolving activity in comparison with the Pt(bpy)Cl2 derivatives. In this study, we have synthesized a new type of photo-hydrogen-evolving molecular device by replacing the Pt(bpy)Cl2 moiety in A with a [Pt(ppy)Cl2]- derivative (2) (Scheme 3). We have also prepared an analog of 2 (i.e., 3) in which one of the two chloride ions bound to the Pt(II) ion of 2 is replaced by a DMSO molecule; Pt(ppy)(DMSO)Cl (DMSO = dimethylsulfoxide). Although these systems have been found to be ineffective towards the HER as a single-molecular device, the present study further ascertains that the driving force for the intramolecular electron transfer must be accurately controlled in the development of such photosynthetic molecular devices.
Scheme 3.

Chemical structures of the new Ru(II)Pt(II) dimers investigated.
2. Results and Discussion
2.1. Syntheses
The new Ru(II)Pt(II) dimers 2 and 3 were prepared according to Scheme 4. In the first step, cis-RuCl2(bpy)2 was reacted with N-(1,10-phenanthrolin-5-yl)-4-(2-pyridyl)benzamide (HL) [25] in ethanol, giving complex ligand 1 in a high yield (85%). In this reaction, none of an undesirable ruthenium complex ligated with a phenylpyridinate chelate in HL was produced, which was confirmed by 1H-NMR and ESI-TOF MS. In the second step, the monoruthenium precursor 1 was platinated by either (Bu4N)2[PtCl4] (Bu4N+ = tetra(n-butyl)ammonium) or cis-PtCl2(DMSO)2 in methanol to give the corresponding Ru(II)Pt(II) dimer (ca. 40% for 2 and ca. 70% for 3), where the synthetic method and the yield of 3 are similar to those previously reported for Pt(ppy)(DMSO)Cl [26]. An important feature is that the use of a pressure-resistant vial allowed us to carry out the synthesis at a relativity high temperature (140 °C), which is much higher than the boiling point of methanol under the ambient pressure (65 °C). The target Ru(II)Pt(II) dimers have been characterized by elemental analysis, ESI-TOF MS, IR, and 1H-NMR.
Scheme 4.

Synthetic routes to complexes 1-3.
2.2. Spectroscopic study
The spectroscopic and photophysical data of 1-3 are listed in Table 1. UV-visible absorption spectra of 1-3 in DMF are shown in Figure 1.
Table 1.
Absorption and emission properties of 1-3.a
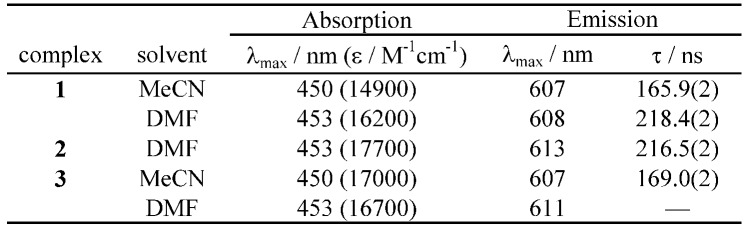 |
a Measured in air at room temperature
Figure 1.

Absorption spectra of [1](PF6)2·3H2O (red line), [2]Cl·6H2O (blue line), and [3](PF6)2·2H2O (green line) in DMF at 20 °C in air.
Each complex displays a strong absorption band in the visible region at ca. 450 nm, unambiguously assigned to the metal-to-ligand charge transfer (MLCT) transition, d(Ru) → π*(bpy). No significant difference is seen in the spectra of 1-3 at the π → π* transitions around 300 nm. On the other hand, the absorptivities of 2 and 3 around 300-400 nm are higher than that of 1, obviously due to the overlap of the MLCT band of the Pt(ppy)ClX moiety (X = Cl- or DMSO) at these wavelengths [12,27,28].
Figure 2 shows the steady-state emission spectra of 1-3, where the photon flux absorbed by each system is controlled as equal, by giving a common absorbance at the excitation wavelength. The luminescence intensity at ca. 610 nm for DMF solutions of 2 and 3 are similar to that of the precursor complex 1. The emission decay profiles of 1-3 in air at room temperature all show a good fit to a monoexponential function.
Figure 2.

Luminescence spectra of [1] (PF6)2·3H2O (red line), [2]Cl·6H2O (blue line), and [3](PF6)2·2H2O (green line) in DMF at 20 °C in air, where the excitation wavelength was fixed at 425 nm and all the solutions had an equal absorbance at 425 nm (0.1).
The emission lifetimes of 2 and 3 are same as that of the precursor complex 1 (see Table 1), indicating that the triplet character of the [Ru(bpy)2(phen)]2+ unit in these Ru(II)Pt(II) dimers is preserved even after the platination. We recently pointed out that the platination-induced quenching ratio (the luminescence intensity ratio of the platinated product and the non-platinated precursor compound) is correlated with the photo-hydrogen-evolving activity of the molecular catalysts [10,22,23,24]. These results suggest that the intramolecular electron or energy transfer is not enhanced at all in these new Ru(II)Pt(II) dimers 2 and 3, implying that desirable photoinduced intramolecular electron transfer leading to HER may not be enhanced in these systems.
