

Abstract
Background
Ambient particulate matter (PM) exposure has been associated with respiratory function decline in epidemiological studies. We hypothesize that a possible underlying mechanism is the perturbation of airway microbiome by PM exposure.
Methods
During October 2016 – October 2017, on two human cohorts (n = 115 in total) in Shanghai China, we systematically collected three categories of data (1) respiratory functions, (2) airway microbiome from sputum and (3) ambient PM2.5 (PM of ≤ 2.5 micron in diameter) – level in ambient air. We investigated the impact of PM2.5 on airway microbiome as well as the link between airway microbiome and respiratory functions using mix linear regression models.
Results
The respiratory function of our primary interest includes forced vital capacity (FVC) and forced expiratory volume in 1st second (FEV1). FEV1/FVC, an important respiratory function trait and key diagnosis criterion of COPD, was significantly associated with airway bacteria load (p = 0.0038); and FEV1 was associated with airway microbiome profile (p = 0.013). Further, airway microbiome was significantly influenced by PM2.5 exposure (p = 4.48E-11).
Conclusions
To our knowledge, for the first time, we demonstrated the impact of PM2.5 on airway microbiome, and reported the link between airway microbiome and respiratory functions. The results expand our understanding on the scope of PM2.5 exposure’s influence on human respiratory system, and pointed to novel etiological mechanism of PM2.5 exposure induced diseases.
Keywords: airway microbiome, ambient particulate matter (PM) exposure, respiratory functions, next generation sequencing, chronic obstructive pulmonary disease (COPD)
Introduction
Air pollution is identified as a leading cause of global disease burden, especially in low-income and middle-income countries (Cohen et al., 2017). China is one of the regions in the world of severe ambient air pollution (Wang et al., 2017a; Xu et al., 2016). As a major component of airborne pollutants, particulate matter (PM) has been linked to pulmonary disorders (Bahr et al., 2013; Beelen et al., 2014; Bell et al., 2013), including chronic obstructive pulmonary disease (COPD) (Wang et al., 2017b). Particulate matter of ≤ 2.5 micron in diameter is termed as PM2.5, and its adverse effect on respiratory functions has been reported by twenty years of epidemiology studies (Xing et al., 2016). The “Harvard Six Cities Study” demonstrated that PM2.5 exposure was causally associated with non-accidental death (Schwartz et al., 1996). Respiratory diseases account for a large proportion of the non-accidental deaths caused by airborne pollutant exposure (Schwartz et al., 1996). Studies also indicated the causal role of PM2.5 in asthma, respiratory inflammation, and decline in respiratory functions (https://developer.apple.com/healthkit/; Lewis et al., 2005; Ostro et al., 2006). It was estimated that the prevalence of respiratory diseases increased by 2.07% and hospitalization rate elevated by 8% when daily PM2.5 concentration increased by 10 µg/m3 (Dominici et al., 2006; Zanobetti et al., 2009). Further, the impact of PM2.5 exposure is more profound in the elderly, pregnant women, adolescents, infants, patients with a history of cardiopulmonary problems and other susceptible populations (de Oliveira et al., 2012; Huynh et al., 2006; Martinelli et al., 2012).
The exact mechanism that PM2.5 exposure reduces respiratory function is unknown, and there are multiple molecular etiology pathways possible: (1) Injury from free radical peroxidation. Components of PM2.5 can induce free radical production, oxidize lung cells and directly lead to injury (Donaldson et al., 1996; Greenwell et al., 2002; Kelly, 2003; Rahman and MacNee, 1996). The reactive oxygen species (ROS) generated by particles, particularly by water soluble particles, produce hydroxyl radical (• OH) by activating metals (Valavanidis et al., 2005). The PM2.5 surface is rich in iron, copper, zinc, manganese, and other transition elements, as well as polycyclic aromatic hydrocarbons (PAH), which are able to increase free radical production in the lung, consume antioxidant ingredients and cause oxidative stress (Donaldson et al., 1996). (2) Injury from inflammatoryresponse. PM2.5 is related to inflammatory cytokines as it upregulate multiple inflammation-inducing transcription factor (TF), which leads to increase in the number of neutrophils, eosinophils, T cells and mastocytes in bronchoalveolar lavage (BAL) fluid (Gripenback et al., 2005; Sigaud et al., 2007). The interactions between inflammatory cells and cytokines can damage lung cells synergistically (Xing et al., 2016). COPD is a leading cause of morbidity and mortality worldwide (http://www.goldcopd.org; Wang et al., 2017b). COPD has a complex etiology, where inflammation plays an important role (Barnes, 2014). Oxidative stress may further amplify the inflammation (Rahman, 2005), and it is established that PM2.5 contribute to the oxidative stress (Wong, 2017). (3) Changes in airway microbiome. Humans and microbes have evolved together for millions of years. Humans harbor 10 times more microbes inside or on their bodies than the total number of their somatic and germ cells (Wang et al., 2017b). Human Microbiome Project (HMP)(Blaser, 2010) showed that we greatly underestimated the diversity, prevalence, and persistence of the bacteria in our body, partly because majority of the bacteria cannot be cultured in the laboratory (Hayashi et al., 2002; Suau et al., 1999). The development of culture-independent techniques for microbiological analysis (e.g., 16S rRNA sequencing) showed the airway and bronchial tree is not sterile even in healthy individuals (Huang et al., 2013). Instead, the lung is a complex microbial ecosystem, the microbiota resides in the bronchial tree and parenchymal tissues, which could be important for colonization resistance, epithelial integrity, and immune-regulation. Different taxonomic composition was reported in sputum microbiome samples from eight COPD patients and ten “healthy” smokers (Cameron et al., 2016). The changes of microbiome composition and functional capacity were associated with COPD severity (FEV1% of predicted) (Cameron et al., 2016).
