

Abstract
Heart failure is associated with mitochondrial dysfunction so that restoring or improving mitochondrial health is of therapeutic importance. Recently, reduction in NAD+ levels and NAD+-mediated deacetylase activity has been recognized as negative regulators of mitochondrial function. Using a cardiac specific KLF4 deficient mouse line that is sensitive to stress, we found mitochondrial protein hyperacetylation coupled with reduced Sirt3 and NAD+ levels in the heart before stress, suggesting that the KLF4-deficient heart is predisposed to NAD+-associated defects. Further, we demonstrated that short-term administration of Nicotinamide Mononucleotide (NMN) successfully protected the mutant mice from pressure overload-induced heart failure. Mechanically, we showed that NMN preserved mitochondrial ultrastructure, reduced ROS and prevented cell death in the heart. In cultured cardiomyocytes, NMN treatment significantly increased long-chain fatty acid oxidation despite no direct effect on pyruvate oxidation. Collectively, these results provide cogent evidence that hyperacetylation of mitochondrial proteins is critical in the pathogenesis of cardiac disease and that administration of NMN may serve as a promising therapy.
Keywords: NAD, Protein hyperacetylation, Mitochondria, Heart failure, Pressure overload
1. Introduction
The adult mammalian heart requires a continuous supply of ATP to sustain contraction. The majority of the heart’s ATP (> 95%) is produced from mitochondria through oxidative phosphorylation (OXPHOS) [1,2]. Given the fact that cardiac ATP reserve is very limited, a robust uninterrupted ATP supply from mitochondrial is critical to maintain cardiac function and failure to do so contributes to disease states such as heart failure [3–5]. The importance of this adaptation is underscored by the severe cardiomyopathy seen in patients with inborn errors of the mitochondrial fatty acid oxidation (FAO) pathway [6]. In adulthood, abnormalities in FAO and other mitochondrial functions also occur with aging and a variety of acquired pathologic conditions (ischemia, hypertension, valvular disease, etc.) leading to heart failure [2,7,8]. Accumulating experimental evidence from animal studies identifies reduced cardiac oxidative metabolism as a signature of heart dysfunction and failure [9]. Clinical studies also find the myocardial [PCr]/[ATP] ratio correlates with heart failure severity and is a strong predictor of cardiovascular mortality [10]. To date, mitochondrial dysfunction has been recognized as critical for the pathogenesis of cardiac diseases and a promising therapeutic target [11,12].
In addition to inborn genetic defects, mitochondrial dysfunction can result from accumulated damage at the mitochondrial genomic DNA (mtDNA), protein, and lipid levels due to high oxidative environment inside the mitochondria. However, clinical trials with a variety of antioxidant strategies have failed to show beneficial effects [13]. In the last decade, mitochondrial protein acetylation has emerged as an important mechanism that affects mitochondrial homeostasis and organ function. Lysine-acetylation is a revisable posttranslational modification of proteins that is determined by the balance between acetyltransferase and deacetylase activity. In mitochondria, there are three NAD+-dependent deacetylases, namely Sirt3, Sirt4 and Sirt5, forming a Sirtuin network that mediates the acetylation of mitochondrial proteins and subsequently regulates mitochondrial function [14]. Given that the reducing equivalents NADH (and FADH2) drives the mitochondrial electron transfer chain (ETC), the equilibrium of NADH/NAD+ is critical for mitochondrial OXPHOS. Therefore, NAD+ is at center of OXPHOS and protein acetylation, making it an attractive target for heart failure treatment [12].
