

Abstract
Research regarding histone deacetylase (HDAC) inhibitors (HDACis) has garnered interest for the treatment of multiple myeloma (MM). In addition, the high expression of nuclear factor (NF)-κB in MM cells is considered an important factor in the occurrence and development of MM. The present study aimed to determine the short-term effects of HDACis, chidamide and valproic acid (VPA), on MM cells, their effects on NF-κB and the underlying mechanisms. The present study measured HDAC activity, and the proliferation and apoptosis of U266 and RPMI8226 MM cells following treatment with various concentrations of chidamide and VPA for 6 and 48 h. Western blotting was used to detect the expression levels of phosphorylated (p)-IκB kinase (IKK)α/β, NF-κB p65 and inhibitor of NF-κB (IκBα) in U266 and RPMI8226 cells at various time points following treatment with chidamide and VPA (0, 2, 4 and 6 h). The results revealed that chidamide and VPA had no significant effect on the HDAC activity, proliferation and apoptosis of cells at 6 h; however, cell HDAC activity and proliferation were inhibited, and apoptosis was induced at 48 h. Furthermore, the expression levels of IκBα were gradually increased over time, whereas the expression levels of NF-κB p65 gradually decreased. These findings indicated that long-term (48 h) treatment with the HDACis chidamide and VPA inhibited the proliferation and promoted the apoptosis of MM cells; however, these HDACis had little effect on cell proliferation and apoptosis in the short term (6 h). Notably, in the short term (2-6 h), hyperactivation of NF-κB was inhibited via the IκBα-NF-κB p65 pathway. These findings indicated that cell growth may be inhibited and drug susceptibility may be promoted by blocking the NF-κB pathway at an early stage, when HDACis are combined with other drugs in the treatment of MM.
Key words: multiple myeloma, histone deacetylase inhibitor, chidamide, valproic acid, NF-κB
Introduction
Multiple myeloma (MM) is a monoclonal, malignant, proliferative tumor derived from plasma cells, which is characterized by monoclonal proteins in the serum or urine, and clonal plasma cells in the bone marrow (1). MM accounts for ~13% of hematological malignancies worldwide (2). With the introduction of novel therapeutic drugs and methods, the 5-year survival rate of patients with MM has been increased to 45% (3); however, the majority of these patients experience relapse and refractory disease. The treatment of patients with relapsed/refractory MM remains a complex issue (4).
Due to the abnormal expression pattern of epigenetic genes in MM, including abnormal methylation, the overproduction of misfolded proteins and the successful application of panobinostat in the clinical treatment of relapsed and refractory MM, histone deacetylase (HDAC) inhibitors (HDACis) have become a focus of research in the treatment of MM (5,6). HDACis can be classified as members of five classes of compounds: i) Hydroxamic acids, ii) short chain fatty acids, iii) benzamides, iv) cyclic tetrapeptides and v) sirtuin inhibitors (7). HDACis bind to the catalytic region of HDACs, reduce their activity, and exert their antitumor effects by altering epigenetic modifications (8).
Chidamide is a novel benzamide class of HDACi, which was developed in China. It exerts its cell death-inducing effects on several types of tumor through its high selective inhibition of HDAC1, 2, 3 and 10 (9), and has been approved by the China Food and Drug Administration for its clinical use in the treatment of peripheral T-cell lymphoma. In vitro, negative effects on MM cells can be exerted by blocking cell cycle progression, and inducing caspase-3-mediated apoptosis and DNA damage (9). Furthermore, a previous study reported the successful treatment of a patient with relapsed/refractory MM with chidamide, bortezomib and dexamethasone in China (10).
Valproic acid (VPA) is type of clinical antiepileptic drug, which belongs to the short-chain fatty acids class of HDACis, which can exert antitumor effects by acting on class I and IIa HDACs (11). In vitro, it has been reported that VPA inhibits the development of MM by blocking cell cycle progression (12), inducing apoptosis (13), suppressing angiogenesis (14,15), inhibiting the Notch pathway (16) and regulating autophagy (17,18). In addition, VPA has been revealed to inhibit the nuclear factor (NF)-κB pathway in malignant myeloblasts in the short term (2-4 h), and this effect remains following denucleation of cells. It has also been reported that the inhibitory effects on the NF-κB pathway are not regulated by histone acetylation, but are a result of direct effects on NF-κB pathway-associated proteins (19).
NF-κB is an important nuclear transcription factor, which comprises RelA (p65), RelB, c-Rel, NF-κB1 (p50/p105) and NF-κB2 (p52/p100) subunits (20). In addition, NF-κB is involved in classical and non-classical pathways. The NF-κB classical pathway predominantly refers to the p50/p65 heterodimer, which is ubiquitous in mammalian cells. The p50/p65 heterodimer coexists in the cytoplasm with inhibitor of NF-κB (IκB) in an inactive state (21). IκB kinase (IKK) is phosphorylated by the action of inflammatory factors or apoptotic genes, which leads to the degradation of IκB, after which, NF-κB is released from IκB, translocates to the nucleus, induces target gene production and participates in various cellular processes (22). The high activation state of NF-κB in MM is an important cause of MM development and drug resistance (23,24). Downregulating the activity of NF-κB in MM cells can inhibit cell proliferation and drug resistance (25), thus providing novel therapeutic targets.