To date, the link between airway microbiome and respiratory functions, particularly in non-COPD individuals, has not been fully studied. There are no empirical data assessing the impact of PM2.5 exposure on airway microbiome. As results, the roles of airway microbiome, in terms of mediating the adverse effect of PM2.5 exposure on respiratory functions and biomarkers for airway micro-environment damage caused by PM2.5 exposure, remain unknown. In order to fill in such knowledge gaps, three categories of data were collected on human cohorts of this study: (1) respiratory functions, (2) airway microbiome from sputum and (3) levels of PM2.5 exposure. We investigated the association between airway microbiome and respiratory functions, as well as, the perturbation caused by PM2.5 exposure.
Materials and Methods
COPD risk cohort
Seventy-four participants from a community-based epidemiological study, termed as COPD Risk Cohort (Fig 1 and Table 1), were examined at the Department of Respiratory Medicine, Shanghai Tenth People’s Hospital, Tongji University, Shanghai, China. All subjects reside in Shanghai, China, and provided written informed consent. None of the subjects were diagnosed with COPD, but satisfying the criteria: history of cigarette smoking or a history of exposure to second-hand smoke; post-bronchodilator FEV1/forced vital capacity (FVC) ratio < 70% and FEV1% predicted value >80% in the absence of a bronchodilator or an inhaled corticosteroid treatment. The exclusion criteria included asthma, bronchiectasis, infectious disease, psychiatric disease, cognitive disorders, malignancies, severe kidney or liver dysfunction. An annual on-site follow-up were conducted on June 4th and 11th, 2017. We collected sociodemographic characteristics, such as age, gender, height, weight, smoking status, street address and comorbidities. All subjects completed the COPD Assessment Test (CAT), the modified British Medical Research Council (mMRC) questionnaire, and the Clinical COPD Questionnaire (CCQ); in addition, all subjects conducted the spirometry test and contributed sputum sample. Sputum is the coughed-up material (phlegm) from the lower airways (trachea and bronchi), and contain large number of white blood cells. The study was approved by the Institutional Review Boards at Shanghai Tenth People’s Hospital, Tongji University, Shanghai, China.
Figure 1. Study design and flow chart.
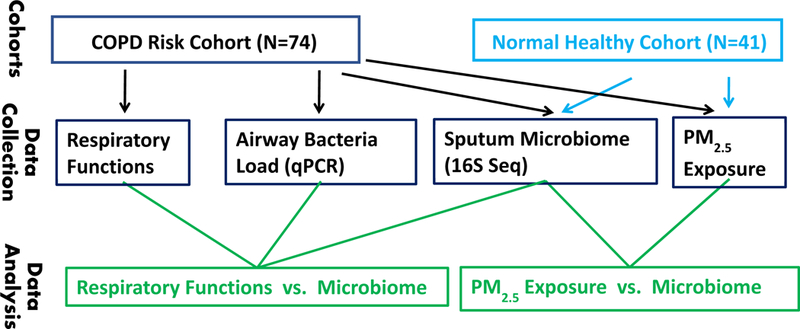
Table 1.
Demographical and respiratory function characteristics of two human cohorts.
| Variable | COPD risk cohort (n = 74) |
Normal healthy cohort (n = 41) |
|---|---|---|
| Agea | 59 ± 8 | 26 ± 3g |
| Gender (Female)b | 47 (63.5%) | 19 (46.3%) |
| Height (m) | 1.63 ± 0.08 | 1.69 ± 0.09 (1) |
| Weight (kg) | 63.7 ± 8.3 | 60.7 ± 9.7 (1) |
| Smoke (Yes) | 14 (18.9%) (7) | 2 (4.9%) |
| FEV1 (L) | 2.28 ± 0.40 (3) | |
| FEV1% predicted | 91.2 ± 12.3 (3) | |
| FVC (L) | 2.72 ± 0.50 (3) | |
| FVC% predicted | 88.2 ± 14.8 (3) | |
| FEV1/FVC ratio (%) | 84.1 ± 6.4 (3) | |
| CATc | 11.0 ± 3.4 (5) | |
| mMRCd | 0.3 ± 0.5 (6) | |
| CCQe | 3.0 ± 4.8 (5) | |
| Airway bacteria loadf | 119.5 ± 188.2 | |
Values shown for continuous variables are mean ± S.D. The number of missing data is shown in parentheses if there is any.
Values shown for categorical variables are the number of samples along with percentage shown in the parentheses. The number of missing data is shown in the subsequent parentheses if there is any.
CAT: COPD Assessment Test score.
mMRC: modified Medical Research Council dyspnea scale.
CCQ: Clinical COPD Questionnaire score.
defined in Formula 1.
Since the sputum sample for 16S sequencing in normal healthy cohort were collected at multiple time points during 2016–2017, the age for each subject is calculated as the mean value across time points.