2.3. Electrochemistry
The redox potentials for the electrochemical processes observed for 1-3 in either MeCN or DMF are summarized in Table 2. The cyclic voltammograms for 1-3 are shown in Figure 3.
Table 2.
Electrochemical dataa of 1-3 and A-C.
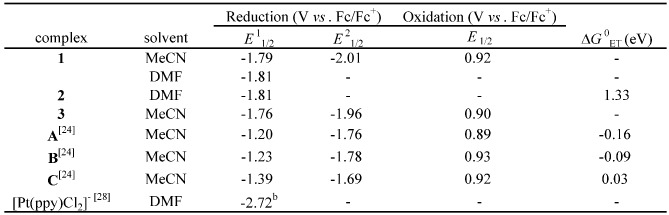 |
Figure 3.
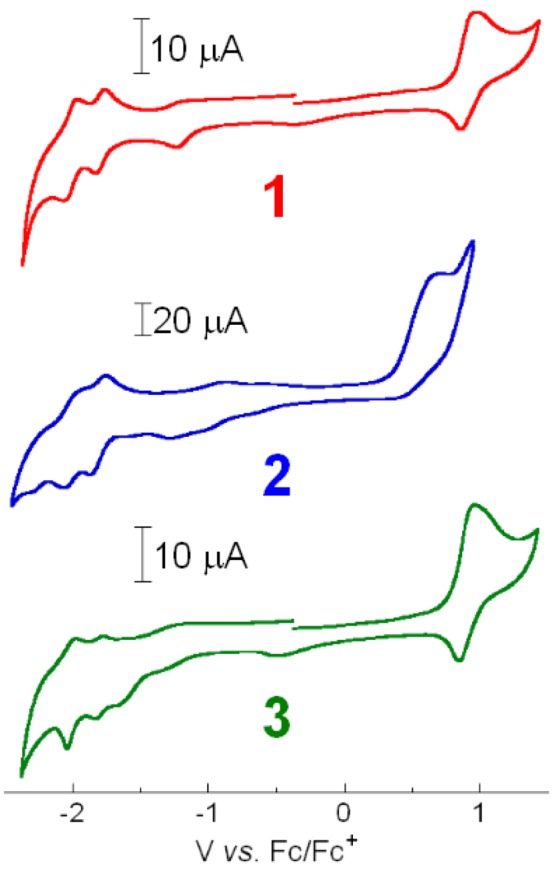
Cyclic voltammograms of [1](PF6)2·3H2O (red), [2]Cl·6H2O (blue) and [3](PF6)2·2H2O (green) in either 0.1 M TBAP/dry MeCN (1, 3) or 0.1 M TBAP/dry DMF (2), recorded at a scan rate of 100 mVs-1.
The Ru(II)/Ru(III) couple in the non-platinated precursor complex 1 is observed at 0.92 V vs. Fc/Fc+. Upon platination of 1 into 3, the Ru(II)/Ru(III) couple is slightly shifted to the negative side (ΔE1/2 = -0.02 V), as previously observed for A-C [24] (see Table 2). On the other hand, the first reduction peak for 1-3 occurs at around -1.8 V vs. Fc/Fc+ (Table 2), indicating that the first reduction of 1-3 occurs at the bpy or phen coordinated to the Ru(II) ion. Importantly, our recent studies on the related Ru(II)Pt(II)-based molecular devices A-C revealed that the first reduction must occur at the bpy bound to the Pt(II) ion and must be positioned at a potential more positive than ca. -1.2 V vs. Fc/Fc+ in order to serve as an active photo-hydrogen-evolving molecular device [24]. This was reasonably explained by comparing the driving forces for the intramolecular electron transfer from the [Ru*(bpy)2(phen)]2+ moiety to the π*(bpy attached to Pt) orbitals. As summarized in Table 2, molecular devices A and B exhibit activity towards photoinduced HER because of their downhill characters for the photoinduced electron transfer process, while C does not due to the uphill character (ΔG0ET = 0.03 eV). On the other hand, since the reduction at the ppy bound to Pt does not occur at potentials close to -1.2 V vs. Fc/Fc+, the photoinduced electron transfer from the [Ru*(bpy)2(phen)]2+ moiety to the π*(ppy attached to Pt) orbitals is not thermodynamically favorable. It must be noted here that the present study was originally started in our laboratory prior to our approach to A-C, and thereby we were not aware of the fact that the intramolecular electron transfer from the [Ru*(bpy)2(phen)]2+ moiety to the π*(bpy or ppy attached to Pt) orbitals is a key factor to drive the photoinduced HER in these molecular devices. In other words, we decided to focus on the Pt(ppy)Cl2 moiety merely to improve the H2-evolving activity of the catalyst center when we started working on this study. Using the observed electrochemical and spectroscopic parameters, the driving force (ΔG0ET) for the intramolecular electron transfer from the 3MLCT excited state of the [Ru(bpy)2(phen)]2+ moiety to the tethering Pt(ppy) moiety is estimated based on the Rehm-Weller equation (1) [29]:
| ΔG0ET = Eox – Ered – ET – e2/εd | (1) |
where Eox is the first oxidation potential of the Ru center, Ered is the first reduction potential of the ppy moiety bound to the Pt(II) ion, ET is the energy of the 3MLCT exited state, ε is the dielectric constant of DMF, and d is the distance between the donor and acceptor. Since ε values are high for polar solvents like DMF, the last term is negligible. The value of ET is estimated from a tangent to the high-energy side of the emission spectra of 2 in DMF (2.31 eV) [30]. In addition, from the reported reduction potentials of the ppy in [Pt(ppy)Cl2]- (-2.72 V vs. Fc/Fc+; see also Table 2) [28] as well as the oxidation potential for the Ru(II)/Ru(III) couple for 1 (0.92 V vs. Fc/Fc+), the driving force for the intramolecular electron transfer from the [Ru*(bpy)2(phen)]2+ moiety to the π*(ppy attached to Pt) orbitals in 2 can be estimated as 1.33 eV, revealing that this is an amazingly uphill process. This was an obviously unexpected result for us, and the present study reconfirms the importance of having a negative value of ΔG0ET to realize photo-hydrogen-evolving activity for such systems.