Kruppel-like factors (KLF) are a subclass of the zinc-finger transcription factors that bind a consensus 5′-C(A/T)CCC-3′ motif in the promoters and enhancers of various genes and regulate critical cellular processes, such as cell proliferation, differentiation, survival, apoptosis, metabolism and stemness. Members of the KLF family play important roles in multiple cell types within the cardiovascular system [15–20]. In particular, our recent studies have identified Kruppel-like factor 4 (KLF4) as a critical regulator of cardiac mitochondrial homeostasis [20]. We previously showed that mice bearing cardiac deficiency of KLF4 were sensitized to pressure overload-induced heart failure due to disruption of mitochondrial homeostasis upon stress [18,20]. However, the underlying initial defects leading to such rapid cardiac dysfunction remain unknown. Although mitochondrial function is reduced in the KLF4-deficient hearts at baseline, transcriptomic studies did not reveal big changes at transcriptional level (82 genes with FDR < 0.05 at baseline) [20]. These results strongly suggest that the posttranslational mechanisms are important contributors to the mitochondrial defects. Here we show that hyperacetylation of mitochondrial proteins in the KLF4-deficient hearts might be the underlying defects at baseline and administration of Nicotinamide Mononucleotide (NMN), a precursor of NAD+, successfully preserved mitochondrial homeostasis and rescued heart function during pressure overload.
2. Methods
2.1. Animal models
Mice with cardiac-specific deficiency of Klf4 (Myh6-Cre:Klf4fl/fl mice, designated CM-K4KO) have been described previously, and the Myh6-Cre line (designated MHC-Cre) was used as genetic control [20]. All mice are on a C57BL/6J background. Mice were housed in a temperature- and humidity-controlled specific pathogen–free facility with a 12-hour-light/dark cycle and ad libitum access to water and standard laboratory rodent chow. Transverse aortic constriction (TAC) model and echocardiography were performed as described previously [20]. In brief, mice were anaesthetized by ketamine xylazine cocktail (100 μL i.p.) for TAC surgery and buprenorphine was administrated daily (i.p.) for first 3-days post-TAC. For echocardiography, mice were anaesthetized by inhalation of 1% isoflurane vaporized in 100% oxygen. In mice receiving NAD+ supplement, NMN (β-Nicotinamide Mononucleotide, Sigma, N3501) was injected intraperitoneally (i.p.) at 500 mg/kg/day (in PBS). Vehicle group received equal volume of PBS.
2.2. RNA extraction and qPCR
Tissue samples were homogenized in QIAzol lysis reagent (Qiagen, 79306) with a TissueLyser (Qiagen). Cell samples were directly dissolved in QIAzol lysis reagent. Total RNA was extracted, treated with DNase I (Life Technologies, 18068015), purified using the RNeasy MinElute Cleanup Kit (Qiagen, 74204), and reverse transcribed to complementary DNA using the iScript Reverse Transcription Kit (Bio-Rad, 170–8841). qPCR was performed with either the TaqMan method (Roche Universal ProbeLibrary System) or the SYBR green method on a ViiA 7 Real-Time PCR System (Applied Biosystems). Relative expression was calculated using the ΔΔCt method with normalization to Gapdh.
2.3. Mitochondrial isolation and Blue-Native PAGE
Cardiac mitochondria were isolated using differential centrifugation as described previously [20]. In brief, heart from 3–4-month-old mouse was excised, washed in ice-cold Chappell-Perry buffer, homogenized with a polytron homogenizer and subjected to two-step differential centrifugation (500 g and 3000 g). Mitochondrial pellet from 3000 g centrifugation was washed and subjected to further experiments. Mitochondria from cultured cells were isolated using the Mitochondria Isolation Kit for Culture Cells (ThermoFisher Scientific, 89874). To assess the integrity of mitochondrial respiration complexes, mitochondrial protein was solubilized with 1% DDM and separated in 4–16% NativePAGE™ Bis-Tris Gel (ThermoFisher Scientific, BN2005, BN1002BOX).
2.4. Immunoprecipitation and Western blot
Immunoprecipitation of mitochondrial proteins was performed using a Mitochondrial Protein Immunoprecipitation Kit (Sigma MTP001). Mitochondria were dissolved in Mitochondrial Protein IP Buffer containing 1% DDM. Immunoprecipitation of acetylated protein was done with acetyl-Lysine antibody (Cell Signaling, 9441) and Protein A + G magnetic beads (Millipore, 16–663), followed by Western blot analysis. Immunoprecipitation samples were boiled in 1× Laemmli Sample Buffer and subjected to SDS-PAGE and Western blot analysis for SOD2 (Abcam, ab13533), CypD (Santa Cruz, sc-376061) and LCAD (Abcam, ab128566). Total mitochondrial protein was solubilized with 2% CHAPS (Sigma-Aldrich, C5070), boiled in 1× Laemmli Sample Buffer (Bio-Rad), and subjected to SDS-PAGE and Western blot analysis with antibodies for acetyl-Lysine and Sirt3 (Cell Signaling, 9441, 5490). Tom20, VDAC1 or CoxIV was blotted as loading control for mitochondrial samples (Cell Signaling, 42460, 4661, 4s850).