Normal healthy cohort
During October 2016 – October 2017, 41 healthy volunteers were examined at the Department of Respiratory Medicine, Shanghai Tenth People’s Hospital, Tongji University, Shanghai, China (Fig 1 and Table 1). The inclusion criteria were: age ≥ 18 years, no use of antibiotics or immunosuppressive medications in the past two months, and no evidence of acute upper respiratory symptoms for the previous four weeks. The exclusion criteria were: history of asthma, other lung disease, heart failure, stroke, pregnancy or lactation, and infectious disease. Each subject was followed up every two months, contributed sputum sample and completed a questionnaire including anthropometric characteristics, education, street address, smoking status, medications used, and outdoor exercise time. The CAT, mMRC, and CCQ (see Abbreviations) are instrument for assessment of COPD status and severity, therefore, not applied to the health volunteers. All subjects provided written informed consent, and the study was approved by the Institutional Review Boards at Shanghai Tenth People’s Hospital, Tongji University, Shanghai, China.
Assessment of respiratory function
Spirometry was performed by clinicians according to standards established by the American Thoracic Society (ATS), using the Jaeger Spirometry Analyzer (Master Screen PFT, Hoechberg, Germany). The respiratory function parameters measured were: FVC and FEV1 (see Abbreviations).
Sputum sample collection and DNA extraction
We collected sputum samples following previous described protocol (Brightling et al., 2001). Oral washes (OW) were performed with 10 ml sterile 0.9% saline before sputum induction. Sputum was induced as given inhaled sequences of 3%, 4%, and 5% saline for 7–15minutes via an ultrasonic nebulizer. The sputum induction procedure was terminated after the expectoration of sputum. Sputum is collected into sterile Petri dishes with phosphate-buffered saline, transferred to sterile collection tubes, and then stored at −80ºC for further analysis. DNA was extracted from sputum using PowerSoil® DNA Isolation Kit (Qiagen, Germantown, MD, USA) according to manufacture provided protocol. In each batch of sputum collection, we placed negative control, which was the sterile Petri dish with phosphate-buffered saline. The negative controls underwent identical procedure as patients’ sample, including storage, DNA extraction, and PCR.
Assessment of Bacteria abundance in sputum
Quantitative polymerase chain reaction (qPCR) using SYBR Green qPCR kit (MilliporeSigma, Burlington, MA, USA) was conducted to assess the abundance of bacteria in sputum samples. Quantifying the absolute count of bacteria is difficult and labor-intensive; instead, we assess the abundance of bacteria relative to the human cells. Two pairs of primers were added into the qPCR system, one pair specific to bacteria 16S rRNA gene, and the other pair specific to human β-actin gene. The ΔΔCt method (Livak and Schmittgen, 2001) was used to calculate the ratio of bacteria 16S rRNA genes relative to human β-actin gene in the DNA extracted from sputum.
Profiling airway microbiome by 16S rRNA sequencing
The 16S rRNA gene amplicon sequencing of the V4 hypervariable region was performed using Illumina HiSeq Fast Run mode (paired-end 250bp). For each PCR reaction, 4 ng of purified template DNA was amplified in a reaction volume of 20ul. Primers were designed to target the V4 region of the 16S rDNA (position 515 of the bacterial 16S rRNA gene to position 806). Each reaction was conducted with denaturation at 98°C for 30 sec, 26 cycles of (98°C × 10 sec, 50°C × 30 sec, 72°C × 30 sec), and a final extension at 72°C for 2 min. PCR amplicon were purified from agarose gel and sequenced on an Illumina HiSeq instrument at a loading concentration of 10pM.
To assess data reproducibility, we created duplicate pairs. In detail, aliquots of the same sputum sample were separated into different tubes, and performed DNA extraction, PCR and sequencing independently, hence, created technical replicates. Importantly, aliquots of each duplicate pair were sequenced in different batches in order to factor in the uncertainty or systematical difference sequencing conditions in our reproducibility assessment.
Data processing and quality control
The raw bacterial 16S rRNA sequencing data (i.e. fastq files) was processed within each batch (i.e. sequencing library). The pair-end reads (i.e., R1 and R2 reads) were merged. The merged reads was then processed for microbiome profiling by QIIME 1.9.1 (Hiergeist et al., 2016). For each sample, the reads were clustered into operational taxonomic units (OTUs). A training dataset for taxonomic assignments was created using a modified NCBI taxonomy from the ‘Isolated named strains 16S’ in the Greengenes database (http://greengenes.secondgenome.com/). This dataset was manually curated by (1) removing strains in ‘Isolated named strains 16S’ that had non-standard taxonomy or that were not members of the domain bacteria, and (2) grouping strain level taxonomy from Greengenes assignments under a single NCBI species assignment. The Ribosomal Database Project (RDP) version 2.4 classifier (http://gordonlab.wustl.edu/SuppData.html) was used to assign taxonomy and OTUs.
We performed quality control (QC) to filter out OTUs and samples of poor quality. At OTU level, we excluded the OTUs with extremely low abundance, i.e. the median proportion across all samples is < 0.001%. As result, 78 OTUs were kept for downstream analysis, including a category of unassigned OTU, which cannot be assigned to any known taxonomic unit. At sample level, we performed principle component analysis (PCA) on the 78 OTU profile data and did a visual check on the first two PCs (Fig S1). A total of 6 outliers (1 in COPD risk cohort and 5 in normal healthy cohort) were excluded. We excluded an additional sample of COPD risk cohort, because it contained much more low abundance OTUs (ie, OTU proportion < 0.001%) than other samples and appeared as an outlier. We ended up with 16S sequencing data for 72 samples in COPD risk cohort, for which only one sample was collected from each subject at one time point, and 242 samples in normal healthy cohort, for which multiple samples were collected for each subject at multiple time points.