2.4. DFT studies
To better understand the electronic structures of the new Ru(II)Pt(II) dimers, the structures of the compounds were computed by the DFT method. First, the four possible conformers of 2 were developed and the structures in aqueous media (polarizable continuum model; PCM) were fully optimized at the B3LYP level of DFT (conformers 2a-d in Figure 4). The relative energies of structures 2a-d are listed in Table 3, showing that the maximum shift is only 2.5 kcal/mol.
Figure 4.

Optimized structures of 2 for the four possible conformers 2a-d in water, computed at the B3LYP level of DFT using the LanL2DZ base set, using the PCM method.
Table 3.
Relative energies and dihedral angles for the four possible conformers of 2.
 |
aDihedral angles between the ppy and the phen planes
For the conformer having the lowest energy (i.e., conformer 2a), some of the relevant frontier molecular orbitals are depicted in Figure 5. The HOMO consists of pz(Cl), dxz(Pt), and π(ppy) orbitals, where the z axis is taken perpendicular to the Pt coordination plane and the x axis is take along the C(ppy)−Pt−Cl axis. These orbitals are coupled in such a manner that both the C(ppy)−Pt and Pt−Cl bonds bear the anti-bonding characters. The HOMO-2 corresponds to the dz2(Pt) orbital, while the HOMO-1, HOMO-3, and HOMO-4 the d(Ru) orbitals. On the other hand, the major contributions to the four lowest unoccupied MOs (i.e., LUMO-LUMO+3) are either π*(bpy) or π*(phen) orbitals. The LUMO+4, which is ca. 0.47 eV higher in energy than the LUMO, is the first lowest unoccupied MO with a major contribution from the π*(ppy) orbitals. These confirm that the first reduction of 2 and 3 occurs at either bpy or phen rather than at ppy.
Figure 5.

Frontier molecular orbitals of a fully optimized structure of conformer 2a for 2 in water, where the structure was computed at the B3LYP level of DFT using the LanL2DZ base set, using the PCM method.
2.5. Photolysis experiments
In order to evaluate the photo-hydrogen-evolving activity of the new Ru(II)Pt(II)-based compounds 2 and 3, the amounts of H2 evolved during the photoirradiation of the compounds in aqueous media were monitored by gas chromatography under the experimental conditions same to those adopted in our previous studies on A-C [13,24]. The pH of each solution was adjusted at 5.0 (acetate buffer) and EDTA was added as a sacrificial electron donor, as previously described. As discussed above, 2 and 3 do not drive the photoinduced HER at all (see Figure 6, lines a,c). However, important observations are that an effective amount of H2 evolves in the presence of methylviologen (MV2+) (see Figure 6, lines b,d). The total amount of H2 evolved after 300-min irradiation was 0.18 mL for the EDTA/2/MV2+ system and 0.05 mL for the EDTA/3/MV2+ system. The turnover number (TON), estimated from the total amount of H2 evolved and the amount of Ru(II)Pt(II) dimer employed (1 μmol) was TON = ca. 8 (after 5 h) for the EDTA/2/MV2+ system and TON = ca. 2 (after 5 h) for the EDTA/3/MV2+ system. The former is effectively higher than the value of TON = 2.4 (after 10 h) reported for the EDTA/A system [13]. These results clearly indicate that both 2 and 3 involve an active photosensitizer unit, [Ru(bpy)2(phen)]2+, as well as a catalyst center active enough to promote the HER. It is thereby reasonable to consider that the 3MLCT excited state of the [Ru(bpy)2(phen)]2+ unit in 2 or 3 is intermolecularly quenched by the coexisting electron relay (MV2+) to once afford MV+, which is then consumed to drive the HER as a dark reaction, as recently reported by the authors [32]. Moreover, photoirradiation of a system consisting of EDTA, [Ru(bpy)3]2+, and a Pt(II)-based catalyst does not lead to HER [7,8], which suggested that [Ru(bpy)3]+ {i.e., [Ru(bpy)2(bpy-·)]+} is not given during the photolysis (reductive quenching of [Ru*(bpy)3]2+ by EDTA does not proceed under those conditions). This further explains the reason why no hydrogen evolves with use of the EDTA/2 and EDTA/3 systems, in which oxidative quenching of the [Ru*(bpy)3]2+ moiety by the H2-evolving catalyst site cannot be promoted in an intramolecular fashion.