2.5. Primary cardiomyocyte culture and Seahorse assay
Neonatal rat ventricular myocytes (NRVM) were isolated from day-2 Sprague Dawley rats and cultured as previously described [20]. After 36 h of initial culture, cells were transferred to serum free DMEM containing 1% ITS Liquid Media Supplement (Sigma I3146) and treated with 2 mmol/L NMN for 24–48 h. Mitochondrial respiration rate was assessed using an Agilent Seahorse XFp Extracellular Flux Analyzer with the Mito Stress Test Kit (Agilent). Pyruvate + glucose was used as standard substrates to interrogate mitochondrial respiration capacity. BSA-conjugated palmitate (Agilent, 102720–100) was used as long-chain fatty acid substrate to interrogate the fatty acid oxidation pathway.
2.6. Transmission electron microscopy (EM)
Small pieces of tissue from the LV free wall were fixed by sequential immersion in triple aldehyde-DMSO, ferrocyanide-reduced osmium tetroxide, and acidified uranyl acetate; dehydrated in ascending concentrations of ethanol; passed through propylene oxide; and embedded in Poly/Bed resin (Polysciences Inc., 21844-1). Thin sections were sequentially stained with acidified uranyl acetate, followed by a modification of Sato’s triple lead stain, and examined with a JEOL 1200EX electron microscope.
2.7. Histology and biochemistry
Cardiac tissue samples were fixed in 10% neutralized formalin and embedded with paraffin following standard protocol. TUNEL staining was performed with the ApopTag Peroxidase In Situ Apoptosis Detection Kit following manufacturer’s protocol (Millipore, S7100) [18]. DHE staining was performed with OCT compound-embedded fresh frozen sections in argon-buffered oxygen-free chambers [20]. Microscopic images were analyzed using ImagePro software for quantification. To measure myocardial NAD+ levels, hearts were freeze-clamped and powderized in liquid nitrogen before subjected to the NAD + assay using a NAD/NADH Quantitation Colorimetric Kit (BioVision, K337).
2.8. Statistics
Results are presented as mean ± SEM. Two-tailed Student’s t-test was used to compare the differences between two groups. One-way ANOVA and Bonferroni post-test was used for multiple comparisons. Statistical significance was defined as P < 0.05.
2.9. Study approval
The animal experiments were performed conform the NIH guide-lines (Guide for the care and use of laboratory animals). All animal studies were approved by the Institutional Animal Care and Use Committee of Case Western Reserve University.
3. Results
3.1. Hyperacetylation of mitochondrial proteins in KLF4-deficient hearts
We previously reported that cardiac KLF4-deficient mice (CM-K4KO) developed acute heart failure and death in response to pressure overload induced by transverse aortic constriction (TAC) due to disrupted mitochondrial homeostasis [18,20]. Consistently, the transcriptomic study revealed ∼4000 differentially expressed genes (FDR < 0.05) between the MHC-Cre and CM-K4KO groups after 3-day of TAC, underscoring the sharp difference observed in cardiac function upon stress [20]. However, at baseline, only ∼80 genes were identified as being differentially expressed between the MHC-Cre and CM-K4KO groups suggesting minimal changes at the transcriptional level [20]. However, we did observe reduced cardiac mitochondrial function at baseline despite overall heart function is normal in those young adult mice (age: 8–16 weeks) [20]. Furthermore, these mutant mice gradually developed age-related cardiomyopathy coupled with mitochondrial degeneration [20]. These data suggest that posttranslational mechanisms might contribute to the mitochondrial defects.