To evaluate the quality of 16 sequencing data, a subset (n=40) of the samples in the normal healthy cohort were technically sequenced multiple times with exactly the same protocol. We calculated the Spearman correlation coefficients of the OTU abundance profile for 40 replicated pairs. The correlations were computed across all the 78 OTUs, common OTUs (median proportion > 0.1%; n = 26) and rare OTUs (median proportion < 0.1%; n = 52).
Estimating PM2.5 exposure level
We downloaded PM2.5 data collected at 10 MEP (China Ministry of Ecology and Environment) stations in Shanghai, China, in 2016 and 2017 (http://beijingair.sinaapp.com/), which matched to cohort study period. The latitude and longitude of each station is also obtained. We calculated the mean of hourly measured PM2.5 concentrations as the daily measurement at each station. The street address of the participant was retrieved from the questionnaire and translated into latitude and longitude using google map. PM2.5 concentration of the closest station to each individual was treated as his/her exposure level. We considered the average daily PM2.5 concentrations of the 14 days prior to the sputum collection date (including the collection day) in this study.
Association between OTU abundance and PM2.5 exposure
To explore whether PM2.5 exposure influenced the relative abundance (i.e. proportion) of the OTUs, we perform an association study on the pooled data of both COPD risk and normal healthy cohorts with the R package variancePartition (Hoffman and Schadt, 2016):
| (model A1) |
where E is a vector of 14 variables corresponding to PM2.5 exposure of 1~14 days prior to sputum collection date and E is treated as fixed effect; XR and XF are vectors of covariates: XR is the vector of covariates coded as random effects, including cohort, batch, individual, sex, smoking status, collection date, and PM2.5 station; XF is the vector of covariates coded as fixed effects including age, height and weight; ε is the error term. The variances explained by different factors were visualized by violin plot (Fig S2). For each OTU, the p-value of the overall influence of the PM2.5 exposure during the 14 days prior to sputum collection was derived from a likelihood ratio test of 14 degree of freedom (LRT14df) using nested models, where model A1 was compare to the model A0:
Overall significance of PM2.5 exposure’s effect on OTUs was evaluated using a one-side Kolmogorov-Smirnov (K-S) test, where the distribution of the p-values generated for term E in nest models was compared to a uniform distribution. Given strong correlations between the proportions of the OTUs, the p-values were not independent. An iterative pruning procedure was performed to produce a list of independent p-values in order to ensure a valid K-S test. We compute the Pearson correlation between all pair of OTUs adjusted for covariates (using model A0), and then started the pruning. First, we define an empty vector, C. Second, the OTUs were sorted based on their LRT14df p-values (derived from nested models: model A1 vs model A0) ascendingly, and the list is termed as S. Third, the actually pruning was executed in following steps: 1) picked up the OTU with smallest p-value in S, and term it as OTUC; 2) append OTUC to C; 3) remove all the OTUs in S (if any) whose Pearson correlation coefficient with OTUc ≥ 0.2; repeat 1) to 3) if S is not empty. After the pruning, C contains independent OTUs, whose LRT14df p-values subsequently entered the K-S test.
Association between respiratory function traits and OTU abundance
In COPD risk cohort, we considered the effect of 78 OTUs on respiratory function traits, including FEV1, FEV1% predicted, FVC, FVC% predicted, FEV1/FVC ratio, CAT score, mMRC score and CCQ score. In a linear mixed model (LMM), each trait was regressed on each OTU proportion.
| (model B1) |
where Traiti is the ith respiratory function trait, OTUj is the jth OTU’s proportion. The denotation of XR, XF and ε are the same as in model A1. The p-value for each OTU was derived from a likelihood ratio test of 1 degree of freedom (LRT1df) using nested models, where model B1 was compared to model B0: Traiti = XR + XF + ε. For each respiratory function trait, the overall significance of the effect of all the OTUs was also evaluated by a K-S test (after OTU pruning).
Results
Demographic characteristics and bacteria load in bronchial tree
A total of 74 individuals and 41 individuals from the COPD risk cohort and normal healthy cohort, respectively was included in this study (Table 1). The average age of COPD risk cohort was 59 years old, much older than normal healthy cohort. Because of such systematical difference, we did not directly compare the two cohorts in this study in terms of sputum microbiome profile.
The abundance of bacteria in sputum relative to human cells (termed as R) was quantified by qPCR
| (Formula 1). |
The average R value was 119.5 (Table 1), and R was significantly associated with FEV1/FVC ratio (p = 0.0038, Table 2), which is an essential respiratory function trait for COPD diagnosis. Interestingly, the association is positive, indicating individuals with better lung function (i.e., higher FEV1/FVC ratio) had higher bacteria load in bronchial tree.
Table 2.
Association of respiratory function traits with airway bacteria load
| Crude model | Adjusted modela | |||||
|---|---|---|---|---|---|---|
| Traitb | Estimate | Std. Err. | P-value | Estimate | Std. Err. | P-value |
| FEV1 (L) | 0.00031 | 0.00026 | 0.23 | 0.00025 | 0.00015 | 0.11 |
| FEV1% predicted | 0.0026 | 0.0079 | 0.74 | 0.0081 | 0.0065 | 0.22 |
| FVC (L) | −0.00043 | 0.0018 | 0.81 | −0.00083 | 0.0019 | 0.65 |
| FVC% predicted | −0.0040 | 0.0095 | 0.68 | −0.0011 | 0.0089 | 0.91 |
| FEV1/FVC ratio (%) | 0.0076 | 0.004 | 0.061 | 0.010 | 0.0035 | 0.0038 |
| CAT | −0.00071 | 0.0021 | 0.74 | −0.00065 | 0.0019 | 0.74 |
| mMRC | −0.00025 | 0.00034 | 0.46 | −0.00015 | 0.00032 | 0.64 |
| CCQ | −0.0028 | 0.003 | 0.37 | −0.0032 | 0.0028 | 0.26 |
The association test was performed in the COPD risk cohort (n = 74). In the linear mixed model, age, gender, height and weight were adjusted as fixed effect while collection date was adjusted as random effect.