Figure 6.

Photochemical H2 production from an aqueous acetate buffer solution (0.03 M CH3CO2H, 0.07 M CH3CO2Na, pH 5.0, 10 mL) containing 30 mM EDTA in the presence of (a) 0.1 mM [2]Cl·6H2O, (b) 0.1 mM [2]Cl·6H2Oand 2 mM MV(NO3)2, (c) 0.1 mM [3](PF6)2·2H2O, and (d) 0.1 mM [3](PF6)2·2H2Oand 2 mM MV(NO3)2.
3. Conclusions
The new heterodinuclear Ru(II)Pt(II) complexes 2 and 3 possessing Pt(ppy)ClX units have been synthesized and characterized. Unfortunately, these dimers do not exhibit catalytic activity as photo-hydrogen-evolving molecular devices in a single-component fashion due to the largely uphill character for the photoinduced electron transfer process. This study reconfirms the importance of controlling the driving force for the electron transfer from the photosensitizing center to the catalyst center to promote the HER within such a photosynthetic molecular device. We now realize that the molecular design strategy must, at the same time, fulfill two factors; one is to improve the H2-evolving activity of the Pt(II)-based moiety, while the other is to have at least one electron acceptor unit in the close vicinity of the catalyst center. Such attempts are now in progress in our laboratory.
4. Experimental
4.1. Materials and measurements
K2PtCl4 was purchased from Tanaka Kikinzoku Kogyo and used as received. All solvents and reagents were of the highest qualities available and were used as received without further purification. N-(1,10-phenanthrolin-5-yl)-4-(2-pyridyl)benzamide (HL) [25], (Bu4N)2[PtCl4] [27], cis-RuCl2(bpy)2 [33], and cis-PtCl2(DMSO)2 [34] were synthesized as previously described.
UV-visible spectra were recorded on a Shimadzu UV-2450 spectrophotometer. IR spectra were recorded on a Perkin Elmer Spectrum One equipped with a diamond ATR (attenuated total reflection) system. 1H-NMR spectra were measured on a JEOL JNM-AL300 spectrometer. Luminescence spectra were recorded on a Shimadzu RF5300PC spectrofluorophotometer. Emission lifetime data were determined using a customized apparatus equipped with an Iwatsu DS-4262 digital oscilloscope and a Hamamatsu R928/C3830 photomultiplier tube coupled to a Horiba H-20-VIS grating monochromator. The excitation source was an N2 laser (337 nm) (Usho KEN-1520). Electrochemical measurements were carried out in argon-purged acetonitrile (MeCN) or N,N-dimethylformamide (DMF) solutions at a sweep rate of 100 mVs-1 using a Pt disk working electrode, a Pt wire counter electrode, and an Ag wire reference electrode (490 mV vs. NHE). The supporting electrolyte was 0.1 M tetra(n-butyl)ammonium perchlorate (TBAP). ESI-TOF mass spectra (ESI-TOF MS) were recorded on a JEOL JMS-T100LC mass spectrometer. All the ESI-TOF MS measurements were performed in the positive-ion mode, at a cone voltage of 20 V, and the mobile phase was acetonitrile or methanol. Typically, a solution of each sample was introduced onto the spectrometer at a flow rate of 10 μL min-1 using a syringe pump.
4.2. DFT calculations
Density functional theory (DFT) calculations were performed using the Gaussian 03 package of programs [35]. The structure was fully optimized using the B3LYP method which uses hybrid Becke's three-parameter exchange functional [36] with the correlation energy functional of Lee, Yang and Parr [37]. Calculations were performed using the standard double-ζ type LanL2DZ basis set [38,39,40] implemented in Gaussian 03, without adding any extra polarization or diffuse function. All the calculations were performed using the polarizable continuum model (PCM) [41] to compute the structures in aqueous media. We experienced that the results obtained by the PCM method are clearly different from those obtained for the structures in their gaseous states, and is considered as more realistic with respect to the energy level of the molecular orbitals.