We first analyzed the mitochondrial electron transport chain (ETC) complexes by Blue-Native-PAGE (BN-PAGE) and found no difference in ETC complex assembly between the CM-K4KO and MHC-Cre groups at baseline (Fig. 1A). As protein acetylation has been shown to negatively affect mitochondrial function, we subjected cardiac mitochondrial proteins to Western blot analysis for acetyl-Lysine to assess overall protein acetylation levels in the mitochondria. As shown in Fig. 1B, there was marked increase of protein acetylation in the CM-K4KO group. Pressure overload induced by transverse aortic constriction (TAC) further enhanced mitochondrial protein acetylation and the same hyperacetylation in CM-K4KO group persisted even after TAC (Fig. 1C). Such hyperacetylation of mitochondrial proteins was also associated with TAC-induced heart failure in C57BL6 WT mice (Fig. 1D), suggesting a plausible relationship between hyperacetylation of cardiac mitochondrial proteins and cardiac dysfunction.
Fig. 1.

Hyperacetylation of mitochondrial proteins in KLF4-deficient hearts.
(A) Blue-Native PAGE showing mitochondrial ETC complexes.
(B–D) Mitochondxrial protein acetylation assessed by anti-acetyl-Lysine Western blot. Cardiac mitochondria isolated from baseline 4-month old animals (B), animals that received 5 days TAC (C) and animals that developed heart failure (EF < 30%, data not shown) after 5-weeks of high intensity TAC (D).
(E) Acetylation of SOD2, CypD and LCAD in cardiac mitochondria. Mitochondrial protein was immunoprecipitated with anti-acetyl-Lysine antibody and probed for specific proteins as indicated.
To gain a further understanding of these hyper-acetylated proteins, we immunoprecipitated all acetylated mitochondrial proteins by the anti-acetyl-Lysine antibody and probed for Superoxide dismutase 2 (SOD2), Cyclophilin D (CypD) and long chain Acyl-CoA dehydrogenase (LCAD), all of which are known Sirt3 targets and can be acetylated under certain conditions [12,21–23]. As shown in Fig. 1E, all three acetylated proteins were enriched in the CM-K4KO group. Previous studies have reported that hyperacetylation of these proteins are associated with mitochondrial dysfunction and heart failure [12,21–23]. As such, our data suggested that mitochondria in the KLF4-deficient cardiomyocytes were likely predisposed to hyperacetylation-related defects. Previously we showed impaired mitochondrial metabolic function in the KLF4-deficient heart [20], which can be partly attributed to protein hyperacetylation.
Collectively, these data demonstrate that cardiac KLF4-deficiency led to hyperacetylation of mitochondrial proteins, likely due to reduced deacetylation function. Such posttranslational modification of mitochondrial proteins can profoundly impair mitochondrial metabolic function at baseline, which may predispose the CM-K4KO hearts vulnerable to stress [18,20].
3.2. Reduction of Sirt3, NAD+ and NAMPT in the KLF4-deficient heart
Mitochondrial protein acetylation is mainly regulated mainly by micotinamide adenine dinucleotide (NAD+) and the Sirtuin family of NAD+-dependent deacetylases. Among all known mammalian Sirtuin genes, we found the mRNA levels of Sirt3 and Sirt5 were reduced in CM-K4KO heart (Fig. 2A). Of note, Sirt3 is the key deacetylase in the mitochondrial matrix while Sirt5 is more of a deacylase rather than a deacetylase [24]. Hence, we focused on Sirt3 and found its protein levels were also reduced in the CM-K4KO mitochondria (Fig. 2B). Although it is hard to determine if such subtle decrease of Sirt3 protein is rate-limiting, undoubtably Sirt3 insuffciency can contribute to mitochondrial protein hyperacetylation [25].
Fig. 2.

Myocardial Sirt3, NAD+ and NAMPT levels.
(A) Expression of mammalian Sirtuin genes in the heart. n = 5–7, *p < 0.05.
(B) Sirt3 protein in cardiac mitochondria isolated from 4-month old animals.
(C) Myocardial NAD+ levels before and after 3-day TAC. n = 4, *p < 0.05.
(D) Outline of NAD+ synthesis pathway.
