

Abstract
A series of dinuclear octahedral PtIV complexes trans,trans,trans-[{Pt(N3)2(py)2(OH)(OC(O)CH2CH2C(O)NH)}2R] containing pyridine (py) and bridging dicarboxylate [R = −CH2CH2– (1), trans-1,2-C6H10– (2), p-C6H4– (3), −CH2CH2CH2CH2– (4)] ligands have been synthesized and characterized, including the X-ray crystal structures of complexes 1·2MeOH and 4, the first photoactivatable dinuclear PtIV complexes with azido ligands. The complexes are highly stable in the dark, but upon photoactivation with blue light (420 nm), they release the bridging ligand and mononuclear photoproducts. Upon irradiation with blue light (465 nm), they generate azidyl and hydroxyl radicals, detected using a 5,5-dimethyl-1-pyrroline N-oxide electron paramagnetic resonance spin trap, accompanied by the disappearance of the ligand-to-metal charge-transfer (N3 → Pt) band at ca. 300 nm. The dinuclear complexes are photocytotoxic to human cancer cells (465 nm, 4.8 mW/cm2, 1 h), including A2780 human ovarian and esophageal OE19 cells with IC50 values of 8.8–78.3 μM, whereas cisplatin is inactive under these conditions. Complexes 1, 3, and 4 are notably more photoactive toward cisplatin-resistant ovarian A2780cis compared to A2780 cells. Remarkably, all of the complexes were relatively nontoxic toward normal cells (MRC5 lung fibroblasts), with IC50 values >100 μM, even after irradiation. The introduction of an aromatic bridging ligand (3) significantly enhanced cellular uptake. The populations in the stages of the cell cycle remained unchanged upon treatment with complexes in the dark, while the population of the G2/M phase increased upon irradiation, suggesting that DNA is a target for these photoactivated dinuclear PtIV complexes. Liquid chromatography–mass spectrometry data show that the photodecomposition pathway of the dinuclear complexes results in the release of two molecules of mononuclear platinum(II) species. As a consequence, DNA binding of the dinuclear complexes after photoactivation in cell-free media is, in several respects, qualitatively similar to that of the photoactivated mononuclear complex FM-190. After photoactivation, they were 2-fold more effective in quenching the fluorescence of EtBr bound to DNA, forming DNA interstrand cross-links and unwinding DNA compared to the photoactivated FM-190.
Short abstract
Novel all-trans dinuclear PtIV complexes bridged by a dicarboxylate linker, highly stable in the dark, generate azidyl and hydroxyl radicals upon irradiation with blue light. They are photocytotoxic to human cancer cells, whereas cisplatin was inactive under these conditions and more photoactive toward cisplatin-resistant ovarian cancer cells compared to wild-type cells. Remarkably, the dinuclear complexes were relatively nontoxic toward normal human cells. Cell cycle and DNA binding experiments suggested that DNA is a target.
Introduction
Photoactivation is a promising effective and noninvasive chemotherapeutic strategy.1−4 Photochemotherapy using the appropriate Pt complexes has the potential to overcome some of the limitations of conventional cisplatin therapy, including poor pharmacokinetics, dose-limiting side effects, restricted spectrum of anticancer activity, high incidence of resistance, and lack of normal-cell discrimination.5,6 The application of O2-dependent photodynamic therapy is limited because of the low concentration of O2 in hypoxic tumors, together with the need for diffusion of O2 from adjacent tissues.7 Photoactivated chemotherapy, in contrast, is less dependent on O2 for cytotoxicity and provides a new avenue to anticancer drug design.
Octahedral, low-spin 5d6 PtIV complexes are usually considered to be prodrugs because they are kinetically more inert than their PtII counterparts under biological conditions.8−10 Photoactivatable PtIV prodrugs can exhibit high dark stability and potential photocytotoxicity;11−13 examples include diazidoplatinum(IV) complexes containing various nonleaving amines and axial substituents.14−20 Among them, trans,trans,trans-[Pt(N3)2(OH)2(py)2] (FM-190) is highly potent,21,22 has good aqueous solubility, is relatively stable in cell culture media and toward reactions with abundant intracellular tripeptide glutathione (GSH; γ-l-Glu-l-Cys-Gly), and can be activated by UVA, blue (420 nm) and green (500 ± 30 nm) light, achieving low micromolar IC50 values even in cisplatin-resistant cell lines, with a high phototoxicity index. Not only does the Pt center become reactive upon photoreduction, but, in addition, azidyl radicals can be released by FM-190 in a manner controllable by the amino acid l-Trp.23
Axial ligands in diazidoplatinum(IV) complexes normally act as leaving groups upon reduction to PtII and greatly influence the reduction potential of PtIV. Derivatization of an axial ligand can be used to improve the pharmacological properties without interfering with the ultimate mode of action of the active PtII species or the potential production of other reactive species. Several studies have been carried out previously on the conjugation of FM-190 to functional fragments, including αvβ3 and αvβ5 integrin-selective arginine–glycine–aspartic acid(RGD)-containing peptides,24 RNA-binding guanidinoneomycin,25 2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) radical,26 upconversion-luminescent nanoparticles,27 and G-quadruplex G4K+ borate hydrogels.28 For the former two conjugates, the selectivity and cellular uptake toward specific cancer cells was remarkably enhanced, while for the prodrug with TEMPO, azidyl and TEMPO radicals were released upon irradiation with blue light (420 nm), accompanied by the formation of toxic PtII species, resulting in improved cytotoxicity. Upconversion-luminescent nanoparticles enable photoactivation with near-IR light, and hydrogels allow the treatment of surface cancers with minimal damage to normal tissues.
Polynuclear Pt complexes have attracted much attention because of their unique biological properties, including coordinative binding to DNA bases and interaction of the phosphodiester linker through electrostatic and hydrogen-bonding effects.29 The preassociation of polynuclear Pt complexes with DNA prior to coordinate bond formation can significantly affect the kinetics of cross-link formation,30−33 which distinguishes them from their mononuclear counterparts. The trinuclear complex [{trans-PtCl(NH3)2}2(μ-trans-Pt(NH3)2{NH2(CH2)6NH2}2)]4+ (BBR3464) underwent phase I and II human clinical trials as a promising anticancer Pt drug.34−36
In order to investigate the structure–activity relationships related to modification of the axial ligands of diazidoplatinum(IV) complexes and improve the antiproliferative activity of photoactivatable PtIV prodrugs, we synthesized and characterized a series of dinuclear PtIV complexes of the type trans,trans,trans-[{Pt(N3)2(py)2(OH)(OC(O)CH2CH2C(O)NH)}2R] [R = −CH2CH2– (1), trans-1,2-C6H10– (2), p-C6H4– (3), −CH2CH2CH2CH2– (4)]. The X-ray crystal structures of complexes 1·2MeOH and 4 were determined. These appear to be the first photoactivatable dinuclear PtIV complexes containing azides to be reported. Their photodecomposition, photoreactions with 5′-guanosine monophosphate (5′-GMP), interaction with DNA, photocytotoxicity, cellular accumulation, and effect on the cancer cell cycle distribution were studied. The chemical and biological behavior of these complexes containing two Pt centers and four azides in one molecule are compared to related mononuclear diazido complexes, including the influence of the nature of the bridging ligand.
Experimental Section
Materials and Instruments
O-(Benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate (TBTU) was purchased from Merck. Pyridine was purchased from Fischer Scientific UK. Calf thymus DNA (ct-DNA), K2PtCl4, NaN3, H2O2 (30%), succinic anhydride, and other chemicals were purchased from Sigma-Aldrich and used without further purification.
NMR spectra were recorded on Bruker Avance III HD 400 MHz and Bruker 500 MHz spectrometers; the residual signal of the solvent was used as a reference. Electrospray ionization mass spectrometry (ESI-MS) spectra were recorded on an Agilent 6130B single quadrupole detector instrument, and high-resolution ESI-MS data were collected on a Bruker microTOF instrument at 298 K with a scan range of m/z 50–2000 for positive ions. Samples were prepared in methanol or aqueous solution. Electronic absorption spectra were recorded on a Varian Cary 300 UV–vis spectrophotometer in a quartz cuvette and referenced to neat solvent. The spectral width was 200–800 nm, the bandwidth was 1.0 nm, and the scan rate was set to 600 nm/min. Analytical reversed-phase high-performance liquid chromatography (HPLC) analyses were carried out on an Agilent ZORBAX Eclipse XDB-C18 column (250 × 4.6 mm, 5 μm, flow rate 1 mL/min) by using linear gradients of 0.1% trifluoroacetic acid (TFA) in water (solvent A) and 0.1% TFA in acetonitrile (solvent B). Liquid chromatography–mass spectrometry (LC–MS) was carried out on a Bruker Amazon X connected online with HPLC. The light sources used for photoactivation were an LZC-ICH2 photoreactor (Luzchem Research Inc.) equipped with a temperature controller, eight Luzchem LZC-420 lamps without light filtration, and a KiloArcTM broad-band arc lamp monochromator supplied with the appropriate filters to cut off any unwanted light. A light-emitting-diode (LED) light source with λmax = 465 nm was used for in vitro growth inhibition and cell cycle assays. Platinum contents were analyzed on a 5300DV inductively coupled plasma optical emission spectrometer (ICP-OES, PerkinElmer) or a 7500cx inductively coupled plasma mass spectrometer (ICP-MS, Agilent). The emission wavelengths detected for Pt were 265.945, 214.423, 299.797, 204.937, and 193.700 nm.