There are numerous proteins involved in the NF-κB pathway, and inhibition of NF-κB is predominantly an additional inhibitory effect of other targeted drugs, such as arsenic trioxide (26) and VPA (19); the latter acts rapidly on P39 MDS/AML cells, and its effects may not be regulated by histone acetylation. Therefore, the present study aimed to determine whether short-term treatment with chidamide and VPA, both of which act via epigenetic modification, can induce inhibitory effects on the NF-κB pathway in MM cells. In addition, the study aimed to determine whether these treatments could affect the proliferation and apoptosis of MM cells in the short term.
Materials and methods
Cell lines and culture
Human MM cell lines U266 and RPMI8226 were purchased from the Tumor Cell Bank of Chinese Academy of Medical Sciences (Beijing, China). U266 cells were cultured in RPMI-1640 (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.) and 1% penicillin/streptomycin, and RPMI-8226 cells were cultured in RPMI-1640 supplemented with 15% FBS and 1% penicillin/streptomycin, at 37°C in a humidified incubator containing 5% CO2.
Cell Counting kit (CCK)‑8 cell proliferation assay
Cells (5×103-1×104) were seeded in 96-well plates with culture medium. U266 cells were cultured with chidamide (0.5, 1, 2, 4 and 8 µM) or VPA (0.5, 1, 2 and 4 mM) for 6 and 48 h at 37°C, whereas RPMI-8226 cells were cultured with chidamide (0.5, 1, 2, 4 and 8 µM) or VPA (0.25, 0.5, 1, 2 and 4 mM) for 6 and 48 h. Simultaneously, the chidamide control group was treated with an equal volume of dimethyl sulfoxide (DMSO), and the VPA control group was treated with an equal volume of culture medium. In addition, the blank control well contained no cell culture solution. Each group was treated in triplicate at the same time. Following treatment, a 10-µl aliquot of CKK-8 (Dojindo Molecular Technologies, Inc., Kumamoto, Japan) was added to each well, and the cells were incubated for a further 4 h. The plate was read at a wavelength of 450 nm using a microplate reader and cell proliferation was calculated using the following equation: Cell proliferation inhibition rate (%)=[1-(OD450 of the experimental group-OD450 of the blank control group)/(OD450 of the control group-OD450 of the blank control group)] ×100%. OD refers to optical density. Each experiment was independently repeated three times.
HDAC activity analysis
Chidamide (1 µM) and VPA (1 mM) were added to the MM cells and incubated at 37°C for 72 h. HDAC activity was detected using the Colorimetric HDAC Activity Assay kit (BioVision, Inc., Milpitas, CA, USA), according to the manufacturer’s protocol. Initially, cells were lysed with a lysis buffer [20 mM Tris-HCl (pH 8.0), 150 mM NaCl, 2 mM EDTA, 100 mM NaF, 1% NP40, 1 µg/ml leupeptin, 1 µg/ml antipain dihydrochloride and 1 mM phenylmethylsulfonyl fluoride]. Each reaction (100 µl) contained nuclear protein extracts (50 µg) from RPMI-8226 and U266 cells. The HDAC activities were measured using a microplate reader (SpectraMax M5; Molecular Devices, LLC, Sunnyvale, CA, USA) at 405 nm. The positive control (i.e., only the nuclear extract) was set at 100% and double-distilled water containing 10 µM Trichostatin A (cat. no. S1045; Selleck Chemicals, Houston, TX, USA) a strong HDACi, was used as a negative control and was set at 0%.
Flow cytometry and apoptosis
Cells (1×105) were seeded in 24-well plates with culture medium. U266 cells were incubated with chidamide (1, 2, 4 and 8 µM) or VPA (0.5, 1, 2 and 4 mM) for 6 and 48 h, whereas RPMI8226 cells were incubated with chidamide (1, 2, 4 and 8 µM) or VPA (0.25, 0.5, 1 and 2 mM) for 6 and 48 h. The chidamide control group was treated with an equal volume of DMSO, and the VPA control group was treated with an equal volume of culture medium. Annexin V-fluorescein isothiocyanate/propidium iodide (BD Pharmingen; BD Biosciences, Franklin Lakes, NJ, USA) double staining was used to quantify apoptosis, according to the manufacturer’s protocol, and cell apoptosis was analyzed by flow cytometry using BD FACSDiva software 8.0 (BD Biosciences).