FEV1, forced expiratory volume in 1 second; FVC, forced vital capacity; CAT, COPD Assessment Test; mMRC, modified British Medical Research Council; CCQ, Clinical COPD Questionnaire.
Sputum microbiome profile
16S rRNA-Seq data for 72 sputum samples from COPD risk cohort and 242 sputum samples from normal healthy cohort passed QC filters, and 78 OTUs were identified (Fig 2A and Table S1). Majority of the OTUs was of very small proportion (i.e., ≤1%) in airway microbiome, and the most abundant genera were Veillonella, Prevotella and Neisseria. Substantial inter-individual variation was observed in airway microbiome (Fig 2A). The OTU proportion of duplicate pairs were highly correlated, where median Spearman r2= 0.9295 (Fig 2B). On relative common OTUs (ie, median proportion ≥ 0.1%), the duplicate pairs were correlated with median Spearman r2= 0.9750 (Fig 2B).
Figure 2. Airway microbiome profile.

(A). OTU proportions in each sample. Each stacked color bar indicates the percentages of OTUs in each sample. Sample IDs from COPD risk cohort and normal healthy cohort are indicated by orange and blue labels beneath the color bars. The OTUs of low abundance (median percentage < 1%) are collapsed into one category “other.otus”. More detailed summary statistics of all the OTUs for the two cohorts are reported in Table S1. (B). Reproducibility of OTU abundance in technical replicates from normal healthy cohort. The reproducibility was measured as Spearman correlation coefficient between technical replicates across all OTUs (n = 78), common OTUs (median percentage > 0.1%; n = 26) and rare OTUs (median percentage < 0.1%; n = 52).
PM2.5 exposure level
For each sputum sample, we quantified the subject’s exposure level for 14 days prior to the follow-up date. The PM2.5 level in Shanghai varied greatly during the one-year study period (Fig 3A), where the average daily PM concentration was high in winter than in summer. The follow-up dates of normal healthy cohort (blue asterisks, Fig 3A) were well-spread, and both good and poor air quality periods were sampled. The follow-up of COPD risk cohort were conducted on two days (orange asterisks, Fig 3A). The PM2.5 concentrations measured by MEP station in Shanghai were highly correlated (Fig 3B and 3C), for example, the correlation of Hong-Kou and Yang-Pu stations was as high as 97%. In consequence, the COPD risk cohort subjects were estimated to have very similar exposure level with very limited inter-individual variation, further the sample size of the COPD risk cohort was relatively small, hence, we did not examine the association between PM2.5 exposure and respiratory functions.
Figure 3. PM2.5 measured at 5 surveillance stations in Shanghai.

(A) Average daily PM2.5 concentration at 5 surveillance stations in Shanghai, China, from October 1, 2016 to September 30, 2017. The dates on which sputum samples were collected for COPD risk cohort and normal health cohort are marked by orange and blue stars respectively. (B) The name, latitude and longitude of each station. (C) The Pearson correlation coefficients of PM 2.5 measurements between pairs of the surveillance stations across the time period shown in (A) are visualized by heatmap. (C) The name, latitude and longitude of each station.
Airway OTU profile was significantly associated with PM2.5 exposure
Using variancePartition methods (Hoffman and Schadt, 2016), we evaluated the influence of PM2.5 exposure (on each date up to 14 days prior to sputum collection) and covariates (Fig S2). The most influential factor was sequencing batch followed by exposure occurred 7 and 6 days prior to sputum collection. Interestingly, age, height, weight, sex, sputum collection date and smoking status had less influence (Fig S2). Focusing on each of the 14 days’ exposure level (Fig 4A), exposure occurred ≥ 6 days prior to collection had much stronger impact on airway microbiome that exposure occurred day 1 to day 3 prior to sputum collection. Further, the effect of PM2.5 was long lasting. The exposure occurred 14 days before sputum collection still had substantial effect on airway microbiome, even controlled the exposure of day 1 to day 13 in the multi-variate model (model A1). Overall, airway OTUs were significantly associated with PM2.5 exposure (K-S test p=4.48e-11, Fig 4B and Table S2), and the affected OTUs were not the most abundant one (Fig S3).
Figure 4. Association of OTU proportions with PM2.5 exposure.

(A) The variance of each OTU proportion (n = 78) explained by the PM2.5 exposure of the 14 days prior to sputum collection day (inclusive) with a linear mixed model, which was fitted based on the combined data from both COPD risk cohort and normal health cohort (n = 314 samples in total after QC). The results are visualized by violin plot. A violin plot is a method of visualizing the distribution of numeric data, similar to box plot with a rotated kernel density plot on each side. Inside the violin plot is the regular box plot with outliers indicated by dots. (B) Q-Q plot of the p-values (on -log10 scale) in the association test between the OTU proportion and PM2.5 exposure. The overall significance of the enrichment of small p-values was evaluated by a one-side Kolmogorov–Smirnov (K-S) test with respect to a uniform distribution on [0, 1] as null. The blue line has a slop of 1. The dashed line indicates the significance level after Bonferroni correction (0.05/78 = 6.41×10−4). The OTUs that passed the significance level are highlighted in red.