Synthesis and Characterization
Caution! Heavy-metal azides can be shock-sensitive detonators. We did not encounter any problems during the work reported here, but due care and attention with appropriate precautions should be taken in their synthesis and handling. All synthesis and purifications were carried out in the dark with minimal light exposure.
General Synthesis Procedure for Complexes 1–4
To a solution of trans,trans,trans-[Pt(N3)2(OH)(succinate)(py)2] (24.4 mg, 42.8 μmol) and TBTU (11.8 mg, 36.8 μmol) in freshly degassed anhydrous N,N-dimethylformamide (DMF; 2 mL) N,N-diisopropylethylamine (DIPEA; 100 μL) was added. After stirring for 3 min, a solution of 0.5 mol equiv of the corresponding diamine, DIPEA (60 μL) and DMF (1 mL), was added to the resulting mixture dropwise. The reaction mixture was stirred overnight at 298 K under a N2 atmosphere. After evaporation to dryness, the oily residue was collected and purified by column chromatography on aluminum oxide (5% methanol + 95% dichloromethane).
trans,trans,trans-[{Pt(N3)2(py)2(OH)(OC(O)CH2CH2C(O)NH)}2(CH2)2] (1)
Diamine = ethylenediamine. 1H NMR (DMSO-d6, 400 MHz): δ 8.81 (dd, J = 5.5 Hz, J195Pt–1H = 26.6 Hz, 8H, Hα py), 8.26 (t, J = 7.5 Hz, 4H, Hγ py), 7.82 (t, J = 7.0 Hz, 8H, Hβ py), 7.75 (t, J = 5.7 Hz, 2H, CONH), 3.66 (s, 2H, OH), 3.05 (s, 4H, CH2), 2.46 (t, J = 7.6 Hz, 4H, CH2), 2.22 (t, J = 7.3 Hz, 4H, CH2). 13C NMR (DMSO-d6, 125 MHz): δ 175.27 (COO), 172.08 (CONH), 149.83 (Cαpy), 142.42 (Cγ py), 126.66 (Cβ py), 38.80 (CH2), 32.39 (CH2), 32.21 (CH2). ESI-MS (M + Na+): m/z 1189.2160. Anal. Calcd for C30H36N18O8Pt2: C, 30.88; H, 3.11; N, 21.61. Found: C, 30.43; H, 3.10; N, 20.69.
trans,trans,trans-[{Pt(N3)2(py)2(OH)(OC(O)CH2CH2C(O)NH)}2(trans-1,2-C6H10)] (2)
Diamine = trans-1,2-cyclohexanediamine. 1H NMR (DMSO-d6, 400 MHz): δ 8.82 (dd, J = 5.5 Hz, J195Pt–1H = 26.5 Hz, 8H, Hα py), 8.25 (t, J = 7.5 Hz, 4H, Hγ py), 7.82 (t, J = 6.9 Hz, 8H, Hβ py), 7.52 (d, J = 7.3 Hz, 2H, CONH), 3.66 (s, 2H, OH), 3.47 (s, 2H, CH2 cyclohexyl), 2.48–2.33 (m, 4H, CH2), 2.26–2.10 (m, 4H, CH2), 1.74 (s, 2H, CH2 cyclohexyl), 1.62 (s, 2H, CH2 cyclohexyl), 1.18 (s, 4H, CH2 cyclohexyl). 13C NMR (DMSO-d6, 125 MHz): δ 175.23 (COO), 171.68 (CONH), 149.84 (Cα py), 142.38 (Cγ py), 126.65 (Cβ py), 55.38 (CH2 cyclohexyl), 52.50 (CH cyclohexyl), 49.06 (CH cyclohexyl), 32.74 (CH2), 32.58 (CH2), 32.10 (CH2 cyclohexyl), 24.87 (CH2 cyclohexyl). ESI-MS (M + Na+): m/z 1243.2631. Anal. Calcd for C34H42N18O8Pt2: C, 33.45; H, 3.47; N, 20.65. Found: C, 33.84; H, 3.48; N, 19.14.
trans,trans,trans-[{Pt(N3)2(py)2(OH)(OC(O)CH2CH2C(O)NH)}2(1,4-C6H4)] (3)
Diamine = p-phenylenediamine. 1H NMR (DMSO-d6, 400 MHz): δ 9.79 (s, 2H, CONH), 8.82 (dd, J = 5.5 Hz, J195Pt–1H = 26.7 Hz, 8H, Hα py), 8.25 (t, J = 7.3 Hz, 4H, Hγ py), 7.79 (t, J = 7.1 Hz, 8H, Hβ py), 7.50 (s, 4H, CH benzyl), 3.67 (s, 2H, OH), 2.67 (t, J = 7.3 Hz, 4H, CH2), 2.33 (t, J = 7.6 Hz, 4H, CH2). 13C NMR (DMSO-d6, 125 MHz): δ 175.20 (COO), 170.71 (CONH), 149.83 (Cα py), 142.39 (Cγ py), 135.10 (C benzyl), 126.64 (Cβ py), 119.77 (CH benzyl), 33.09 (CH2), 32.13 (CH2). ESI-MS (M + Na+): m/z 1237.2138. Anal. Calcd for C34H36N18O8Pt2·Et2O: C, 35.41; H, 3.60; N, 19.56. Found: C, 35.54; H, 3.76; N, 19.96
trans,trans,trans-[{Pt(N3)2(py)2(OH)(OC(O)CH2CH2C(O)NH)}2(CH2)4] (4)
Diamine = 1,4-butylenediamine. 1H NMR (DMSO-d6, 400 MHz): δ 8.82 (dd, J = 5.5 Hz, J195Pt–1H = 26.9 Hz, 8H, Hα py), 8.27 (t, J = 7.6 Hz, 4H, Hγ py), 7.83 (t, J = 7.0 Hz, 8H, Hβ py), 7.71 (t, J = 5.5 Hz, 2H, CONH), 3.65 (s, 2H, OH), 3.00 (m, 4H, CH2), 2.45 (t, J = 7.3 Hz, 4H, CH2), 2.23 (t, J = 7.4 Hz, 4H, CH2), 1.34 (s, 4H, CH2). 13C NMR (DMSO-d6, 125 MHz): δ 175.28 (COO), 171.72 (CONH), 149.84 (Cα py), 142.41 (Cγ py), 126.66 (Cβ py), 38.69 (CH2), 32.51 (CH2), 32.21 (CH2), 27.10 (CH2). ESI-MS (M + Na+): m/z 1217.2461. Anal. Calcd for C32H40N18O8Pt2: C, 32.16; H, 3.37; N, 21.10. Found: C, 32.44; H, 3.36; N, 20.30.
X-ray Crystallography
Single crystals of 1·2MeOH and 4 were grown from methanol/ether at ambient crystal was selected and mounted on a Mitegen head with Fomblin oil and placed on an Xcalibur Gemini diffractometer with a Ruby charge-coupled-device (CCD) area detector. The crystal was kept at 150(2) K during data collection. Using Olex2,37 the structure was solved with the ShelXT(38) structure solution program using intrinsic phasing and refined with the ShelXL(39) refinement package using least-squares minimization.
Photodecomposition in Aqueous Solution
Photodecomposition of complexes 1–4 in aqueous solution was monitored by UV–vis spectroscopy or LC–MS at different time intervals after irradiation with blue light (420 nm) at ambient temperature.
Cell Culture
A number of human cell lines, parental (A2780) and cisplatin-resistant (A2780cis) ovarian carcinoma, esophageal adenocarcinoma (OE19), and normal fibroblast cells (MRC5), were obtained from the European Collection of Animal Cell Culture, Salisbury, U.K. All cell lines used in this work were grown in Roswell Park Memorial Institute media (RPMI-1640), which was supplemented with 10% (v/v) of fetal calf serum, 1% (v/v) of 2 mM glutamine, and 1% (v/v) penicillin/streptomycin. The adherent monolayers of cells were grown at 310 K in a humidified atmosphere containing 5% CO2 and passaged regularly at ca. 80% confluence.
Photo/dark Cytotoxicity
Approximately 10000 cells were seeded per well in 96-well plates. Independent duplicate plates were used, one for dark experiments and the other for irradiation experiments. Complexes were first dissolved in dimethyl sulfoxide (DMSO) and then diluted in phenol-red-free RPMI-1640 to make the stock solution of the drug. These stock solutions were further diluted using a phenol-red-free cell culture medium until working concentrations were achieved, in these solutions, the maximum DMSO concentration was in all cases <0.5% (v/v). Cells were exposed to the drugs with different concentrations for 1 h. Then one plate was irradiated for 1 h using blue light (4.8 mW cm–2 per LED at 465 nm), while the dark plate was kept in the incubator. After irradiation, supernatants of both plates were removed by suction and washed with a phosphate-buffered saline (PBS) buffer. The photocytotoxicity was determined after another 24 h of recovery at 310 K in a drug-free phenol-red-containing medium by comparison to untreated controls, which were only exposed to a vehicle. Untreated controls were also compared between the irradiated and nonirradiated plates to ensure that the differences in cell survival were not statistically relevant, hence guaranteeing that the differences in cell viability observed were not due to the light source. Sulforhodamine B (SRB) assay was used to determine the cell viability.40 Absorbance measurements of the solubilized dye (on a Promega microplate reader, 510 nm) allowed the determination of viable treated cells compared to untreated controls. The IC50 values (concentrations that caused 50% of cell death) were determined as the average of triplicates, and their standard deviations were calculated. Stock concentrations for all metal complexes used in these biological assays were adjusted/verified after ICP-OES metal quantification.