Western blot analysis
U266 cells were treated with chidamide (2 µM) or VPA (1 mM), and RPMI8226 cells were treated with chidamide (2 µM) or VPA (0.5 mM) at different time points (0, 2, 4 and 6 h). For HDAC activity analysis. U266 and RPMI8266 cells were treated with chidamide (1 µM) and VPA (1 mM) for 72 h at 37°C. Subsequently, cells were resuspended in lysis buffer containing radioimmunoprecipitation assay buffer and 10 µg/ml phenylmethylsulfonyl fluoride. Protein concentrations were determined using the bicinchoninic acid protein assay kit (Pierce; Thermo Fisher Scientific, Inc. Proteins (20 µg) were separated by 8% SDS-PAGE and were transferred onto nitrocellulose membranes, which were blocked with 5% non-fat milk in Tris-buffered saline containing 0.1% Tween-20. The membranes were then incubated with the following primary antibodies at 4°C overnight: Anti-IκBα (1:1,000; cat. no ab32518; Abcam, Cambridge, MA, USA), anti-NF-κB p65 (1:50,000; cat. no. ab32536; Abcam), anti-IKKα/β (1:1,000; cat. no. ab178870; Abcam), anti- phosphorylated (p)-IKKα/β (1:1,000; cat. no. 2697; Cell Signaling Technology, Inc., Danvers, MA, USA), anti-acetyl-histone H3 (1:1,000; cat. no. 9649; Cell Signaling Technology, Inc.) and anti-GAPDH (1:1,000; cat. no. ab9485; Abcam) as an internal control. Subsequently, membranes were visualized using an enhanced chemiluminescence (ECL) plus reagent (Western Lightning Plus-ECL; cat. no. NEL104001EA; PerkinElmer, Inc., Waltham, MA, USA), following the addition of a horseradish peroxidase-conjugated secondary antibody at room temperature for 1 h (1:2,500; cat. no. TA140002; OriGene Technologies, Inc., Beijing, China), under a chemiluminescence and fluorescence imaging system (LumiX®/ChemiX®/FluorChemiX®; Federal Bioproducts, Inc., London, United Kingdom).
Statistical analysis
Each experiment was repeated three times. All data are expressed as the means ± standard deviation, and were analyzed using SPSS19.0 (SPSS, Inc., Chicago, IL, USA). Pairwise comparisons were conducted using Student’s t-test. Multiple comparisons were performed by one-way analysis of variance followed by Dunnett’s post hoc test. P<0.05 was considered to indicate a statistically significant difference.
Results
Chidamide and VPA enhance histone H3 acetylation and inhibit HDAC activity
To explore whether chidamide and VPA might influence histone H3 acetylation in MM cells, the present study detected histone H3 acetylation levels in cells following exposure to chidamide or VPA. Following treatment with chidamide or VPA for 48 h, the acetylation levels of histone H3 were increased compared with in cells treated for 0 or 6 h. There were no marked alterations in acetylation levels between the 0 and 6 h chidamide or VPA groups (Fig. 1A). The results of a HDAC activity analysis indicated that treatment of cells with chidamide or VPA for 48 h resulted in a greater HDAC inhibition compared with in the 0 and 6 h culture groups (Fig. 1B). These results indicated that, in the 48 h chidamide and VPA culture groups, levels of histone H3 acetylation were increased and HDAC activity was inhibited compared with in the 0 and 6 h culture groups. There was no significant difference between the 0 and 6 h culture groups when MM cells were exposed to chidamide or VPA.
Figure 1.
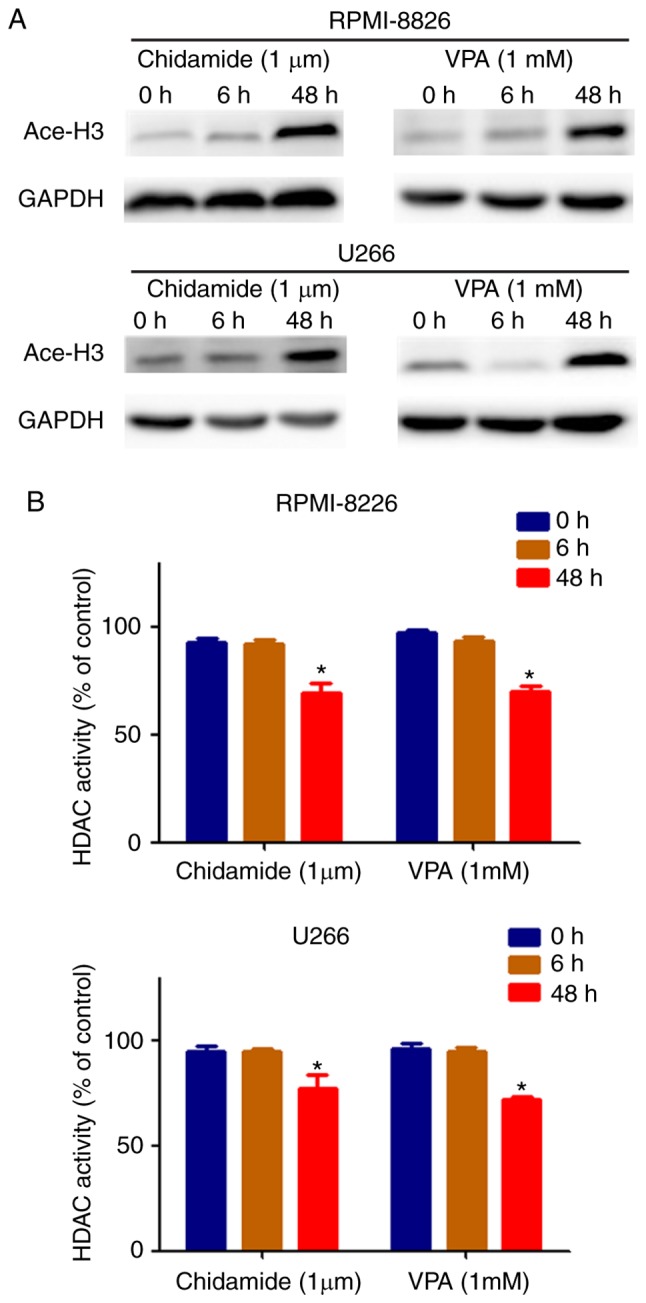
Chidamide and VPA enhance histone H3 acetylation and inhibit HDAC activity in mM cell lines at 48 h, but not at 0 and 6 h. (A) Chidamide and VPA promoted an increase in the levels of histone H3 acetylation in human MM cells (RPMI-8226 and U266). (B) Chidamide and VPA inhibited HDAC activity in human mM cells (RPMI-8226 and U266). *P<0.05 vs. the 0 h culture group. Ace-H3, acetyl-histone H3; HDAC, histone deacetylase; MM, multiple myeloma; VPA, valproic acid.