Respiratory function traits were associated with airway microbiome
We surveyed the association between each of the respiratory function traits and overall airway microbiome using K-S test. Firstly, we focused on key characters in COPD diagnosis: FEV1% predicted and FEV1/FVC ratio. FEV1% predicted was significantly associated with airway microbiome (p = 0.013, Fig 5A and Table S3). In contrast, FEV1/FVC ratio was not associated with OTU proportion (Fig 5B), although it was significantly associated with airway bacteria load (Table 2). Secondly, several additional respiratory function measures (Fig S4) were in significant association with airway microbiome, including FEV1 (p = 0.017) and CAT (p = 0.0042).
Figure 5. Q-Q plot of the p-values in the association test between respiratory function traits and OTUs.
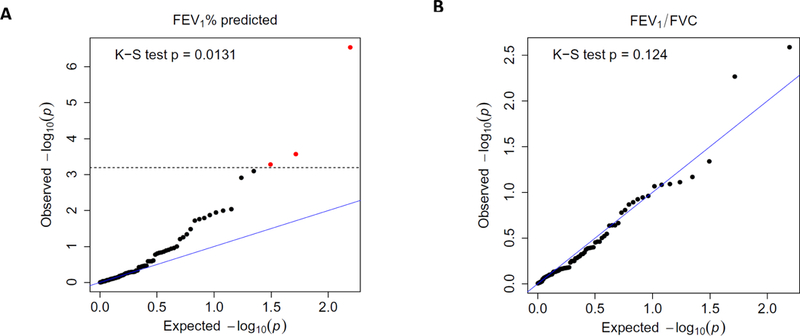
The x- and y-axis of the plot present the p-values of the association test between respiratory function trait and OTUs. The x-axis is the expected p-value under the null hypothesis that there is no association, and the y-axis is the p-value observed on the actual data. All p-values are –log10 transformed. (A) FEV1% predicted; (B) FEV1/FVC ratio (%). In the linear mixed model, age, gender, height, weight, and smoke were adjusted as fixed effect while 16S sequencing batch and sputum collection date were adjusted as random effect. The overall significance of the enrichment of small p-values was evaluated by a one-side Kolmogorov–Smirnov (K-S) test with respect to a uniform distribution on [0, 1] as null. The blue line has a slop of 1. The dashed line indicates the significance level after Bonferroni correction (0.05/78 = 6.41×10−4). The OTUs that passed the significance level are highlighted in red.
Discussions
In the past two decades, multiple epidemiology studies have linked PM2.5 exposure and declines in respiratory functions (Xing et al., 2016). Microbiome is a constitutive component of human’s airway micro-environment and closely interacts with host immune systems. To our knowledge, we are the first to demonstrate the impact of PM2.5 on airway microbiome, and reported the link between airway microbiome and respiratory functions. Key findings of this study: (1) FEV1/FVC, an important respiratory function trait and a diagnosis criterion of COPD, is positively associated with sputum bacteria load (p = 0.0038). (2) Multiple respiratory function traits are associated with airway microbiome profile, for example, FEV1% predicted (Fig 5A, p = 0.013). (3) Airway microbiome was profoundly influenced by PM2.5 exposure (Fig 4B, p = 4.48e-11). Importantly, the strongest impact attributed to exposure occurred ≥ 6 days ago (Fig 4A and Fig S2). Further, PM2.5 exposure has long-lasting effect, and exposure occurred 14 days ago was still associated with the airway microbiome (Fig 4A and Fig S2).
We compared published respiratory tract microbiome to that found in our sample. On 64 individuals, Morris et al used 16S rDNA sequencing technology to profile bacteria from BAL which mainly represents lower lung (Morris et al., 2013). Eight genera (Streptococcus, Veillonella, Prevotella, Fusobacterium, Neisseria, Actinomyces, Porphyromonas, Gemella) were universally found in BAL sample (Dickson et al., 2015), and importantly, all these genera were detected in our data. Dickson et al studied BAL from 15 healthy individuals and detected 12 unique bacteria family, among which 9 were found in our sample. In this study, the 10 most abundant genera are Veillonella, Prevotella, Neisseria, Streptococcus, Haemophilus, Porphyromonas, Fusobacterium, Leptotrichia, Rothia, Actinomyces (Table S1), and all these 10 genera were reported by at one of the BAL microbiome studies (Dickson et al., 2015; Morris et al., 2013).
There are varieties of bacteria on ambient particulate matters (Cao et al., 2014). But the alternation of sputum microbiome we detected is unlikely be simply addition of PM-attached bacteria onto the indigenous airway microbiota. In fact, the PM-attached bacteria are distinct from those of any body site, as detailed by HMP (Blaser, 2010). The PM-attached bacteria are primarily from soil, followed by fecal, freshwater and marine sources (Cao et al., 2014), and not directly related to bacteria that habitat in human airway. PM in Chinese cities contains sulfate, ammonium, nitrate, and organic matter, and altogether they represent 80% of PM2.5 (w/w) (Cao et al., 2014). A plausible mechanism that PM2.5 exposure perturbs airway microbiome could be that the component of PM2.5 (organic- and inorganic matters and bacteria) influence the micro-environment inside the bronchial tree, and in turn affect the microbiome.
A major confounder in human microbiome study is medication (especially antibiotic use). Microbiome study on COPD patients is particularly difficult, given majority of COPD patients receive medication (including antibiotics) to maintain and manage symptoms, and such medication profoundly impacts the microbiome ecosystems in the airway. Unfortunately, this confounder is also hard to control for, since the healthy controls have distinct medication profile comparing to patients. In other words, the confounder perfectly correlates with endpoint (case/control status), making it impossible to distinguish between the effect of disease and that of the confounder. Herein, we only studied non-COPD individuals who do not receive medication in a regular basis.