Pt Accumulation in Cancer Cells
For Pt cellular accumulation studies, ca. 4.5 × 106 A2780, A2780cis, and OE19 cells were plated in 100 mm Petri dishes and allowed to attach for 24 h. Then the plates were exposed to complexes at equipotent concentrations equal to the photoactive IC50 values in the corresponding cell line. Additional plates were incubated with a medium alone as a negative control. After 1 h of incubation in the dark at 310 K, the cells were rinsed three times with cold PBS and harvested by trypsinization. The number of cells in each sample was counted manually using a hemocytometer. Then the cells were centrifuged to obtain the whole cell pellet for ICP-MS analysis. All experiments were conducted in triplicate.
ICP-MS Sample Preparation
The whole cell pellets were dissolved in concentrated 72% (v/v) nitric acid (200 μL), and the samples were then transferred into Wheaton V vials (Sigma-Aldrich) and heated in an oven at 343 K overnight. The vials were then allowed to cool, and each cellular sample solution was transferred into a vial and diluted with Milli-Q water (3.8 mL) to obtain a final HNO3 concentration of ca. 3.6% (v/v).
Photoreactions with 5′-GMP
An amount of 2, 4, or 8 mol equiv of 5′-guanosine monophosphate disodium salt hydrate (5′-GMP-Na2) was mixed with 30 μM complex 4 in 5:95 (v/v) methanol/water. The solution was incubated at 310 K for 1 h, then irradiated for 1 h, and analyzed immediately. HPLC analysis was carried out on an Agilent ZORBAX Eclipse XDB-C18 column (250 × 4.6 mm, 5 μm, flow rate 1 mL/min). The mobile phases for HPLC were A (0.1% formic acid in HPLC-grade water, volume percentage) and B (0.1% formic acid in HPLC-grade acetonitrile, volume percentage). The Pt adducts were isolated and analyzed by a Bruker Amazon X connected online with the high-performance liquid chromatography (HPLC).
Electron Paramagnetic Resonance (EPR) Spectroscopy
The EPR spectra were recorded on a Bruker EMX (X-band) spectrometer at 298 K. Samples (ca. 100 μL) in aqueous solution were prepared, transferred using a plastic syringe with metal needle to a standard quality quartz tube with an inner diameter of 1.0 mm and an outer diameter of 2.0 mm (Wilmad LabGlass), and sealed with parafilm. A y-incremental sweep mode of 120 with an accumulation of five scans in the x dimensions was used. Typical key EPR spectrometer settings were 2.0 G modulation amplitude, 1.37 mW microwave power, 1.0 × 105 receiver gain, 5.12 ms conversion time, 5.12 ms time constant, and 200 G sweep width. The LED465E light irradiation source was mounted within the EPR magnet, supported by a foam sponge, to maintain its position throughout the EPR measurements. The distance from the tip of the irradiation light bulb to the EPR cavity was ca. 3 cm. Data were processed by Matlab R2016b with Easyspin 5.1.12 through a multicomponent fit.
Flow Cytometry
All flow cytometry experiments were carried out using a Becton Dickinson FACScan flow cytometer in the School of Life Sciences at Warwick University. Typically, cells were seeded in 6-well plates using 1.5 × 106 cells/well. Experiments included 24 h of preincubation in drug-free media at 310 K in a CO2 humidified atmosphere, followed by 1 h of drug exposure under the same conditions. After this, samples were irradiated for 1 h. For comparison, the dark plates were kept in the incubator for another 1 h. Supernatants were removed, cells were washed with PBS. Samples were then collected after trypsinization, washed with PBS, and stained in the dark with a mixture of propidium iodide and RNase. After 30 min of staining, the cell samples were washed and set up for flow cytometry reading on the red channel FL-2.
Kinetics of Binding to ct-DNA
ct-DNA was mixed with complexes in 10 mM NaClO4, immediately irradiated (visible light, λmax = 455 nm) for 60 or 120 min, and then kept at 310 K in the dark. The ri value was 0.05–0.08. Aliquots were removed at various time intervals and quickly filtered using a Sephadex G-50 column to remove free (unbound) Pt. The Pt content in these DNA samples (rb, defined as the number of the molecules of the Pt complex coordinated per nucleotide residue) was determined by atomic absorption spectroscopy (AAS) and the concentration of DNA by UV–vis spectroscopy.
Characterization of DNA Adducts of Photoactivated Complexes by EtBr Fluorescence
ct-DNA was incubated with the Pt complexes under irradiation conditions (visible light, λmax = 455 nm for 2 h and subsequently incubated in the dark for an additional 23 h) at various rb values in 10 mM NaClO4 at 310 K. Fluorescence measurements of ct-DNA modified by Pt complexes in the presence of EtBr were performed at an excitation wavelength of 546 nm, and the emitted fluorescence was analyzed at 590 nm. The fluorescence intensity was measured in NaCl (0.4 M) to avoid secondary binding of EtBr to DNA. The concentrations were 0.01 mg/mL for DNA and 0.04 mg/mL for EtBr, which corresponded to the saturation of all binding sites for EtBr in DNA. The fluorescence spectra were recorded by a Varian Cary Eclipse spectrofluorophotometer using a 0.5 cm quartz cell at room temperature.
Formation of Interstrand Cross-links
Solutions of the plasmid pUC19 linearized by EcoRI (50 μg/mL) were incubated with the Pt complex under irradiation conditions (for 1 h and subsequently incubated in the dark for an additional 23 h) at various ri values in NaClO4 (10 mM) at 310 K. Autoradiograms were recorded of denaturing 1% agarose gel of linearized DNA, which was 3′-end-labeled and nonmodified or modified by FM-190, 1, and 4 photoactivated by visible light.
Transcription Mapping of Pt-DNA Adducts
Transcription of the (NdeI/HpaI) restriction fragment of pSP73KB DNA modified by cisplatin, transplatin, or 1, 4, and FM-190 in the dark or photoactivated by visible light (λmax = 455 nm for 1 h and subsequently incubated 23 h in the dark, 310 K, 10 mM NaClO4) with DNA-dependent T7 RNA polymerase and electrophoretic analysis of the transcripts was performed according to the protocols recommended by the manufacturer (Promega Protocols and Applications, 43–46, 1989/90) and described in detail previously.19 The concentration of DNA in this assay was 1.54 × 10–4 M (relative to the monomeric nucleotide content). The ri values for platination reactions were chosen so as to obtain an rb value in the range 0.001–0.01. Pt complexes not bound to DNA were removed by ethanol precipitation.
Unwinding of Negatively Supercoiled DNA
Negatively supercoiled pSP73KB plasmid was treated with FM-190, 1, or 4, photoactivated by visible light (λ = 420 nm) for 1 h, and incubated for 23 h at 310 K in the dark. The degree of supercoiling was monitored using electrophoresis in 1% native agarose gel. The unwinding angle is given by Φ = −18σ/rb(c), where σ is the superhelical density (−0.063) and rb(c) is the value of rb at which the supercoiled and nicked forms comigrate.
DNase I Footprinting
Supercoiled pSP73 plasmid was digested with HindIII restriction endonuclease and 3′-end-labeled by treatment with Klenow exo- and [α-32P]-deoxy-ATP. After radioactive labeling, the DNA first cleaved with HindIII was further digested with NdeI to yield 158 and 2306 base-pair (bp) fragments. The 158 bp fragment was purified by 1% agarose gel electrophoresis and isolated from the gel by a Promega Wizard SV Gel cleanup system.
Radioactively labeled DNA (5.4 × 10–6 M) and the Pt complex (ri = 0.1, 0.2, and 0.4) in 10 mM NaClO4 were irradiated by λ = 420 nm for 1 h and incubated for 23 h at 310 K in the dark. Radioactively labeled DNA and the Pt complex at ri = 0.4 were also incubated in the dark for 24 h at 310 K. The reaction mixture was then lyophilized and dissolved in a 9 μL solution containing a 1.11 × TKMC buffer (10 mM Tris, pH 7.9, 10 mM KCl, 10 mM MgCl2, and 5 mM CaCl2) and 3 μg of ct-DNA. Cleavage was initiated by the addition of 0.25 U of DNase I (one unit of RQ1 RNase-free DNase is defined as the amount required to completely degrade 1 μg of lambda DNA in 10 min at 310 K in 50 μL of a buffer containing 40 mM Tris-HCl (pH 7.9), 10 mM NaCl, 6 mM MgCl2, and 10 mM CaCl2) and allowed to react for 30 s at 298 K before quenching with 2.5 μL of a DNase stop solution (3 M NaOAc and 0.25 M ethylenediaminetetraacetic acid). This was then precipitated with ethanol, lyophilized, and resuspended in a formamide loading buffer. DNA cleavage products were resolved by poly(acrylamide) (PAA) gel electrophoresis under denaturing conditions (8%/8 M urea PAA gel). The autoradiograms were visualized and quantified by using a bioimaging analyzer. Assignment of the cleavage to a particular base has been made so that it corresponds to cleavage of the phosphodiesteric bond on the 5′ side of that base.