4.3. Photolysis experiments
Photochemical hydrogen production from water was analyzed using the automatic H2 monitoring system developed in our group. In this system, a continuous flow of Ar (10.0 mL min-1, controlled by an STEC SEC-E40/PAC-D2 digital mass flow controller) was bubbled through each photolysis solution (10 mL) contained in a Pyrex vial (ca. 20 mL). The vent gas from the vial was introduced into a 6-way valve which allowed the automatic injection of the sample gas onto a gas chromatograph (Shimadzu GC-14A equipped with a 5Å molecular sieve column of 2.5 m × 3 mm i.d., thermostatted at 30 °C). The injection of sample gas was driven by a control software operating on a Windows system. The signal output from the thermal conductivity detector was also monitored using the same software. Photolysis solutions were deaerated with Ar for at least 30 min prior to the photolysis. The photoirradiation was carried out by an Ushio Xe short arc lamp UXL500D-O (operated at 350 W). The photolysis vial was immersed in a water bath thermostatted at 20 °C to remove IR radiation and to eliminate the temperature effect.
4.4. Syntheses
[1](PF6)2·3H2O. A solution of cis-RuCl2(bpy)2·3H2O (0.13 g, 0.25 mmol) and HL·1.5H2O (0.094 g, 0.23 mmol) in ethanol (10 mL) was refluxed for 15 h. After the solution was cooled down to room temperature, the solution was filtered for the removal of insoluble materials. Then, water (15 mL) was added to the filtrate, followed by removal of ethanol by evaporation. To this solution was added a saturated aqueous solution of NH4PF6 (ca. 1 mL). The orange powder deposited was collected by filtration and dried in vacuo. Yield: 0.24 g (85%). Calculated for C44H38N8O4P2F12Ru: C, 46.41; H, 3.38; N, 9.73. Found: C, 46.34; H, 3.07; N, 9.73; 1H-NMR (300.53 MHz, DMSO-d6), ppm: 11.04 (s, 1H), 8.90-8.80 (m, 6H), 8.74 (d, 1H, J = 4.8 Hz), 8.62 (s, 1H), 8.34 (d, 2H, J = 8.3 Hz), 8.25-8.07 (m, 8H), 8.00-7.85 (m, 5H), 7.61-7.57 (m, 5H), 7.47-7.36 (m, 3H); ESI-TOF MS: m/z 395.00 [M - 2PF6 - 3H2O]2+; FT-IR (cm-1): 3649 (w), 1675 (m), 1630 (m), 1605 (m), 1587 (m), 1528 (m), 1467 (m), 1434 (m), 1425 (m), 1385 (m), 1314 (m), 1276 (m), 1254 (m), 1162 (m), 1015 (m), 836 (s), 802 (m), 760 (s), 731 (m), 557 (s).
[2]Cl·6H2O. [1](PF6)2·3H2O (0.057 g, 0.050 mmol) was dissolved in a 1:1 H2O-ethanol mixture and was treated with an anion-exchange resin (Amberlite IRA-400) followed by removal of solvents to give the dichloride salt of the complex ([1]Cl2) in a quantitative yield. The residue (i.e., [1]Cl2) and (Bu4N)2[PtCl4] (0.050 g, 0.061 mmol) in methanol (12 mL) was then sealed in a pressure-resistant vial and was stirred at 140 °C for 4 h. After the solution was cooled down to room temperature, the solution was filtered for the removal of insoluble materials (Caution! Do not open the vial while it is hot, since the solution splashes out because of the violent boiling phenomenon upon a sudden decrease in pressure.). The filtrate was concentrated by evaporation under reduced pressure to a total volume of ca. 1 mL. Addition of acetone (20 mL) to the solution resulted in prompt deposition of the product as an orange powder, which was collected by filtration and dried in vacuo. Yield: 0.022 g (37%). Calculated for C44H43N8O7Cl3RuPt: C, 44.10; H, 3.62; N, 9.35. Found: C, 44.18; H, 3.60; N, 9.38; ESI-TOF MS: m/z 1054.85 [M - Cl - 6H2O]+; FT-IR (cm-1): 3369 (m), 3073 (m), 1650 (m), 1627 (m), 1602 (m), 1515 (m), 1480 (m), 1463 (m), 1445 (m), 1423 (m), 1382 (m), 1312 (m), 1270 (m), 1245 (m), 1160 (m), 1108 (m), 1065 (m), 882 (m), 762 (s), 725 (m), 417 (m).