The study protocol showed high reproducibility (median r2=92.95%, Fig 2B). It should be noted, the association between airway microbiome and respiratory functions does not imply causality. Nevertheless, the microbiome at least can serve as biomarkers reflecting the health status of airway ecosystem. Most importantly, we showed the ecosystem can be perturbed by PM2.5 exposure. Since microbiome is in close inter-talk with airway immune system, the perturbation induced by PM2.5 exposure could be an etiological pathway towards respiratory function decline. Airway microbiome can be assess from sputum or BAL (Wang et al., 2017b). Sputum is the coughed-up material (phlegm) from the lower airways (trachea and bronchi), and may also contain materials from upper airways. The pattern of airway microbiome is “transient but not resident (TBNR)” (Saeedi et al., 2015), reflecting its complex and dynamic nature. The migration of microbes along the airway is bidirectional given there are no physical barriers. 16S rRNA profiling on sputum describes the bacteria ecosystem of the entire airway. In contract, BAL requires much more invasive procedure to collect (comparing to sputum) and has low bacteria content (Charlson et al., 2012; Erb-Downward et al., 2011), hence, is less suitable for airway microbiome study of moderate-to-large sample size.
The current study reported the short-term effect of PM2.5 exposure. With the cohort established and still on-going, we are in a position to examine the long term impact of PM2.5 on airway microbiome in the future. Besides PM, a variety of pollutants (e.g, O3) exists in ambient air, but less studied in terms of their adverse effects on respiratory functions and influence on airway microbiome.
In summary, to our knowledge, this paper for the first time reported the impact of PM2.5 exposure on airway microbiome, as well as the link between airway microbiome and respiratory functions. Given microbiota colonizes and inter-talks with the host cells in the bronchial tree, it may modulate to epithelial integrity and immune-regulation. The results advanced the understanding on the impact of PM2.5 exposure on airway ecosystem, and shed light on possible biomarkers for pollution-induced airway injury.
Supplementary Material
Highlights.
On human cohorts, we demonstrate the impact of PM2.5 exposure on respiratory tract microbiome.
The strongest impact is attributed to exposure occurred ≥ 6 days prior to microbiome collection.
This study reports the link between airway microbiome and human respiratory functions.
Acknowledgements
This work is partially supported by National Natural Science Foundation of China (Grant No. 21477087, 91643201, 21876134), the Ministry of Science and Technology of China (Grant No. 2016YFC0206507) and NIH-NIEHS (1R01ES029212–01).
Abbreviations
- COPD
chronic obstructive pulmonary disease
- ROS
reactive oxygen species
- PAH
polycyclic aromatic hydrocarbons
- TF
transcription factor
- HMP
human microbiome project
- BAL
bronchoalveolar lavage
- FVC
forced vital capacity
- FEV1
forced expiratory volume in 1st second
- OUT
operational taxonomic units
- CAT
COPD assessment test
- mMRC
British Medical Research Council questionnaire
- CCQ
clinical COPD questionnaire
- PCR
polymerase chain reaction
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests
No competing interests declared
References
- Bahr TM, et al. , 2013. Peripheral blood mononuclear cell gene expression in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 49, 316–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes PJ, 2014. Cellular and molecular mechanisms of chronic obstructive pulmonary disease. Clin Chest Med 35, 71–86. [DOI] [PubMed] [Google Scholar]
- Beelen R, et al. , 2014. Effects of long-term exposure to air pollution on natural-cause mortality: an analysis of 22 European cohorts within the multicentre ESCAPE project. Lancet 383, 785–95. [DOI] [PubMed] [Google Scholar]
- Bell ML, et al. , 2013. Associations of PM Constituents and Sources with Hospital Admissions: Analysis of Four Counties in Connecticut and Massachusetts (USA) for Persons >/= 65 Years of Age. Environ Health Perspect [DOI] [PMC free article] [PubMed]
- Blaser MJ, 2010. Harnessing the power of the human microbiome. Proc Natl Acad Sci U S A 107, 6125–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brightling CE, et al. , 2001. Induced sputum and other outcome measures in chronic obstructive pulmonary disease: safety and repeatability. Respir Med 95, 999–1002. [DOI] [PubMed] [Google Scholar]
- Cameron SJ, et al. , 2016. Metagenomic Sequencing of the Chronic Obstructive Pulmonary Disease Upper Bronchial Tract Microbiome Reveals Functional Changes Associated with Disease Severity. PLoS One 11, e0149095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao C, et al. , 2014. Inhalable microorganisms in Beijing’s PM2.5 and PM10 pollutants during a severe smog event. Environ Sci Technol 48, 1499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlson ES, et al. , 2012. Lung-enriched organisms and aberrant bacterial and fungal respiratory microbiota after lung transplant. Am J Respir Crit Care Med 186, 536–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AJ, et al. , 2017. Estimates and 25-year trends of the global burden of disease attributable to ambient air pollution: an analysis of data from the Global Burden of Diseases Study 2015. Lancet 389, 1907–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira BF, et al. , 2012. Risk assessment of PM(2.5) to child residents in Brazilian Amazon region with biofuel production. Environ Health 11, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson RP, et al. , 2015. Spatial Variation in the Healthy Human Lung Microbiome and the Adapted Island Model of Lung Biogeography. Ann Am Thorac Soc 12, 821–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominici F, et al. , 2006. Fine particulate air pollution and hospital admission for cardiovascular and respiratory diseases. JAMA 295, 1127–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson K, et al. , 1996. Free radical activity associated with the surface of particles: a unifying factor in determining biological activity? Toxicol Lett 88, 293–8. [DOI] [PubMed] [Google Scholar]