Results and Discussion
Synthesis and Characterization of Complexes 1–4
The synthetic route for dinuclear PtIV complexes 1–4 is summarized in Scheme 1. The mononuclear complexes trans,trans,trans-[Pt(N3)2(OH)2(py)2] (FM-190) and trans,trans,trans-[Pt(N3)2(OH)(OC(O)CH2CH2C(O)OH)(py)2] (FMSA) were prepared by procedures similar to those reported.26 The two PtIV centers with axial succinate carboxylates were coupled through amide bond formation using bridging diamines containing ethylene (1), 1,2-cyclohexyl (2), 1,4-phenylene (3), or butyl (4) linkers. Complexes 1–4 are air-stable at ambient temperature, both in the solid state and in aqueous solution. They gave satisfactory elemental analysis and were further characterized by ESI-MS, NMR, and UV–vis spectroscopy and, for complexes 1 and 4, by X-ray crystallography.
Scheme 1. Synthetic Route for Photoactive Dinuclear Diazidoplatinum(IV) Complexes 1–4.

The 1H and 13C NMR spectra of complexes 1–4 in DMSO-d6 contain the expected peaks (Figures S1–S8). In the 1H NMR spectra, the doublets with 195Pt satellites at ca. 8.82 ppm (J = 5.5 Hz; J195Pt–1H = 26.6 Hz) and the triplets at ca. 8.26 ppm (J = 7.5 Hz) and 7.82 ppm (J = 7.0 Hz) are assigned to the Hα, Hγ, and Hβ of pyridine, respectively. The peaks at ca. 3.66 ppm are assignable to the resonances of the hydroxyl protons and the peaks at 7.52–9.79 ppm to the amide protons. The 13C NMR resonances of complexes 1–4 at ca. 149.83, 142.40, and 126.66 ppm are attributable to the Cα, Cγ, and Cβ of pyridine, respectively. The complexes all display peaks at ca. 175.27 and 172.00 ppm for the carboxyl and amide C atoms, respectively. The electronic absorption spectrum of complex 1 is similar to that of its mononuclear analogue FM-190. In a water/methanol (2:3) solution, complex 1 shows a high-energy band at 262 nm (ε = 25260 M–1 cm–1) and a maximum absorption band at 300 nm (34109 M–1 cm–1), which are assigned to a ligand-to-metal charge-transfer (LMCT; OH → Pt and N3 → Pt) transition. In addition, a weak band is observed at 420 nm that has mixed 1LMCT/1IL (interligand) character and involves N3 and OH ligands and Pt.21 This band enables the photoactivation of complex 1 using blue light. Complexes 2–4 show electronic absorption spectra similar to that of complex 1, except that the high-energy band (at 260 nm, ε = 49189 M–1 cm–1) of complex 3 is more intense than those of its analogues (ε ≈ 25000 M–1 cm–1) because of absorption of the phenyl fragment.
X-ray Crystallography
Crystals suitable for X-ray diffraction studies of complexes 1·2MeOH and 4 were obtained through the diffusion of diethyl ether into corresponding solutions in methanol. The structures of complexes 1·2MeOH and 4 are shown in Figure 1. The crystallographic data are summarized in Table S1, and selected bond distances and angles are listed in Table 1. Complex 1·2MeOH crystallized in the triclinic space group P1̅ with one complex and two methanol molecules in the unit cell. Complex 4 crystallized in the monoclinic space group P21/n with two molecules in the unit cell.
Figure 1.

X-ray crystal structures of 1·2MeOH (A) and 4 (B). The complexes sit on inversion centers, and only key atoms are labeled. Thermal ellipsoids are drawn at the 50% probability level.
Table 1. Selected Bond Lengths (Å) and Bond Angles (deg) for 1·2MeOH and 4.
| 1·2MeOH | |||
| Pt1–O1 | 1.9857(18) | O1–Pt1–O13 | 174.49(7) |
| Pt1–N1 | 2.030(2) | N21–N20–Pt1 | 114.52(18) |
| Pt1–N7 | 2.038(2) | N22–N21–N20 | 175.0(3) |
| Pt1–O13 | 2.0185(18) | N24–N23–Pt1 | 113.68(18) |
| Pt1–N20 | 2.053(2) | N25–N24–N23 | 175.6(3) |
| Pt1–N23 | 2.052(2) | ||
| N20–N21 | 1.211(3) | ||
| N21–N22 | 1.140(4) | ||
| N23–N24 | 1.220(3) | ||
| N24–N25 | 1.138(4) | ||
| 4 | |||
| Pt1–O1 | 1.969(3) | O1–Pt1–O13 | 170.35(11) |
| Pt1–N1 | 2.030(3) | N22–N21–Pt1 | 114.3(3) |
| Pt1–N7 | 2.033(3) | N23–N22–N21 | 175.6(5) |
| Pt1–O13 | 2.059(3) | N25–N24–Pt1 | 120.4(3) |
| Pt1–N21 | 2.048(4) | N26–N25–N24 | 174.4(4) |
| Pt1–N24 | 2.052(3) | ||
| N21–N22 | 1.183(5) | ||
| N22–N23 | 1.163(6) | ||
| N24–N25 | 1.217(5) | ||
| N25–N26 | 1.137(5) | ||
Complexes 1·2MeOH and 4 are neutral and consist of two mononuclear PtIV fragments bridged by an aliphatic linker of a diamine that forms amide bonds to the axial succinate carboxylates. The two Pt centers are symmetric, with the diamine linker sitting on an inversion center. The geometries of the Pt centers are similar to that of their mononuclear analogue FM-190: octahedral with [N4O2] coordination. The equatorial plane is defined by four N atoms, two from the trans-pyridine molecules and two from the trans-azides. An O atom from a hydroxido ligand is located in an axial position, while the other axial position is occupied by an O atom from one of the succinate carboxylate groups.
Compared with the fully symmetrical complex FM-190, complexes 1·2MeOH and 4 exhibit distortions from ideal octahedral geometry. The axial bond angles between PtIV and trans donor atoms are <180°, and the Pt–O bond [hydroxide, 1.9857(18) Å] is slightly shorter than the Pt–O bond [succinate, 2.0185(18) Å], which can be attributed to the steric hindrance caused by the relatively bulky carboxylate groups. The azido ligands are almost linear, with the Pt–N(α)–N(β) angles slightly smaller than 120°. The terminal azide N–N bond [1.140(4) and 1.138(4) Å] is slightly shorter than the azide N–N bond [1.211(3) and 1.220(3) Å] closest to Pt, which is typical of the azide ligands because of their resonance structure. The Pt–N(pyridine) bond [2.030(2) and 2.038(2) Å] is shorter than the Pt–N(azide) bond [2.053(2) and 2.052(2) Å]. The amide C=O distances are 1.240(3) and 1.236(5) Å, while those in the ester group are 1.216(3) and 1.217(4) Å, which resemble typical carbonyl groups in their mononuclear analogues.25 Weak hydrogen bonds are observed in complexes 1·2MeOH and 4 (Table S2 and the detailed description in the Supporting Information).
Photoactivation
Photodecomposition of complexes 1–4 in aqueous solution was monitored by UV–vis spectroscopy at various time intervals after irradiation with blue light (420 nm) at ambient temperature. Figure 2 shows the decrease in the absorbance maximum of complex 1 at ca. 300 nm, assigned as the N3 → Pt LMCT band. The decrease indicates that release of the azide ligands was complete within 1 h. This photodecomposition behavior is similar to that of the mononuclear Pt counterpart. Similar results were obtained for the other three complexes (Figure S9). Photoreactions of the dinuclear complex 4 were investigated by LC–MS (Figure S10 and Table S3). The HPLC peak assigned to complex 4 disappeared within 10 min of irradiation with blue light (420 nm). Peaks assignable to dinuclear photoproducts increased in intensity gradually, peaked after 5 min, and then decreased over 30 min with a concomitant increase in the formation of mononuclear photoproducts [{PtII(py)2(OC(O)H)2}+Na]+ (m/z 466.05) and {PtII(CH3CN)(N3)(py)2}+ (m/z 436.10). It appears that complex 4 loses one azide from both Pt centers initially, followed by dissociation of a Pt center and the associated bridging ligand (Figure S10).
Figure 2.
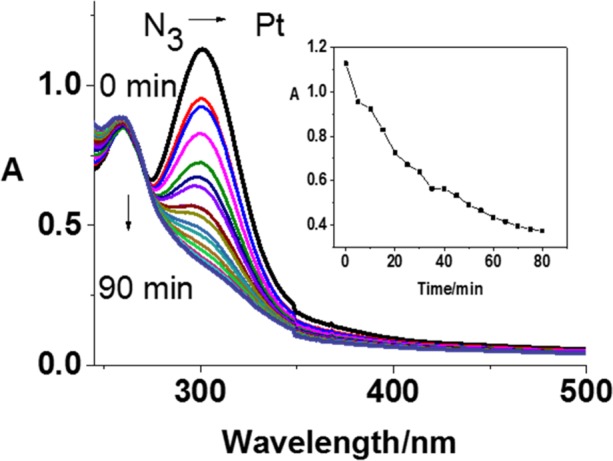
Photochemical decomposition of 1 [5 × 10–5 M; in 1:2 (v/v) water/methanol] upon irradiation with blue light (420 nm) determined by UV–vis spectroscopy. Inset: Time dependence of the absorbance at 300 nm.
Formation of Azidyl Radicals
The formation of azidyl radicals from photodecomposition of these PtIV complexes was monitored by EPR using DMPO as a spin trap. EPR spectra were recorded continuously for a 5 mM solution of complex 4 and 40 mM DMPO in 5% DMSO/95% water upon irradiation with blue light (465 nm, 20 mW) at 298 K. Both azidyl and hydroxyl radical adducts of DMPO (DMPO–N3• and DMPO–OH•) were detected upon irradiation, as a 1:2:2:1 quartet of triplets and a 1:2:2:1 quartet, respectively (Figure 3). The simulation was carried out using the parameters listed in Figure 3, which is in accordance with ref (23).