Chidamide and VPA inhibit MM cell proliferation after 48 h
In order to study the effects of chidamide and VPA on the activity of U266 and RPMI8226 cells during a short time period, cell viability was measured after 6 h of drug action. In addition, cell viability was measured after 48 h as a comparison. The experimental results revealed that there was no obvious effect on cell proliferation after 6 h (P>0.05); however, chidamide and VPA inhibited cell proliferation after 48 h compared with in the 6 h group (P<0.05; Fig. 2). In addition, the inhibition of cell proliferation was increased alongside drug concentration. These results indicated that chidamide and VPA may affect the proliferation of U266 and RPMI8226 cells in the long term.
Figure 2.

Effects of chidamide and VPA on the proliferation of multiple myeloma cells, as determined by Cell Counting kit-8 assay. Inhibition rate of (A) U266 and (B) RPMI8226 cell proliferation in response to various concentrations of chidamide for 6 and 48 h. Inhibition rate of (C) U266 and (D) RPMI8226 cell proliferation in response to various concentrations of VPA for 6 and 48 h. VPA, valproic acid. *P<0.05.
Chidamide and VPA induce MM cell apoptosis after 48 h
Cell apoptosis was measured following treatment with chidamide and VPA for 6 and 48 h using flow cytometry. The results revealed that chidamide and VPA exerted no significant effect on cell apoptosis after 6 h (P>0.05; Fig. 3). Conversely, after 48 h, chidamide (2, 4 and 8 µM) and VPA (1, 2 and 4 mM) induced apoptosis of U266 cells, and chidamide (1, 2, 4 and 8 µM) and VPA (0.25, 0.5, 1 and 2 mM) induced apoptosis of RPMI8226 cells compared with in the control group (P<0.05; Fig. 4). The apoptotic effect was increased with increasing drug concentrations. These findings suggested that chidamide and VPA may affect the apoptosis of U266 and RPMI8226 cells in the long term.
Figure 3.

Effects of chidamide and VPA on the apoptosis of multiple myeloma cells after 6 h, as determined by flow cytometry using Annexin V-FITC/PI staining. Effects of various concentrations of chidamide on the apoptosis of (A) U266 and (B) RPMI8226 cells. Effects of various concentrations of VPA on the apoptosis of (C) U266 and (D) RPMI8226 cells. (E) Chidamide and VPA had no effect on the apoptosis of U266 and RPMI8226 cells after 6 h. FITC, fluorescein isothiocyanate; PI, propidium iodide; VPA, valproic acid.
Figure 4.

Effects of chidamide and VPA on the apoptosis of multiple myeloma cells after 48 h, as determined by flow cytometry using Annexin V-FITC/PI staining. Effects of various concentrations of chidamide on the apoptosis of (A) U266 and (B) RPMI8226 cells. Effects of various concentrations of VPA on the apoptosis of (C) U266 and (D) RPMI8226 cells. (E) Chidamide and VPA could promote apoptosis of U266 and RPMI8226 cells after 48 h; the apoptotic rate was increased with increasing drug concentrations. *P<0.05, **P<0.01 vs. the untreated control group. FITC, fluorescein isothiocyanate; PI, propidium iodide; VPA, valproic acid.