[3](PF6)2·2H2O. A solution of [1](PF6)2·3H2O (0.057 g, 0.050 mmol) and cis-PtCl2(DMSO)2 (0.025 g, 0.060 mmol) in methanol (10 mL) was sealed in a pressure-resistant vial and was stirred at 140 °C for 4 h. After the solution was cooled down to room temperature, the solution was filtered for the removal of insoluble materials (Caution! Do not open the vial while it is hot, since the solution splashes out because of the violent boiling phenomenon upon a sudden decrease in pressure.). By repeating gradual removal of ethanol by evaporation and by gradual addition of water, the major solvent was exchanged from ethanol to water until the total volume of the solution became ca. 10 mL. Addition of a saturated aqueous solution of NH4PF6 (ca. 0.5 mL) to the former solution resulted in prompt deposition of the product as an orange powder, which was collected by filtration and dried in vacuo. Yield: 0.050 g (71%). Calculated for C44H41N8O4ClSP2F12RuPt: C, 38.81; H, 2.90; N, 7.87. Found: C, 38.64; H, 2.75; N, 7.69; 1H-NMR (300.53 MHz, DMSO-d6), ppm: 10.04 (s, 1H), 9.69 (d, 1H, J = 5.7 Hz), 9.22-9.18 (m, 2H), 8.96 (d, 1H, J = 4.1 Hz), 8.89-8.80 (m, 5H), 8.50 (dd, 1H, J = 5.1 Hz, J' = 1.1 Hz), 8.38 (dd, 1H, J = 5.18 Hz, J' = 1.2 Hz), 8.30-8.14 (m, 8H), 8.00-7.88 (m, 6H), 7.68-7.61 (m, 3H), 7.45-7.40 (m, 2H), 3.71(d, 6H, J = 3.1 Hz); ESI-TOF MS: m/z 548.97 [M - 2PF6 - 2H2O]2+; FT-IR (cm-1): 3649 (w), 1680 (m), 1629 (m), 1606 (m), 1524 (m), 1483 (m), 1466 (m), 1447 (m), 1424 (m), 1380 (m), 1315 (m), 1271 (m), 1246 (m), 1163 (m), 1135 (m), 1123 (m), 1025 (m), 835 (s), 760 (s), 730 (m), 556 (s), 444 (m), 421 (m).
Acknowledgements
This work was partly supported by a Grant-in-Aid for Scientific Research (A) (No. 17205008), a Grant-in-Aid for Specially Promoted Research (No. 18002016), a Grant-in-Aid for Scientific Research (B) (No. 21350036), and a Grant-in-Aid for the Global COE Program ('Science for Future Molecular Systems') from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Footnotes
Sample Availability: Samples of the compounds 1-3 are available from the authors.
References and Notes
- 1.Lehn J.-M., Sauvage J.-P. Chemical storage of light energy catalytic generation of hydrogen by visible light or sunlight. Irradiation of neutral aqueous solutions. Nouv. J. Chim. 1977;1:449–451. [Google Scholar]
- 2.Kirch M., Lehn J.-M., Sauvage J.-P. Hydrogen Generation by Visible Light Irradiation of Aqueous Solutions of Metal Complexes. An Approach to the Photochemical Conversion and Storage of Solar Energy. Helv. Chim. Acta. 1979;62:1345–1384. doi: 10.1002/hlca.19790620449. [DOI] [Google Scholar]
- 3.Kalyanasundaram K., Kiwi J., Grätzel M. Hydrogen Evolution from Water by Visible Light, a Homogeneous Three Component Test System for Redox Catalysis. Helv. Chim. Acta. 1978;61:2720–2730. doi: 10.1002/hlca.19780610740. [DOI] [Google Scholar]
- 4.Grätzel M. Artificial Photosynthesis: Water Cleavage into Hydrogen and Oxygen by Visible Light. Acc. Chem. Res. 1981;14:376–384. doi: 10.1021/ar00072a003. [DOI] [Google Scholar]
- 5.Moradpour A., Amouyal E., Keller P., Kagan H.B. Hydrogen production by visible light irradiation of aqueous solutions of tris(2,2'-bipyridine)ruthenium(2+) Nouv. J. Chim. 1978;2:547–549. [Google Scholar]
- 6.Keller P., Moradpour A., Amouyal E., Kagan H.B. Hydrogen production by visible-light using viologen-dye mediated redox cycles. Nouv. J. Chim. 1980;4:377–384. [Google Scholar]
- 7.Sakai K., Matsumoto K. Photochemical Reduction of Water to Hydrogen Catalyzed by Mixed-Valent Tetranuclear Platinum Complex. J. Coord. Chem. 1988;18:169–172. doi: 10.1080/00958978808080702. [DOI] [Google Scholar]
- 8.Sakai K., Matsumoto K. Homogeneous Catalysis of Platinum Blue Related Complexes in Photoreduction of Water into Hydrogen. J. Mol. Catal. 1990;62:1–14. doi: 10.1016/0304-5102(90)85235-A. [DOI] [Google Scholar]
- 9.Sakai K., Kizaki Y., Tsubomura T., Matsumoto K. Homogeneous Catalysis of Mixed-Valent Octanuclear Platinum Complexes in Photochemical Hydrogen Production from Water. J. Mol. Catal. 1993;79:141–152. doi: 10.1016/0304-5102(93)85097-D. [DOI] [Google Scholar]