Figure 3.

Observed (black) and simulated (red) EPR spectra of complex 4 in aqueous solution with 5% DMSO showing the formation of DMPO–N3• and DMPO–OH• adducts after irradiation (465 nm). The experimental trace is the accumulation of 500 scans (conversion time 5.12 ms, time constant 5.12 ms, and sweep time 10.48 s for each scan) with continuous irradiation (465 nm). Parameters for simulation: DMPO–N3• (g = 2.00583, aNON = 14.5 mT, aβ = 14.2 mT, and aNαN = 0.31 mT); DMPO–OH• (g = 2.0058, aNO = 14.5 mT, and aβH = 14.2 mT).
Photocytotoxicity and Cellular Accumulation Studies
Upon irradiation with blue light (465 nm, 4.8 mW/cm2, 1 h), complexes 1–4 exhibited photocytotoxicity toward several human cancer cell lines, including A2780 and A2780cis ovarian and OE19 esophageal cancer cells. The dose-dependent inhibition of the cell viability determined by SRB colorimetric assay both in the dark and after irradiation of complexes 1–4 is summarized in Table 2. In parental (A2780) and cisplatin-resistant (A2780cis) ovarian carcinoma, complexes 1–4 showed no cytotoxicity in the dark. However, upon irradiation for 1 h, complexes 1–4 exhibited cytotoxicity to varying extents. Complex 4 was the most potent member of the series (IC50 = ca. 17.0 μM for ovarian cancer cells A2780 and A2780cis and 8.8 μM for OE19) with a photocytotoxicity index (PI) toward cancer cells of >6 for ovarian carcinoma and 11 for esophageal adenocarcinoma with light exposure. Complexes 1 and 3 had lower IC50 values toward the cisplatin-resistant A2780cis cell line [IC50 = 29.5 μM (1) and 35.5 μM (3)] compared to the parental A2780 cell line [IC50 = 77.0 μM (1) and 78.3 μM (3)], which suggests a different mechanism of action from cisplatin and the possibility of overcoming the resistance. In contrast, the mononuclear analogue FM-190 was 10 times more toxic to parental A2780 (1.4 μM) compared with cisplatin-resistant A2780cis (14.5 μM) cells upon irradiation with UVA (365 nm).21 Complexes 1–4 displayed cytotoxicity toward OE19 both in the dark and upon irradiation. However, irradiation improved the cytotoxicity slightly for complexes 1–3 and considerably so for 4, with PI > 11 [IC50 = 97.0 μM (dark) and 8.8 μM (light)].
Table 2. IC50 and PI (Photocytotoxicity Index) Values for Complexes 1–4 Obtained after 1 h of Incubation, 1 h of Irradiation (465 nm), and 24 h of Recoverya.
| IC50 (μM)b |
||||||||
|---|---|---|---|---|---|---|---|---|
| cell | 1 | 2 | 3 | 4 | CDDP | CPZ | FM-190 | |
| A2780 | dark | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| irrad | 77.0 ± 2.9 | 33.9 ± 5.2 | 78.3 ± 6.8 | 16.7 ± 3.3 | >100 | 6.0 ± 0.3 | 7.1 ± 0.4 | |
| PI | >1.3 | >2.9 | >1.3 | >6.0 | >16.7 | >14.0 | ||
| A2780cis | dark | >100 | >100 | >100 | >100 | >100 | 51.0 ± 0.2 | |
| irrad | 29.5 ± 11.0 | 67.6 ± 15.7 | 35.5 ± 3.4 | 17.0 ± 2.3 | >100 | 6.0 ± 0.3 | ||
| PI | >3.4 | >1.5 | >2.8 | >5.8 | 8.5 | |||
| OE19 | dark | 41.3 ± 3.2 | 49.8 ± 1.5 | 53.4 ± 0.4 | 97.0 ± 5.7 | >100 | ||
| irrad | 36.2 ± 2.4 | 27.5 ± 5.1 | 43.7 ± 1.9 | 8.8 ± 0.9 | >100 | |||
| PI | 1.1 | 1.8 | 1.2 | 11.0 | ||||
| MRC5 | dark | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| irrad | >100 | >100 | >100 | >100 | >100 | >50 | >100 | |
In contrast to the photocytotoxicity observed toward cancer cell lines, complexes 1–4 were relatively nontoxic toward the normal cell line MRC5, with IC50 values of >100 μM, even after irradiation (Table 2). Because azidyl and hydroxyl radicals generated from PtIV complexes with azides are key species involved in cell death,23 their relative noncytotoxicity toward MRC5 may be due to the lower basal ROS level of healthy cells, with higher radical levels being needed to cause cell death.41 For comparison, cisplatin tested under the same conditions induced little cytotoxicity in all cell lines because of the short incubation time (1 h, IC50 > 100 μM with or without light).
Cellular accumulation is related to the molecular structure as well as the lipophilic nature of the complex and often correlates with the antiproliferative activity of metallodrugs. The extent of cellular uptake of complexes 1–4 was investigated by treating cancer cell lines with IC50 concentrations for 1 h in the dark. The cell accumulation of Pt was then determined by ICP-MS. As shown in Table 3, complex 3 exhibited >5 times higher Pt accumulation than other complexes in all cell lines, attributable to its aromatic bridge linker and higher lipophilicity. For complexes with aliphatic linkers, 1 showed slightly higher levels of Pt accumulation in all cancer cell lines compared with complexes 2 and 4. It is notable that the accumulation of Pt from complex 4 in cisplatin-resistant A2780cis ovarian cancer cells is 2 times that for the the parental A2780 cell line, even though complex 4 is equally toxic to both cell lines. These results appear to support the ability of the dinuclear complexes to overcome resistance to cisplatin.
Table 3. Cell Accumulation of Pt (ng/106 cells) in Cancer Cells after Exposure to Complexes 1–4 and FM-190 (Equipotent IC50 Concentration, 1 h, in the Dark)a.
| platinum
accumulation (ng/106 cells) |
|||
|---|---|---|---|
| complex | A2780 | A2780cis | OE19 |
| 1 | 24 ± 8b | 17 ± 6b | 13 ± 1b |
| 2 | 10 ± 2b | 11 ± 2c | 12 ± 2c |
| 3 | 101 ± 6b | 56 ± 17b | 51 ± 5d |
| 4 | 8 ± 1b | 17 ± 2b | 8 ± 2b |
| FM-190 | 0.8 ± 0.2b | ||
All data were determined from triplicate samples, and their statistical significance was evaluated by a two-tail t test with unequal variances.
p < 0.05.
p < 0.01.
p < 0.005.
Cell Cycle Analysis
The effect of complexes 3 and 4 in the presence of light on the progression of the cell cycle was studied. Cells were exposed to prodrugs at equipotent photocytotoxic IC50 concentrations for 1 h in the dark and then irradiated for 1 h (465 nm, 4.8 mW/cm2, 1 h). After 24 h of recovery, cells were stained in the dark with propidium iodide in the presence of RNase and then analyzed by flow cytometry. The results are summarized in Table S4 and Figure 4.
Figure 4.

Flow cytometry histograms showing cell cycle analysis of A2780 and A2780cis ovarian and OE19 esophageal cancer cells: pink, dark; powder blue, irradiated. In all cases, the experiments involved 1 h of drug exposure followed by 1 h of either irradiation (465 nm) or darkness. PI = propdium iodide.
In all cancer cell lines, the populations of cells in the stages of the cycle were not affected by the complexes in the dark but changed dramatically upon irradiation. In OE19 esophageal cancer cells, a reduction of cells in the G0/G1phase was observed (ca. 45% to 29%), paralleled by an accumulation of cells in the G2/M phases (increasing from ca. 36% to 57%), suggesting that dinuclear complexes 3 and 4 inhibit progression from G2/M to G0/G1 upon irradiation.
Photoactivated complex 4 induced more significant changes in the cell cycle progression compared with 3 (two-tail t test with unequal variances against control; Table S4), consistent with the higher photocytotoxicity of complex 4. Similar changes in the cell cycle populations were found for A2780 and A2780cis ovarian cancer cells (Figure 4). This is in contrast to cisplatin, which arrests cells in the S phase,42 suggesting a different mechanism of action and a lack of cross-resistance between complexes 3 and 4 and cisplatin.
Photoinduced Reactions with 5′-GMP
Because the N7 site of the DNA base guanine is a preferred target for platinum amine anticancer complexes, photoreactions of complex 4 with 5′-GMP were investigated. The photoreactions were carried out by irradiating solutions of complex 4 (30 μM) in 5%/95% (v/v) methanol/water in the presence of 5′-GMP (2, 4, or 8 mol equiv) with blue light (420 nm) at 310 K and monitored by reversed-phase HPLC (Figure S11 and Table S5). The photorelease of the bridging ligand [HOOC(CH2)2C(O)NH(CH2)4NHC(O)(CH2)2COOH+H]+ (m/z 289.21) was confirmed by LC–MS, and the amount released did not change significantly when the ratio of 5′-GMP/Pt was increased. The major photoproduct is assignable as {PtII(CH3CN)(py)2(GMP-H)}+ (m/z 756.09), which increased in proportion at higher 5′-GMP ratios. In addition, two small peaks ascribed to {PtII(N3)(py)2(GMP)}+ (m/z 757.95) and {PtII(OC(O)H)(py)2(GMP)}+ (m/z 761.91) were detected. {PtII(OC(O)H)(py)2(GMP)}+ was formed due to the addition of formic acid to the LC–MS mobile phase. Complex 4 reacted completely within 1 h of irradiation. The time dependence of the formation of photoproducts from the UV–vis spectra and LC–MS data is shown in Figure S12. Even though decomposition of complex 4 can be detected within only 1 min of irradiation, the apparent decrease of the absorbance of 5′-GMP and the formation of {PtII(CH3CN)(py)2(GMP-H)}+ (m/z 756.09) is observed only after 5 min of irradiation. No dinuclear photoproduct with 5′-GMP was detected, which suggests that the Pt centers do not react with 5′-GMP until they are detached from the bridging ligands. The results above agree well with those previously reported for the parent complex and its succinate derivative, suggesting that the two PtIV centers have little effect on each other during the photoreactions with 5′-GMP.