Chidamide and VPA inhibit the p‑IKKα/β‑IκBα‑NF‑κB p65 pathway in the short term in MM cells
NF-κB exhibits high activity in MM cells, and promotes its occurrence and development (27). To explore the effects of chidamide and VPA on the NF-κB signaling pathway in U266 and RPMI8226 cells, western blotting was conduced to detect the expression levels of NF-κB pathway-related proteins at 0, 2, 4 and 6 h following treatment with chidamide and VPA. The expression levels of IκBα were gradually increased in response to increasing drug concentrations. Conversely, the expression levels of p-IKKα/β and NF-κB p65 were gradually decreased; however, there was some effect on total IKKα/β protein expression levels following treatment with chidamide or VPA (Fig. 5). These findings indicated that chidamide and VPA may exert their anti-MM effects through inhibiting the NF-κB pathway, and alterations in the expression levels of pathway-associated protein were observed within the 2-6 h time period.
Figure 5.

Effects of chidamide and VPA on the expression levels of NF-κB pathway-associated proteins, p-IKKα/β, IKKα/β, NF-κB p65 and IκBα, in U266 and RPMI8226 cells, as determined by western blotting. (A) Effects of chidamide (2 µM) on the expression levels of NF-κB pathway-associated proteins in U266 cells. (B) Effects of chidamide (2 µM) on the expression levels of NF-κB pathway-associated proteins in RPMI8226 cells. (C) Effects of VPA (1 mM) on the expression levels of NF-κB pathway-associated proteins in U266 cells. (D) Effects of VPA (0.5 mM) on the expression levels of NF-κB pathway-associated proteins in RPMI8226 cells. IκBα, inhibitor of NF-κB; IKK, IκB kinase; NF-κB, nuclear factor-κB; p, phosphorylated; VPA, valproic acid.
Discussion
As an important nuclear transcription factor, NF-κB is involved in various biological cellular processes (28). Gene expression analysis and immunostaining revealed that the majority of MM cells undergo continuous activation of NF-κB, which can promote the development of MM by affecting cell survival, apoptosis, angiogenesis, cell migration and drug resistance (23,29).
HDACis are considered an important research focus as a potential method to treat MM. Among HDACis, VPA has been reported to negatively effect MM via various mechanisms in vitro. In addition, NF-κB activity of malignant myeloblasts can also be inhibited during the early stage, and this inhibition exists following denucleation (19). As the first HDACi developed in China, chidamide has successfully been used in the clinical treatment of peripheral T-cell lymphoma (30), and it has also been confirmed to kill and inhibit various tumor cells in vitro (31-33); however, research regarding HDACis in MM is rare. Due to the importance of NF-κB in the development of MM, and the ability of VPA to rapidly inhibit the activity of NF-κB in malignant myeloblasts, the present study hypothesized that chidamide and VPA may inhibit NF-κB, and affect MM cell proliferation and apoptosis in the short term.
The present study selected two MM cell lines, U266 and RPMI8226, to ensure the reliability of the experimental results. Exploration and selection of drug concentrations used for the cell proliferation assay in this study were derived from previous studies on chidamide and VPA in MM (9,15) or other tumors (34,35). Chidamide doses (1, 2, 4 and 8 µM) that exhibited 20-90% cytotoxicity in the cell proliferation study were selected for the treatment of U266 and RPMI8226 cells prior to apoptosis assessment. In the VPA groups, VPA (0.5, 1, 2 and 4 mM) was used to treat U266 cells, whereas VPA (0.25, 0.5, 1 and 2 mM) was used to treat RPMI8226 cells. In addition, doses close to the half maximal inhibitory concentrations of chidamide (2.395 µM) and VPA (0.554 mM) were used to treat cells prior to western blotting. Proliferation and apoptosis of cells was detected following treatment with chidamide or VPA for 6 and 48 h. The results revealed that these treatments could inhibit cell proliferation and promote apoptosis of U266 and RPMI8226 cells after 48 h, which was consistent with the findings from previous studies in adenoid cystic carcinoma and myelodysplastic syndromes (34,36); however, no significant effects were detected after 6 h. In addition, the present results revealed that treatment with chidamide and VPA for 48 h led to higher levels of histone H3 acetylation and inhibition of HDAC activity compared with in the 0 and 6 h groups; there was no significant difference between the 0 and 6 h culture groups. Western blotting was used to detect the expression levels of NF-κB pathway-associated proteins, p-IKKα/β, IκBα and NF-κB p65, at different time points (0, 2, 4 and 6 h) following treatment. The expression levels of IκBα were increased after 2 h, whereas p-IKKα/β and NF-κB p65 expression was decreased; the extent of expression alterations was increased with the prolongation of treatment time. However, there was some effect on the total expression of IKKα/β following treatment with chidamide and VPA; therefore, it is unclear as to whether the effects of chidamide and VPA on p-IKKα/β follow alterations in the total expression of IKKα/β. Further studies are required to determine the effects and mechanism of chidamide and VPA on p-IKKα/β and total IKKα/β expression. The present findings indicated that IκBα and NF-κB expression was altered following treatment with chidamide and VPA. Therefore, chidamide and VPA may inhibit the activity of NF-κB in MM cells via the NF-κB p65 pathway during the early stage, which is similar to the effects of VPA and trichostatin on malignant myeloblasts (19). Unlike the effects of VPA on Notch signaling in MM cells at 48 h (16), autophagy of lesioned cortices 24 h after traumatic brain injury (TBI) and HDAC3 expression 72 h after TBI (37), and the effects of chidamide on Mcl-1 expression in pancreatic cancer cells at 24 h (38), the effects of chidamide and VPA on the NF-κB pathway were detected in the short term in the present study.