- 10.Ozawa H., Yokoyama Y., Haga M., Sakai K. Syntheses, Characterization, and Photo-Hydrogen-Evolving Properties of Tris(2,2'-bipyridine)ruthenium(II) Derivatives Tethered to a cis-Pt(II)Cl2 Unit: Insights into the Structure-Activity Relationship. Dalton Trans. 2007:1197–1206. doi: 10.1039/b617617h. [DOI] [PubMed] [Google Scholar]
- 11.Sakai K., Ozawa H. Homogeneous catalysis of platinum(II) complexes in photochemical hydrogen production from water. Coord. Chem. Rev. 2007;251:2753–2766. doi: 10.1016/j.ccr.2007.08.014. [DOI] [Google Scholar]
- 12.Kobayashi M., Masaoka S., Sakai K. Synthesis, Crystal Structure, Solution and Spectroscopic Properties, and Hydrogen-Evolving Activity of [K(18-Crown-6)][Pt(II)(2-phenylpyridinato)Cl2. Photochem. Photobiol. Sci. 2009;8:196–203. doi: 10.1039/b814939a. [DOI] [Google Scholar]
- 13.Ozawa H., Haga M., Sakai K. A Photo-Hydrogen-Evolving Molecular Device Driving Visible-Light-Induced EDTA-Reduction of Water into Molecular Hydrogen. J. Am. Chem. Soc. 2006;128:4926–4927. doi: 10.1021/ja058087h. [DOI] [PubMed] [Google Scholar]
- 14.Rau S., Schäfer B., Gleich D., Anders E., Rudolph M., Friedrich M., Görls H., Henry W., JVos J.G. A Supramolecular Photocatalyst for the Production of Hydrogen and the Selective Hydrogenation of Tolane. Angew. Chem. Int. Ed. 2006;45:6215–6218. doi: 10.1002/anie.200600543. [DOI] [PubMed] [Google Scholar]
- 15.Rau S., Walther D., Vos J.G. Inspired by nature: light driven organometallic catalysis by heterooligonuclear Ru(II) complexes. Dalton Trans. 2007:915–919. doi: 10.1039/b615987g. [DOI] [PubMed] [Google Scholar]
- 16.Elvington M., Brewer K.J. Photoinitiated Electron Collection at a Metal in a Rhodium-Centered Mixed-Metal Supramolecular Complex. Inorg. Chem. 2006;45:5242–5244. doi: 10.1021/ic0602648. [DOI] [PubMed] [Google Scholar]
- 17.Elvington M., Brown J., Arachchige S.M., Brewer K.J. Photocatalytic Hydrogen Production from Water Employing A Ru, Rh, Ru Molecular Device for Photoinitiated Electron Collection. J. Am. Chem. Soc. 2007;129:10644–10645. doi: 10.1021/ja073123t. [DOI] [PubMed] [Google Scholar]
- 18.Fihri A., Artero V., Razavet M., Baffert C., Leibl W., Fontecave M. Cobaloxime-Based Photocatalytic Devices for Hydrogen Production. Angew. Chem. Int. Ed. 2008;47:564–567. doi: 10.1002/anie.200702953. [DOI] [PubMed] [Google Scholar]
- 19.Fihri A., Artero V., Pereira A., Fontecave M. Efficient H2-producing photocatalytic systems based on cyclometalated iridium- and tricarbonylrhenium-diimine photosensitizers and cobaloxime catalysts. Dalton Trans. 2008:5567–5569. doi: 10.1039/b812605b. [DOI] [PubMed] [Google Scholar]
- 20.Arachchige S.M., Brown J.R., Chang E., Jain A., Zigler D.F., Rangan K., Brewer K.J. Design Considerations for a System for Photocatalytic Hydrogen Production from Water Employing Mixed-Metal Photochemical Molecular Devices for Photoinitiated Electron Collection. Inorg. Chem. 2009;48:1989–2000. doi: 10.1021/ic8017387. [DOI] [PubMed] [Google Scholar]
- 21.Wang M., Na Y., Gorlov M., Sun L. Light-driven hydrogen production catalysed by transition metal complexes in homogeneous systems. Dalton Trans. 2009:6458–6467. doi: 10.1039/b903809d. [DOI] [PubMed] [Google Scholar]
- 22.Ozawa H., Kobayashi M., Balan B., Masaoka S., Sakai K. Photo-Hydrogen-Evolving Molecular Catalysts Consisting of Polypyridyl Ruthenium(II) Photosensitizers and Platinum(II) Catalysts: Insights into the Reaction Mechanism. Chem. Asian J. 2010 doi: 10.1002/asia.201000083. [DOI] [PubMed] [Google Scholar]
- 23.Ozawa H., Sakai K. An Effect of Structural Modification in the Photo-hydrogen-evolving RuIIPtII Dimers. Chem. Lett. 2007;36:920–921. doi: 10.1246/cl.2007.920. [DOI] [Google Scholar]
- 24.Masaoka S., Mukawa Y., Sakai K. Frontier Orbital Engineering of Photo-Hydrogen-Evolving Molecular Devices: A Clear Relationship Between the H2-Evolving Activity and the Energy Level of the LUMO. Dalton Trans. 2010;39:5868–5876. doi: 10.1039/c0dt00077a. [DOI] [PubMed] [Google Scholar]