DNA Binding Studies in Cell-Free Media
To further explore the differences between mono- and dinuclear complexes and the effect of bridge linkers, the DNA binding ability of photoactivated dinuclear complexes 1 and 4 was investigated in comparison with mononuclear FM-190.
ct-DNA was mixed with complexes in 10 mM NaClO4, immediately irradiated with blue visible light (λmax = 455 nm) for 1 h, and then kept at 310 K in the dark. Aliquots were removed at various time intervals and quickly filtered using a Sephadex G-50 column to remove free (unbound) Pt. The Pt content in these DNA samples was determined by AAS, and the concentration of DNA was determined by UV–vis to give the kinetics of Pt binding to ct-DNA. The amount of Pt bound to DNA increased with time for all complexes. No significant difference was found between the investigated complexes (Figure 5A).
Figure 5.

DNA binding of photoactivated complexes in cell-free media. (A) Kinetics of the reaction of the investigated complexes photoactivated by visible light (λmax = 455 nm) with ct-DNA. For other details, see the text. (B) Dependence of ethidium bromide fluorescence on rb for double-helical ct-DNA modified by the investigated Pt complexes photoactivated by visible light (λmax = 455 nm) in NaClO4 (10 mM) at 37 °C for 24 h. (C) Formation of interstrand (intramolecular) cross-links by the investigated Pt complexes photoactivated by visible light (λmax = 455 nm) in pUC19 plasmid linearized by EcoRI. Solutions of the linearized plasmid (50 μg mL–1) were incubated with the Pt complex under irradiation conditions (for 1 h and subsequently incubated in the dark for an additional 23 h) at various ri values in NaClO4 (10 mM) at 37 °C. The autoradiogram of denaturing 1% agarose gel of linearized DNA which was 3′-end-labeled and nonmodified and nonirradiated (kept in the dark; lane C) or modified by photoactivated FM-190 (rb = 0.0003 and 0.0006 for lanes 1 and 2, respectively), 1 (rb = 0.00015 and 0.0003 for lanes 3 and 4, respectively), or 4 (rb = 0.00015 and 0.0003 for lanes 3 and 4, respectively). Interstrand cross-linked DNA appears as the top bands (marked as ICL), migrating on the gel more slowly than single-stranded DNA (contained in the bottom bands and marked as ss). (D) Unwinding of supercoiled pSP73KB plasmid DNA by the investigated dinuclear Pt complexes photoactivated by visible light (λmax = 455 nm). The top bands correspond to the nicked form of the plasmid and the bottom bands to the closed, negatively supercoiled plasmid. The plasmid was incubated with FM-190, 1, and 4 under irradiation conditions (for 1 h and subsequently incubated in the dark for an additional 23 h) at various ri values in NaClO4 (10 mM) at 37 °C. Lane C: control, nonplatinated, and nonirradiated DNA (rb = 0). Lanes 1–10: DNA modified by photoactivated complexes so that the resulting rb values were, for FM-190, 0.001, 0.003, 0.005, 0.010, 0.015, 0.020, 0.025, 0.030, 0.035, and 0.040, respectively, for 1, 0.0005, 0.0025, 0.005, 0.0075, 0.01, 0.0125, 0.015, 0.0175, 0.02, and 0.025, respectively, and for 4, 0.001, 0.0015, 0.0025, 0.0035, 0.005, 0.01, 0.015, 0.0175, and 0.02, respectively.
The DNA intercalator EtBr was used as a fluorescent probe to characterize perturbations induced in DNA by the adducts with Pt complexes. Modification of DNA by Pt complexes under irradiation conditions resulted in a decrease of EtBr fluorescence.
The decrease caused by the adducts of photoactivated 1 or 4 was double that induced by the DNA adducts of FM-190 at equivalent rb values (Figure 5B). It was also verified that irradiation of ct-DNA by visible light in the absence of complexes for 2 h had no effect on EtBr fluorescence. The results of these experiments suggest that the conformational distortions induced in DNA by the adducts formed by the treatment of DNA with irradiated 1 and 4 are delocalized and extended over the base pairs around the platination sites to an extent that corresponds to the distortions induced in DNA by two adducts of mononuclear complex FM-190. Thus, these results are consistent with decomposition of the dinuclear complexes 1 and 4 due to irradiation, resulting in the release of two mononuclear fragments similar to those from FM-190.
It was shown previously43 that modification of DNA by irradiated FM-190 also resulted in the formation of DNA interstrand cross-links. Therefore, we investigated the capability of irradiated 1 and 4 to form DNA interstrand cross-links as well. Plasmid pUC19 linearized by EcoRI (50 μg mL–1) and the Pt complex were irradiated with blue light for 1 h and subsequently incubated in the dark for an additional 23 h in NaClO4 (10 mM) at 310 K. The plasmid was modified to the extent corresponding to the rb values of 0.003 and 0.0006 (in the case of FM-190) or 0.00015 and 0.0003 (in the case of 1 and 4) and analyzed by agarose gel electrophoresis under denaturing conditions (Figure 5C). Interstrand cross-linked DNA appears as the top band (marked as ICLs), migrating on the gel more slowly than single-stranded DNA (contained in the bottom bands and marked as ss). Quantitative evaluation of the radioactivity associated with the individual bands showed that irradiated 1 and 4 formed a 2-fold higher amount of interstrand cross-links in DNA than FM-190 (the frequencies of interstrand cross-linking were 12.5 ± 0.5, 24 ± 2, and 24 ± 2 for FM-190, 1, and 4, respectively) at the same level of modification (related to the complex). Hence, the results of DNA interstrand cross-linking experiments also support the hypothesis that irradiation of the dinuclear complexes 1 and 4 leads to their decomposition, resulting in the release of two molecules similar to the photoproduct from mononuclear complex FM-190. Our previous work suggested that the trans-{Pt(py)2}2+ fragment released from FM-190 can form GG interstrand DNA cross-links, including 5′-CG/5′-CG cross-links, 5′-GC/5′-GC cross-links, and cross-links between G and the complementary C.44 It is, therefore, reasonable to envisage that the treatment of DNA by irradiated dinuclear 1 or 4 results in the formation of trans GG interstrand DNA cross-links as in the case of the treatment with mononuclear complex FM-190.
It has been shown that the binding of various antitumor metallodrugs to closed-circular DNA can cause partial unfolding of the DNA. This process lowers the superhelical density of plasmid DNA, which causes a decrease in the rate of migration through an agarose gel. This fact makes it possible to observe and quantify the mean value of unwinding per adduct. In the present study, we investigated the unwinding induced in negatively supercoiled pSP73KB plasmid by 1 and 4 photoactivated by visible light (λ = 420 nm) for 1 h and then incubated for another 23 h at 310 K in the dark. The degree of supercoiling was monitored using electrophoresis in a native agarose gel. We investigated the effect of increasing the amounts of the investigated Pt complexes photoactivated by visible light bound to a mixture of relaxed and supercoiled pSP73KB DNA upon migration of these forms in a native agarose gel (shown in Figure 5D). The unwinding angle is given by Φ = −18σ/rb(c), where σ is the superhelical density and rb(c) is the value of rb (related to the complex) at which the supercoiled and nicked forms comigrate. The DNA unwinding angle determined for DNA modified by dinuclear complexes 1 and 4 photoactivated by visible light was 57 ± 8°. This unwinding angle was 2-fold higher than that found under identical conditions for DNA modified by mononuclear complex FM-190 (28 ± 4°).43 Thus, consistent with the results of the experiments aimed at characterization of the DNA adducts by EtBr fluorescence and DNA interstrand cross-linking, the results of DNA unwinding experiments (Figure 5D) can be interpreted to mean that the consequence of irradiation of the dinuclear complexes 1 and 4 is their decomposition, resulting in the release of two molecules of mononuclear complex FM-190.
A parameter that makes identification of the preferential DNA binding sites of Pt antitumor drugs possible can be obtained from transcription mapping experiments.45 In vitro RNA synthesis by RNA polymerases on DNA templates containing several types of bifunctional adducts of Pt complexes can be prematurely terminated at the level, or in the proximity, of DNA adducts. We prepared a 212-bp fragment by the cutting of pSP73KB DNA by NdeI and HpaI restriction endonucleases. A substantial part of its nucleotide sequence is shown in Figure S13B. This fragment contained a T7 RNA polymerase promotor (in the upper strand close to its 3′ end; Figure S13B). The experiments were carried out using this linear DNA fragment, modified by cisplatin, transplatin (in the dark), or 1 or 4 photoactivated by visible light. The samples were irradiated for 1 h and subsequently incubated in the dark for an additional 23 h. The rb values were in the range of 0.004–0.01 and are indicated in the legend to Figure S13A. The major stop signals for DNA modified by dinuclear complexes 1 and 4 (Figure S13) were identical with those found previously under identical conditions for DNA modified by irradiated FM-190.43
In order to obtain further information on the sequence specificity of DNA binding in the dinuclear 1 and 4 analogues of complex FM-190, a DNase I footprinting experiment was also performed (Figure S14). The details of this experiment are described in the Supporting Information. The results show that the preferential binding sites of dinuclear complexes 1 and 4 are those containing mainly G/A-rich sequences and are identical with those of mononuclear complex FM-190. Collectively, the results of transcription mapping and DNase I footprinting experiments suggest that preferential DNA binding sites of irradiated dinuclear complexes 1 and 4 are similar to those of mononuclear complex FM-190, thus also supporting the hypothesis that irradiation of the dinuclear complexes 1 and 4 results in their decomposition, resulting in the release of two molecules similar to the photoproducts from mononuclear complex FM-190.