The present study hypothesized that HDACi-induced inhibition of the NF-κB pathway in tumor cells depended on non-histone acetylation in the cytoplasm, rather than the epigenetic reprogramming caused by alterations in the nuclear transcriptome. However, the specific targets of HDACis in the cytoplasm require further analysis.
Previous studies have revealed that chidamide and DNA damaging agents can synergistically promote apoptosis in relapsed and refractory acute myeloid leukemia cells and reduce tumor burden in patients (39), and that VPA and bortezomib cooperate to induce pancreatic cancer cell death (40). Although chidamide and VPA did not inhibit MM cell proliferation and promote apoptosis, as expected, in the short term (6 h), which may be associated with the complexity of cell proliferation and development mechanisms, the rapid inhibition of NF-κB may inhibit the continued growth of MM cells in the early stages. It remains to be determined as to whether chidamide or VPA, combined with other drugs, may increase the cell death-inducing effects of these other drugs and increase drug sensitivity. In addition, whether the findings of the present study are associated with the successful treatment of patients with relapsed and refractory MM with chidamide combined with bortezomib and dexamethasone is unclear. Further studies are required regarding whether the combination of HDACis and other drugs can increase anti-MM effects by blocking NF-κB in the early stages.
Acknowledgments
Not applicable.
Abbreviations
- MM
multiple myeloma
- HDAC
histone deacetylase
- HDACi
histone deacetylase inhibitor
- VPA
valproic acid
- DMSO
dimethyl sulfoxide
- IκBα
inhibitor of NF-κB
- NF-κB
nuclear factor-κB
- IKK
IκB kinase
- p-IKK
phosphorylated-IKK
Funding
The present study was supported by the National Natural Science Foundation of China (grant nos. 81570106, 81400088 AbD 81400085), the Tianjin Municipal Natural Science Foundation (grant nos. 14JCYBJC25400, 15JCYBJC24300, 15KG150 and 16ZXMJSY00180), the Youth Incubation Fund of Tianjin Medical University General Hospital (grant no. ZYYFY 2016006) and the Tianjin Institute of Lung Cancer.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Authors’ contributions
ZL and RF designed the research. ZL and QJ carried out most of the experiments, analyzed the data, generated the figures and drafted the manuscript. QJ and YW contributed to cell culture and western blotting, YL, FM and CX contributed to RT-qPCR and flow cytometry. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Kohgo Y. Recent advances in multiple myeloma. Int J Clin Oncol. 2015;20:411–412. doi: 10.1007/s10147-015-0809-4. [DOI] [PubMed] [Google Scholar]
- 2.Palumbo A, Anderson K. Multiple myeloma. Current Opinion in Oncology. 2011;24(Suppl):S1. doi: 10.1097/01.cco.0000410242.38332.78. [DOI] [PubMed] [Google Scholar]
- 3.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 4.Moreau P, Attal M, Facon T. Frontline therapy of multiple myeloma. Blood. 2015;125:3076–3084. doi: 10.1182/blood-2014-09-568915. [DOI] [PubMed] [Google Scholar]
- 5.Laubach JP, San-Miguel JF, Hungria V, Hou J, Moreau P, Lonial S, Lee JH. An advance in myeloma therapy? Expert Rev Hematol. 2017;10:229–237. doi: 10.1080/17474086.2017.1280388. [DOI] [PubMed] [Google Scholar]
- 6.Greig SL. Panobinostat: A review in relapsed or refractory multiple myeloma. Targeted Oncol. 2016;11:107–114. doi: 10.1007/s11523-015-0413-6. [DOI] [PubMed] [Google Scholar]
- 7.Ceccacci E, Minucci S. Inhibition of histone deacetylases in cancer therapy: Lessons from leukaemia. Br J Cancer. 2016;114:605–611. doi: 10.1038/bjc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eckschlager T, Plch J, Stiborova M, Hrabeta J. Histone deacetylase inhibitors as anticancer drugs. Int J Mol Sci. 2017;18:1414. doi: 10.3390/ijms18071414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu L, Tang HL, Gong X, Xin XL, Dong Y, Gao GX, Shu MM, Chen XQ. Inducing effect of chidamide on apoptosis of multiple myeloma cells and its relerance to DNA damage response. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2015;23:450–454. doi: 10.7534/j.issn.1009-2137.2015.02.030. In Chinese. [DOI] [PubMed] [Google Scholar]