- 25.Kobayashi M., Masaoka S., Sakai K. N-(1,10-phenanthrolin-5-yl)-4-(pyridin-2-yl)benzamide monohydrate. Acta Cryst. 2008;E64:o1979. doi: 10.1107/S1600536808029851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kobayashi M., Masaoka S., Sakai K. Chrolo(dimethylsulfoxide)(phenylpyridinato)platinum(II) Acta Cryst. 2008;E64:m1557. [Google Scholar]
- 27.Craig C.A., Garces F.O., Watts R.J., Palmans R., Frank A.J. Luminescence properties of two new Pt(II)-2-phenylpyridine complexes; the influence of metal-carbon bonds. Coord. Chem. Rev. 1990;97:193–208. [Google Scholar]
- 28.Kvam P.-I., Puzyk M.V., Balashev K.P., Songstad J. Spectroscopic and Electrochemical Properties of Some Mixed-Ligand Cyclometalated Platinum(II) Complexes Derived from 2-Phenylpyridine. Acta Chem. Scand. 1995;49:335–343. doi: 10.3891/acta.chem.scand.49-0335. [DOI] [Google Scholar]
- 29.Rehm D., Weller A. Kinetik und Mechanismus der Elektronumbertragung bei der Fluoreszenzloschung in Acetonitril. Ber. Bunsen-Ges. Phys. Chem. 1969;73:834–839. [Google Scholar]
- 30.Islam A., Sugihara H., Singh L.P., Hara K., Katoh R., Nagawa Y., Yanagida M., Takahashi Y., Murata S., Arakawa H. Synthesis and photophysical properties of ruthenium(II) charge transfer sensitizers containing 4,4'-dicarboxy-2,2'-biquinoline and 5,8-dicarboxy-6,7-dihydro-dibenzo[1,10]-phenanthroline. Inorg. Chim. Acta. 2001;322:7–16. doi: 10.1016/S0020-1693(01)00548-5. [DOI] [Google Scholar]
- 31.Huber A., Muller L., Elias H., Klement R., Valko M. Cobalt(II) Complexes with Substituted Salen-Type Ligands and Their Dioxygen Affinity in N,N-Dimethylformamide at Various Temperatures. Eur. J. Inorg. Chem. 2005:1459–1467. [Google Scholar]
- 32.Yamauchi K., Masaoka S., Sakai K. Evidence for Pt(II)-Based Molecular Catalysis in the Thermal Reduction of Water into Molecular Hydrogen. J. Am. Chem. Soc. 2009;131:8404–8406. doi: 10.1021/ja902245e. [DOI] [PubMed] [Google Scholar]
- 33.Sullivan B.P., Salmon D.J., Meyer T.J. Mixed Phosphine 2,2'-Bipyridine Complexes of Ruthenium. Inorg. Chem. 1978;17:3334–3341. doi: 10.1021/ic50190a006. [DOI] [Google Scholar]
- 34.Price J.H., Williamson A.N., Schramm R.F., Wayland B.B. Palladium(II) and Platinum(II) Alkyl Sulfoxide Complexes. Examples of Sulfur-Bonded, Mixed Sulfur- and Oxygen-Bonded, and Totally Oxygen-Bonded Complexes. Inorg. Chem. 1972;11:1280–1284. doi: 10.1021/ic50112a025. [DOI] [Google Scholar]
- 35.Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Montgomery J.A., Jr., Vreven T., Kudin K.N., Burant J.C., Millam J.M., Iyengar S.S., Tomasi J., Barone V., Mennucci B., Cossi M., Scalmani G., Rega N., Petersson G.A., Nakatsuji H., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Klene M., Li X., Knox J.E., Hratchian H.P., Cross J.B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R.E., Yazyev O., Austin A.J., Cammi R., Pomelli C., Ochterski J.W., Ayala P.Y., Morokuma K., Voth G.A., Salvador P., Dannenberg J.J., Zakrzewski V.G., Dapprich S., Daniels A.D., Strain M.C., Farkas O., Malick D.K., Rabuck A.D., Raghavachari K., Foresman J.B., Ortiz J.V., Cui Q., Baboul A.G., Clifford S., Cioslowski J., Stefanov B.B., Liu G., Liashenko A., Piskorz P., Komaromi I., Martin R.L., Fox D.J., Keith T., Al-Laham M.A., Peng C.Y., Nanayakkara A., Challacombe M., Gill P.M. W., Johnson B., Chen W., Wong M.W., Gonzalez C., Pople J.A. Gaussian 03. Gaussian, Inc.; Wallingford, CT, USA: 2004. [Google Scholar]
- 36.Becke A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993;98:5648–5652. doi: 10.1063/1.464913. [DOI] [Google Scholar]
- 37.Lee C., Yang W., Parr R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 38.Dunning T.H., Hay P.J. In: In Modern Theoretical Chemistry. Schaefer H.F. III, editor. Plenum; New York, NY, USA: 1976. pp. 1–28. [Google Scholar]
- 39.Hay P.J., Wadt W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985;82:270–283. doi: 10.1063/1.448799. [DOI] [Google Scholar]
- 40.Hay P.J., Wadt W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985;82:299–310. doi: 10.1063/1.448975. [DOI] [Google Scholar]
- 41.Cossi M., Scalmani G., Rega N., Barone V. New developments in the polarizable continuum model for quantum mechanical and classical calculations on molecules in solution. J. Chem. Phys. 2002;117:43–54. [Google Scholar]