Conclusions
We report the synthesis and characterization of a series of novel dinuclear photoactive PtIV anticancer prodrugs trans,trans,trans-[{Pt(N3)2(py)2(OH)(OC(O)CH2CH2C(O)NH)}2R] containing pyridine (py) and bridging dicarboxylate [R = −CH2CH2– (1), trans-1,2-C6H10– (2), p-C6H4– (3), −CH2CH2CH2CH2– (4)] ligands. We compared their photoreactivity with that of a mononuclear analogue FM-190. Azidyl and hydroxyl radicals and PtII species were generated during photodecomposition of these dinuclear complexes. LC–MS analyses of photoreactions of dinuclear complex 4 revealed the photorelease of the bridging ligand and the formation of mononuclear photoproducts. In the presence of 5′-GMP, the PtII-5′-GMP adduct {PtII(CH3CN)(py)2(GMP-H)}+ (756.09) was formed upon irradiation. Dinuclear complexes exhibited promising photocytotoxicity upon irradiation with low-dose blue light (465 nm, 4.8 mW/cm2, 1 h) toward a series of human cancer cell lines, including A2780, A2780cis, and OE19, with high dark stability. Moreover, the dinuclear complexes were remarkably nontoxic toward normal cells (MRC5), even after irradiation. The introduction of an aromatic bridging ligand significantly enhanced cell uptake for complex 3 compared with the complexes containing an aliphatic bridge. The inhibition of progression from G2/M to G0/G1 stages in the cell cycle induced by dinuclear complexes upon irradiation suggests that DNA is a target. The dinuclear complexes behaved in cell-free media and after photoactivation similarly to the photoactivated mononuclear complex FM-190 in terms of the kinetics of binding to ct-DNA, transcription mapping, and DNase I footprinting of Pt-DNA adducts. However, dinuclear complexes after photoactivation were approximately 2-fold more effective in quenching the fluorescence of EtBr bound to DNA, forming DNA interstrand cross-links, and unwinding DNA compared to the photoactivated mononuclear Pt analogue FM-190. This is consistent with photodecomposition of the dinuclear complexes into two molecules of mononuclear PtII species.
Hence, dinuclear complexes exhibit promising dark stability, photocytotoxicity, and selectivity toward cancer cells, dependent on the nature of the bridging ligand.
Acknowledgments
This research was supported by the EPSRC (Grants EP/G006792 and EP/F034210/1 to P.J.S.), the ERC (Grant 247450 to P.J.S.), a Chancellor’s International Ph.D. Scholarship from the University of Warwick (to H.S.), the Czech Science Foundation (Grant 18-09502S to O.N. and V.B.), Palacky University in Olomouc (Grant IGAPrF2018022 to M.H.), and the Royal Society (Newton International Fellowship and follow-up funding under Grant AL170006\1 for V.V.).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.inorgchem.8b02599.
1H and 13C NMR spectra for all dinuclear complexes, UV–vis spectra, reactions with 5′-GMP, DNA binding and transcription mapping (PDF)
Accession Codes
CCDC 1862975–1862976 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Dolmans D. E.; Fukumura D.; Jain R. K. Photodynamic therapy for cancer. Nat. Rev. Cancer 2003, 3, 380–387. 10.1038/nrc1071. [DOI] [PubMed] [Google Scholar]
- Bednarski P. J.; Mackay F. S.; Sadler P. J. Photoactivatable platinum complexes. Anti-Cancer Agents Med. Chem. 2007, 7, 75–93. 10.2174/187152007779314053. [DOI] [PubMed] [Google Scholar]
- Bonnet S. Why develop photoactivated chemotherapy?. Dalton Trans. 2018, 47, 10330–10343. 10.1039/C8DT01585F. [DOI] [PubMed] [Google Scholar]
- Bjelosevic A.; Pages B. J.; Spare L. K.; Deo K. M.; Ang D. L.; Aldrich-Wright J. R. Exposing “Bright” Metals: Promising Advances in Photoactivated Anticancer Transition Metal Complexes. Curr. Med. Chem. 2018, 25, 478–492. 10.2174/0929867324666170530085123. [DOI] [PubMed] [Google Scholar]
- Wong E.; Giandomenico C. M. Current Status of Platinum-Based Antitumor Drugs. Chem. Rev. 1999, 99, 2451–2466. 10.1021/cr980420v. [DOI] [PubMed] [Google Scholar]
- Pages B. J.; Ang D. L.; Wright E. P.; Aldrich-Wright J. R. Metal complex interactions with DNA. Dalton Trans. 2015, 44, 3505–3526. 10.1039/C4DT02700K. [DOI] [PubMed] [Google Scholar]
- Harris A. L. Hypoxia--a key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- Macquet J. P.; Butour J. L. Platinum-amine compounds: importance of the labile and inert ligands for their pharmacological activities toward L1210 leukemia cells. J. Nat. Cancer Inst. 1983, 70, 899–905. [PubMed] [Google Scholar]
- Van der Veer J. L.; Peters A. R.; Reedijk J. Reaction products from platinum(IV) amine compounds and 5′-GMP are mainly bis(5′-GMP) platinum (II) amine adducts. J. Inorg. Biochem. 1986, 26, 137–142. 10.1016/0162-0134(86)80006-X. [DOI] [PubMed] [Google Scholar]
- Roat R. M.; Reedijk J. Reaction of mer-trichloro (diethylenetriamine)platmum(IV) chloride, (mer-[Pt(dien)Cl3]Cl), with purine nucleosides and nucleotides results in formation of platinum(II) as well as platinum(IV) complexes. J. Inorg. Biochem. 1993, 52, 263–274. 10.1016/0162-0134(93)80030-D. [DOI] [Google Scholar]
- Mitra K. Platinum complexes as light promoted anticancer agents: a redefined strategy for controlled activation. Dalton Trans. 2016, 45, 19157–19171. 10.1039/C6DT03665A. [DOI] [PubMed] [Google Scholar]
- Johnstone T. C.; Suntharalingam K.; Lippard S. J. The Next Generation of Platinum Drugs: Targeted Pt(II) Agents, Nanoparticle Delivery, and Pt(IV) Prodrugs. Chem. Rev. 2016, 116, 3436–3486. 10.1021/acs.chemrev.5b00597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shushakov A. A.; Pozdnyakov I. P.; Grivin V. P.; Plyusnin V. F.; Vasilchenko D. B.; Zadesenets A. V.; Melnikov A. A.; Chekalin S. V.; Glebov E. M. Primary photochemical processes for Pt(IV) diazido complexes prospective in photodynamic therapy of tumors. Dalton Trans. 2017, 46, 9440–9450. 10.1039/C7DT01529A. [DOI] [PubMed] [Google Scholar]
- Müller P.; Schröder B.; Parkinson J. A.; Kratochwil N. A.; Coxall R. A.; Parkin A.; Parsons S.; Sadler P. J. Nucleotide cross-linking induced by photoreactions of platinum(IV)-azide complexes. Angew. Chem., Int. Ed. 2003, 42, 335–339. 10.1002/anie.200390110. [DOI] [PubMed] [Google Scholar]
- Mackay F. S.; Woods J. A.; Heringová P.; Kašpárková J.; Pizarro A. M.; Moggach S. A.; Parsons S.; Brabec V.; Sadler P. J. A potent cytotoxic photoactivated platinum complex. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 20743–20748. 10.1073/pnas.0707742105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westendorf A. F.; Zerzankova L.; Salassa L.; Sadler P. J.; Brabec V.; Bednarski P. J. Influence of pyridine versus piperidine ligands on the chemical, DNA binding and cytotoxic properties of light activated trans, trans, trans-[Pt(N3)2(OH)2(NH3)(L)]. J. Inorg. Biochem. 2011, 105, 652–662. 10.1016/j.jinorgbio.2011.01.003. [DOI] [PubMed] [Google Scholar]
- Mackay F. S.; Moggach S. A.; Collins A.; Parsons S.; Sadler P. J. Photoactive trans ammine/amine diazido platinum(IV) complexes. Inorg. Chim. Acta 2009, 362, 811–819. 10.1016/j.ica.2008.02.039. [DOI] [Google Scholar]
- Zhao Y.; Farrer N. J.; Li H.; Butler J. S.; McQuitty R. J.; Habtemariam A.; Wang F.; Sadler P. J. De Novo Generation of Singlet Oxygen and Ammine Ligands by Photoactivation of a Platinum Anticancer Complex. Angew. Chem., Int. Ed. 2013, 52, 13633–13637. 10.1002/anie.201307505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Woods J. A.; Farrer N. J.; Robinson K. S.; Pracharova J.; Kasparkova J.; Novakova O.; Li H.; Salassa L.; Pizarro A. M.; Clarkson G. J.; Song L.; Brabec V.; Sadler P. J. Diazido mixed-amine platinum(IV) anticancer complexes activatable by visible-light form novel DNA adducts. Chem. - Eur. J. 2013, 19, 9578–9591. 10.1002/chem.201300374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasparkova J.; Kostrhunova H.; Novakova O.; Křikavová R.; Vančo J.; Trávníček Z.; Brabec V. A Photoactivatable Platinum(IV) Complex Targeting Genomic DNA and Histone Deacetylases. Angew. Chem., Int. Ed. 2015, 54, 14478–14482. 10.1002/anie.201506533. [DOI] [PubMed] [Google Scholar]