- 10.Wang DY, Cui YS, Liu YZ, Liu LN, Song YP, Fang BJ. Successful treatment of one case with relapsed refractory multiple myeloma by chidamide in combination with bortezomib and dexamethasone. Zhonghua Xue Ye Xue Za Zhi. 2016;37:463. doi: 10.3760/cma.j.issn.0253-2727.2016.06.004. In Chinese. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Göttlicher M, Minucci S, Zhu P, Krämer OH, Schimpf A, Giavara S, Sleeman JP, Lo Coco F, Nervi C, Pelicci PG, Heinzel T. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. Embo J. 2001;20:6969–6978. doi: 10.1093/emboj/20.24.6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaiser M, Zavrski I, Sterz J, Jakob C, Fleissner C, Kloetzel PM, Sezer O, Heider U. The effects of the histone deacetylase inhibitor valproic acid on cell cycle, growth suppression and apoptosis in multiple myeloma. Haematologica. 2006;91:248–251. [PubMed] [Google Scholar]
- 13.Wang XN, Zhang M. Apoptosis-inducing effect of valproic acid combined with arsenic trioxide on RPMI 8226 cells and its mechanism. Zhongguo Shi Yan Xue Ye Xue Za ZhI. 2014;22:1016–1021. doi: 10.7534/j.issn.1009-2137.2014.04.024. In Chinese. [DOI] [PubMed] [Google Scholar]
- 14.Dong XF, Song Q, Li LZ, Zhao CL, Wang LQ. Histone deacetylase inhibitor valproic acid inhibits proliferation and induces apoptosis in KM3 cells via downregulating VEGF receptor. Neuro Endocrinol Lett. 2007;28:775–780. [PubMed] [Google Scholar]
- 15.Kitazoe KI, Abe M, Hiasa M, Oda A, Amou H, Harada T, Nakano A, Takeuchi K, Hashimoto T, Ozaki S, Matsumoto T. Valproic acid exerts anti-tumor as well as anti-angiogenic effects on myeloma. Int J Hematol. 2009;89:45–57. doi: 10.1007/s12185-008-0226-9. [DOI] [PubMed] [Google Scholar]
- 16.Qiao L, Yang Y, Zhao RJ, Wang LH, Zhao L, Yan LN, Zhang ZH, Hao CL. Effects of VPA on the expression of notch signaling pathway in multiple myeloma RPMI 8226 cell line. Zhongguo Shi Yan Xue Ye XuezazhI. 2016;24:1449–1453. doi: 10.7534/j.issn.1009-2137.2016.05.030. In Chinese. [DOI] [PubMed] [Google Scholar]
- 17.Jin YL, Dong BX, Xu L, Tang HL, Gao GX, Gu HT, Shu MM, Chen XQ. Valproic acid represses autophagy and enhances the anti-myeloma activity of DNA-damaging drugs. Zhongguo Shi Yan Xue Ye Xuezazhi. 2015;23:718–721. doi: 10.7534/j.issn.1009-2137.2015.03.023. In Chinese. [DOI] [PubMed] [Google Scholar]
- 18.Zhang YY, Zhang ZH, Zhao RJ, Li H, Wang TR, Yan LN, Gu CH, Zhao L, Hao CL. Valproic acid activates autophagy in multiple myeloma cell lines RPMI8226 and U266. Zhonghuaxue Ye XuezazhI. 2016;37:478–483. doi: 10.3760/cma.j.issn.0253-2727.2016.06.008. In Chinese. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fabre C, Grosjean J, Tailler M, Boehrer S, Adès L, Perfettini JL, de Botton S, Fenaux P. NFkappaB inhibition in malignant myeloblasts. Cell Cycle. 2008;7:2139–2145. doi: 10.4161/cc.7.14.6268. [DOI] [PubMed] [Google Scholar]
- 20.Dutta J, Fan Y, Gupta N, Fan G, Gélinas C. Current insights into the regulation of programmed cell death by NF-κB. Oncogene. 2006;25:6800–6816. doi: 10.1038/sj.onc.1209938. [DOI] [PubMed] [Google Scholar]
- 21.Oeckinghaus A, Ghosh S. The NF-κB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 2009:a000034. doi: 10.1101/cshperspect.a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen ZJ. Ubiquitination in signaling to and activation of IKK. Immunol Rev. 2012;246:95–106. doi: 10.1111/j.1600-065X.2012.01108.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Annunziata CM, Davis RE, Demchenko Y, Bellamy W, Gabrea A, Zhan F, Lenz G, Hanamura I, Wright G, Xiao W, et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007;12:115–130. doi: 10.1016/j.ccr.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hayden MS, Ghosh S. NF-κB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012;26:203–234. doi: 10.1101/gad.183434.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kannaiyan R, Hay HS, Rajendran P, Li F, Shanmugam MK, Vali S, Abbasi T, Kapoor S, Sharma A, Kumar AP, et al. Celastrol inhibits proliferation and induces chemosensitization through down-regulation of NF-κB and STAT3 regulated gene products in multiple myeloma cells. Br J Pharmacol. 2011;164:1506–1521. doi: 10.1111/j.1476-5381.2011.01449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qu X, Du J, Zhang C, Fu W, Xi H, Zou J, Hou J. Arsenic trioxide exerts antimyeloma effects by inhibiting activity in the cytoplasmic substrates of histone deacetylase 6. PLoS One. 2012;7:e32215. doi: 10.1371/journal.pone.0032215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Z, Yang Z, Peng X, Li Y, Liu Q, Chen J. Nuclear factor-κB is involved in the protocadherin-10-mediated pro-apoptotic effect in multiple myeloma. Mol Med Rep. 2014;10:832–838. doi: 10.3892/mmr.2014.2285. [DOI] [PubMed] [Google Scholar]