- Erb-Downward JR, et al. , 2011. Analysis of the lung microbiome in the “healthy” smoker and in COPD. PLoS One 6, e16384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwell LL, et al. , 2002. Particle-induced oxidative damage is ameliorated by pulmonary antioxidants. Free Radic Biol Med 32, 898–905. [DOI] [PubMed] [Google Scholar]
- Gripenback S, et al. , 2005. Accumulation of eosinophils and T-lymphocytes in the lungs after exposure to pinewood dust. Eur Respir J 25, 118–24. [DOI] [PubMed] [Google Scholar]
- Hayashi H, et al. , 2002. Phylogenetic analysis of the human gut microbiota using 16S rDNA clone libraries and strictly anaerobic culture-based methods. Microbiol Immunol 46, 535–48. [DOI] [PubMed] [Google Scholar]
- Hiergeist A, et al. , 2016. Multicenter quality assessment of 16S ribosomal DNA-sequencing for microbiome analyses reveals high inter-center variability. Int J Med Microbiol 306, 334–42. [DOI] [PubMed] [Google Scholar]
- Hoffman GE, Schadt EE, 2016. variancePartition: interpreting drivers of variation in complex gene expression studies. BMC Bioinformatics 17, 483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beijing air quality historical data http://beijingair.sinaapp.com/
- The Gordon Lab data release http://gordonlab.wustl.edu/SuppData.html.
- The Greengenes Database http://greengenes.secondgenome.com/
- The Global Initiative for Chronic Obstructive Lung Disease (GOLD) http://www.goldcopd.org.
- Apple developer kit https://developer.apple.com/healthkit/
- Huang YJ, et al. , 2013. The role of the lung microbiome in health and disease. A National Heart, Lung, and Blood Institute workshop report. Am J Respir Crit Care Med 187, 1382–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh M, et al. , 2006. Relationships between air pollution and preterm birth in California. Paediatr Perinat Epidemiol 20, 454–61. [DOI] [PubMed] [Google Scholar]
- Kelly FJ, 2003. Oxidative stress: its role in air pollution and adverse health effects. Occup Environ Med 60, 612–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis TC, et al. , 2005. Air pollution-associated changes in lung function among asthmatic children in Detroit. Environ Health Perspect 113, 1068–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD, 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–8. [DOI] [PubMed] [Google Scholar]
- Martinelli N, et al. , 2012. Access rate to the emergency department for venous thromboembolism in relationship with coarse and fine particulate matter air pollution. PLoS One 7, e34831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris A, et al. , 2013. Comparison of the respiratory microbiome in healthy nonsmokers and smokers. Am J Respir Crit Care Med 187, 1067–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostro B, et al. , 2006. Fine particulate air pollution and mortality in nine California counties: results from CALFINE. Environ Health Perspect 114, 29–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman I, 2005. Oxidative stress in pathogenesis of chronic obstructive pulmonary disease: cellular and molecular mechanisms. Cell Biochem Biophys 43, 167–88. [DOI] [PubMed] [Google Scholar]
- Rahman I, MacNee W, 1996. Role of oxidants/antioxidants in smoking-induced lung diseases. Free Radic Biol Med 21, 669–81. [DOI] [PubMed] [Google Scholar]
- Saeedi P, et al. , 2015. The transient but not resident (TBNR) microbiome: a Yin Yang model for lung immune system. Inhal Toxicol 27, 451–61. [DOI] [PubMed] [Google Scholar]
- Schwartz J, et al. , 1996. Is daily mortality associated specifically with fine particles? J Air Waste Manag Assoc 46, 927–39. [DOI] [PubMed] [Google Scholar]
- Sigaud S, et al. , 2007. Air pollution particles diminish bacterial clearance in the primed lungs of mice. Toxicol Appl Pharmacol 223, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suau A, et al. , 1999. Direct analysis of genes encoding 16S rRNA from complex communities reveals many novel molecular species within the human gut. Appl Environ Microbiol 65, 4799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valavanidis A, et al. , 2005. Electron paramagnetic resonance study of the generation of reactive oxygen species catalysed by transition metals and quinoid redox cycling by inhalable ambient particulate matter. Redox Rep 10, 37–51. [DOI] [PubMed] [Google Scholar]
- Wang J, et al. , 2017a. Particulate matter pollution over China and the effects of control policies. Sci Total Environ 584–585, 426–447. [DOI] [PubMed] [Google Scholar]
- Wang L, et al. , 2017b. Role of the Lung Microbiome in the Pathogenesis of Chronic Obstructive Pulmonary Disease. Chin Med J (Engl) 130, 2107–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong TY, 2017. Smog induces oxidative stress and microbiota disruption. J Food Drug Anal 25, 235–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing YF, et al. , 2016. The impact of PM2.5 on the human respiratory system. J Thorac Dis 8, E69–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, et al. , 2016. The meteorological modulation on PM2.5 interannual oscillation during 2013 to 2015 in Shanghai, China. Sci Total Environ 572, 1138–1149. [DOI] [PubMed] [Google Scholar]
- Zanobetti A, et al. , 2009. Fine particulate air pollution and its components in association with cause-specific emergency admissions. Environ Health 8, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.