- Farrer N. J.; Woods J. A.; Salassa L.; Zhao Y.; Robinson K. S.; Clarkson G.; Mackay F. S.; Sadler P. J. A Potent Trans-Diimine Platinum Anticancer Complex Photoactivated by Visible Light. Angew. Chem., Int. Ed. 2010, 49, 8905–8908. 10.1002/anie.201003399. [DOI] [PubMed] [Google Scholar]
- Pracharova J.; Zerzankova L.; Stepankova J.; Novakova O.; Farrer N. J.; Sadler P. J.; Brabec V.; Kasparkova Interactions of DNA with a new platinum(IV) azide dipyridine complex activated by UVA and visible light: Relationship to toxicity in tumor cells. Chem. Res. Toxicol. 2012, 25, 1099–1111. 10.1021/tx300057y. [DOI] [PubMed] [Google Scholar]
- Butler J. S.; Woods J. A.; Farrer N. J.; Newton M. E.; Sadler P. J. Tryptophan Switch for a Photoactivated Platinum Anticancer Complex. J. Am. Chem. Soc. 2012, 134, 16508–16511. 10.1021/ja3074159. [DOI] [PubMed] [Google Scholar]
- Gandioso A.; Shaili E.; Massaguer A.; Artigas G.; González-Cantó A.; Woods J. A.; Sadler P. J.; Marchán V. An integrin-targeted photoactivatable Pt(IV) complex as a selective anticancer pro-drug: synthesis and photoactivation studies. Chem. Commun. 2015, 51, 9169–9172. 10.1039/C5CC03180J. [DOI] [PubMed] [Google Scholar]
- Shaili E.; Fernández-Giménez M.; Rodríguez-Astor S.; Gandioso A.; Sandín L.; García-Vélez C.; Massaguer A.; Clarkson G. J.; Woods J. A.; Sadler P. J.; Marchán V. A photoactivatable platinum(IV) anticancer complex conjugated to the RNA ligand Guanidinoneomycin. Chem. - Eur. J. 2015, 21, 18474–18486. 10.1002/chem.201502373. [DOI] [PubMed] [Google Scholar]
- Venkatesh V.; Wedge C. J.; Romero-Canelón I.; Habtemariam A.; Sadler P. J. Spin-labelled photo-cytotoxic diazido platinum(IV) anticancer complex. Dalton Trans. 2016, 45, 13034–13037. 10.1039/C6DT01382A. [DOI] [PubMed] [Google Scholar]
- Min Y.; Li J.; Liu F.; Yeow E. K. L.; Xing B. Near-infrared light-mediated photoactivation of a platinum antitumor prodrug and simultaneous cellular apoptosis imaging by upconversion-luminescent nanoparticles. Angew. Chem., Int. Ed. 2014, 53, 1012–1016. 10.1002/anie.201308834. [DOI] [PubMed] [Google Scholar]
- Venkatesh V.; Mishra N. K.; Romero-Canelón I.; Vernooij R. R.; Shi H.; Coverdale J. P. C.; Habtemariam A.; Verma S.; Sadler P. J. Supramolecular photoactivatable anticancer hydrogels. J. Am. Chem. Soc. 2017, 139, 5656–5659. 10.1021/jacs.7b00186. [DOI] [PubMed] [Google Scholar]
- Malina J.; Farrell N. P.; Brabec V. DNA condensing effects and sequence selectivity of DNA binding of antitumor noncovalent polynuclear platinum complexes. Inorg. Chem. 2014, 53, 1662–1671. 10.1021/ic402796k. [DOI] [PubMed] [Google Scholar]
- Cox J. W.; Berners-Price S.; Davies M. S.; Qu Y.; Farrell N. P. Kinetic Analysis of the Stepwise Formation of a Long-Range DNA Interstrand Cross-link by a Dinuclear Platinum Antitumor Complex: Evidence for Aquated Intermediates and Formation of Both Kinetically and Thermodynamically Controlled Conformers. J. Am. Chem. Soc. 2001, 123, 1316–1326. 10.1021/ja0012772. [DOI] [PubMed] [Google Scholar]
- Qu Y.; Scarsdale N. J.; Tran M. C.; Farrell N. P. Cooperative effects in long-range 1,4 DNA-DNA interstrand cross-links formed by polynuclear platinum complexes: an unexpected syn orientation of adenine bases outside the binding sites. J. Biol. Inorg. Chem. 2003, 8, 19–28. 10.1007/s00775-002-0383-x. [DOI] [PubMed] [Google Scholar]
- Hegmans A.; Berners-Price S. J.; Davies M. S.; Thomas D.; Humphreys A.; Farrell N. P. Long Range 1,4 and 1,6-Interstrand Cross-Links Formed by a Trinuclear Platinum Complex. Minor Groove Preassociation Affects Kinetics and Mechanism of Cross-Link Formation as Well as Adduct Structure. J. Am. Chem. Soc. 2004, 126, 2166–2180. 10.1021/ja036105u. [DOI] [PubMed] [Google Scholar]
- Malina J.; Farrell N. P.; Brabec V. DNA Interstrand Cross-Links of an Antitumor Trinuclear Platinum(II) Complex: Thermodynamic Analysis and Chemical Probing. Chem. - Asian J. 2011, 6, 1566–1574. 10.1002/asia.201000935. [DOI] [PubMed] [Google Scholar]
- Gourley C.; Cassidy J.; Edwards C.; Samuel L.; Bisset D.; Camboni G.; Young A.; Boyle D.; Jodrell D. A phase I study of the trinuclear platinum compound, BBR 3464, in combination with protracted venous infusional 5-fluorouracil in patients with advanced cancer. Cancer Chemother. Pharmacol. 2004, 53, 95–101. 10.1007/s00280-003-0721-x. [DOI] [PubMed] [Google Scholar]
- Jodrell D. I.; Evans T. R. J.; Steward W.; Cameron D.; Prendiville J.; Aschele C.; Noberasco C.; Lind M.; Carmichael J.; Dobbs N.; Camboni G.; Gatti B.; De Braud F. Phase II studies of BBR3464, a novel tri-nuclear platinum complex, in patients with gastric or gastro-oesophageal adenocarcinoma. Eur. J. Cancer 2004, 40, 1872–1877. 10.1016/j.ejca.2004.04.032. [DOI] [PubMed] [Google Scholar]
- Hensing T. A.; Hanna N. H.; Gillenwater H. H.; Gabriella Camboni M.; Allievi C.; Socinski M. A. Phase II study of BBR 3464 as treatment in patients with sensitive or refractory small cell lung cancer. Anti-Cancer Drugs 2006, 17, 697–704. 10.1097/01.cad.0000215054.62942.7f. [DOI] [PubMed] [Google Scholar]
- Dolomanov O. V.; Bourhis L. J.; Gildea R. J.; Howard J. A. K.; Puschmann H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. 10.1107/S0021889808042726. [DOI] [Google Scholar]
- Sheldrick G. M. SHELXT-Integrated space–group and crystal-structure determination. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3–8. 10.1107/S2053273314026370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick G. M. Crystal structure refinement with SHELXL. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vichai V.; Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. 10.1038/nprot.2006.179. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Oxidative stress and cancer: have we moved forward?. Biochem. J. 2007, 401, 1–11. 10.1042/BJ20061131. [DOI] [PubMed] [Google Scholar]
- Muhammad N.; Sadia N.; Zhu C.; Luo C.; Guo Z.; Wang X. Biotin–tagged platinum(IV) complexes as targeted cytostatic agents against breast cancer cells. Chem. Commun. 2017, 53, 9971–9974. 10.1039/C7CC05311H. [DOI] [PubMed] [Google Scholar]
- Pracharova J.; Zerzankova L.; Stepankova J.; Novakova O.; Farrer N. J.; Sadler P. J.; Brabec V.; Kasparkova J. Interactions of DNA with a New Platinum(IV) Azide Dipyridine Complex Activated by UVA and Visible Light: Relationship to Toxicity in Tumor Cells. Chem. Res. Toxicol. 2012, 25, 1099–1111. 10.1021/tx300057y. [DOI] [PubMed] [Google Scholar]
- Tai H. C.; Brodbeck R.; Kasparkova J.; Farrer N. J.; Brabec V.; Sadler P. J.; Deeth R. J. Combined theoretical and computational study of interstrand DNA guanine–guanine cross-linking by trans-[Pt(pyridine)2] derived from the photoactivated prodrug trans, trans, trans-[Pt(N3)2(OH)2(pyridine)2]. Inorg. Chem. 2012, 51, 6830–6841. 10.1021/ic3005745. [DOI] [PubMed] [Google Scholar]
- Brabec V.; Leng M. DNA interstrand cross-links of trans-diamminedichloroplatinum(II) are preferentially formed between guanine and complementary cytosine residues. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 5345–534. 10.1073/pnas.90.11.5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.