- 28.Alkalay I, Yaron A, Hatzubai A, Orian A, Ciechanover A, Ben-Neriah Y. Stimulation-dependent IκBα phosphorylation marks the NF-κB inhibitor for degradation via the ubiquitin-proteasome pathway. Proc Natl Acad Sci USA. 1995;92:10599–10603. doi: 10.1073/pnas.92.23.10599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bharti AC, Shishodia S, Reuben JM, Weber D, Alexanian R, Raj-Vadhan S, Estrov Z, Talpaz M, Aggarwal BB. Nuclear factor-kappaB and STAT3 are constitutively active in CD138+ cells derived from multiple myeloma patients, and suppression of these transcription factors leads to apoptosis. Blood. 2004;103:3175–3184. doi: 10.1182/blood-2003-06-2151. [DOI] [PubMed] [Google Scholar]
- 30.Shi Y, Dong M, Hong X, Zhang W, Feng J, Zhu J, Yu L, Ke X, Huang H, Shen Z, et al. Results from a multicenter, open-label, pivotal phase II study of chidamide in relapsed or refractory peripheral T-cell lymphoma. Ann Oncol. 2015;26:1766–1771. doi: 10.1093/annonc/mdv237. [DOI] [PubMed] [Google Scholar]
- 31.Liu L, Chen B, Qin S, Li S, He X, Qiu S, Zhao W, Zhao H. A novel histone deacetylase inhibitor Chidamide induces apoptosis of human colon cancer cells. Biochem Biophys Res Commun. 2010;392:190–195. doi: 10.1016/j.bbrc.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 32.Ning ZQ, Li ZB, Newman MJ, Shan S, Wang XH, Pan DS, Zhang J, Dong M, Du X, Lu XP. Chidamide (CS055/HBI-8000): A new histone deacetylase inhibitor of the benzamide class with antitumor activity and the ability to enhance immune cell- mediated tumor cell cytotoxicity. Cancer Chemother Pharmacol. 2012;69:901–909. doi: 10.1007/s00280-011-1766-x. [DOI] [PubMed] [Google Scholar]
- 33.Walkinshaw DR, Yang XJ. Histone deacetylase inhibitors as novel anticancer therapeutics. Curr Oncol. 2008;15:237–243. [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Z, Ding K, Li L, Liu H, Wang Y, Liu C, Fu R. A novel histone deacetylase inhibitor Chidamide induces G0/G1 arrest and apoptosis in myelodysplastic syndromes. Biomed Pharmacother. 2016;83:1032–1037. doi: 10.1016/j.biopha.2016.08.023. [DOI] [PubMed] [Google Scholar]
- 35.Saha SK, Yin Y, Kim K, Yang GM, Dayem AA, Choi HY, Cho SG. Valproic acid induces endocytosis-mediated doxorubicin internalization and shows synergistic cytotoxic effects in hepato-cellular carcinoma cells. Int J Mol Sci. 2017;18:E1048. doi: 10.3390/ijms18051048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang S, Nan P, Li C, Lin F, Li H, Wang T, Zhou C, Zhang X, Meng X, Qian H, et al. Inhibitory effect of chidamide on the growth of human adenoid cystic carcinoma cells. Biomed Pharmacother. 2018;99:608–614. doi: 10.1016/j.biopha.2018.01.110. [DOI] [PubMed] [Google Scholar]
- 37.Chen X, Wang H, Zhou M, Li X, Fang Z, Gao H, Li Y, Hu W. Valproic acid attenuates traumatic brain injury-induced inflammation in vivo: Involvement of autophagy and the Nrf2/ARE signaling pathway. Front Mol Neurosci. 2018;11:1–13. doi: 10.3389/fnmol.2018.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qiao Mu H, Wang Z, Kuai Y, Li Q, Wang C, Jiang Y, Wang X, Li X, He WM, et al. Chidamide inhibits aerobic metabolism to induce pancreatic cancer cell growth arrest by promoting Mcl-1 degradation. PLoS One. 2016;11:e0166896. doi: 10.1371/journal.pone.0166896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Y, Wang Y, Zhou Y, Li J, Chen K, Zhang L, Deng M, Deng S, Li P, Xu B. Cooperative effect of chidamide and chemotherapeutic drugs induce apoptosis by DNA damage accumulation and repair defects in acute myeloid leukemia stem and progenitor cells. Clin Epigenetics. 2017;9:83. doi: 10.1186/s13148-017-0377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.GilardiniMontani MS, Granato M, Santoni C, Del Porto P, Merendino N, D’Orazi G, Faggioni A, Cirone M. Histone deacetylase inhibitors VPA and TSA induce apoptosis and autophagy in pancreatic cancer cells. Cell Oncol (Dordr) 2017;40:1–14. doi: 10.1007/s13402-017-0314-z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.