(E) NAMPT gene expression in the heart. n = 5–7, *p < 0.05.
Since NAD+ is critical for protein acetylation as well as mitochondrial respiration function, we next sought to determine the NAD+ levels in cardiac tissue. NAD+ levels were reduced in CM-K4KO hearts and pressure overload further affect myocardial NAD+ levels (Fig. 2C). Not surprisingly, TAC reduced cardiac NAD+ levels to much lower levels in the CM-K4KO group (Fig. 2C). NAD+ biosynthesis relies on two pathways, namely de novo and salvage pathways (Fig. 2D). Because adult heart does not express TDO (tryptophan 2,3-dioxygenase) or IDO (Indoleamine 2,3-dioxygenase), two rate-limiting enzymes in the de novo pathway, nearly all cardiac NAD+ is generated from the salvage pathway [26]. Nicotinamide phosphoribosyltransferase (NAMPT), the key enzyme in NAD+ salvage pathway, was significantly lower in the CM-K4KO heart and it went further down after TAC (Fig. 2E). These data strongly suggested a NAD+ deficient stage in the CM-K4KO heart, especially after the onset of pressure overload.
The combination of reduced expression of Sirt3 and low NAD+ concentration could result in overall deacetylase activity deficiency in the mitochondria leading to mitochondrial protein hyperacetylation. Moreover, NAD+ is a key coenzyme of the tricarboxylic acid (TCA) cycle and fatty acid β-oxidation, a depleted NAD+ pool would also impair mitochondrial metabolism.
3.3. Administration of NMN corrects mitochondrial acetylation and rescues the heart
Given the mitochondrial hyperacetylation and NAD+ deficiency observed in the CM-K4KO hearts, we asked if NAD+ repletion could be beneficial to these animals. To bypass the NAMPT defect, we chose to use Nicotinamide Mononucleotide (NMN) as the exogenous NAD+ precursor (Fig. 2D). As expected, NMN administration increased cardiac tissue NAD+ level (Fig. 3A). Further, NMN normalized the mitochondrial protein acetylation levels in the hearts (Fig. 3B). Strikingly, administration of NMN at 500 mg/kg/day starting from one day before TAC profoundly preserved the cardiac contractile function and completely rescued the heart failure phenotype in the CM-K4KO group (Fig. 3C). NMN treatment did not affect the expression of hypertrophic genes (i.e. ANF, β-MHC) but blunted the induction of inflammatory genes (i.e. IL1β, CCL2 and IL6) in CM-K4KO hearts (Fig. 3D), indicating that administration of NMN maintained a normal hypertrophic response to pressure overload but reduced cardiac injury. Due to high mortality rate of CM-K4KO mice after TAC [18], the phenotypical comparison between NMN and PBS (Vehicle) administration were performed within 5 days post-TAC but the CM-K4KO animals with TAC + NMN administration exhibited 100% survival rate during the course of study (data not shown).
Fig. 3.

Administration of NMN corrected mitochondrial acetylation and rescued KLF4-deficient heart from TAC-induced heart failure.
(A) Administration of NMN increased myocardial NAD + levels. n = 5, *p < 0.05. NMN administration: 3 days.
(B) Administration of NMN reduced mitochondrial protein acetylation in the heart.
(C) Echocardiography analysis showing cardiac contractility. Left ventricular contractile function shown as fractional shortening (FS).
(D) Expression of hypertrophy marker genes and inflammatory genes in the heart. n = 5–9, *p < 0.05.
TAC and NMN administration: 5 days. NMN was injected intraperitoneally at 500 mg/kg/day. PBS was injected as vehicle control.
3.4. Administration of NMN improves mitochondrial fatty acid oxidation
It has been shown that long-term administration of NMN enhances overall metabolism at the whole organism level. We next tested if short-term administration of NMN in present study could affect mitochondrial respiration. We first treated primary neonatal rat ventricular myocytes (NRVM) with NMN (2 mmol/L) for 24 h and assessed mitochondrial respiration using a Seahorse extracellular flux analyzer. With glucose and pyruvate as substrates, we observed no difference in either basal or maximal oxygen consumption rate (OCR) between NMN-treated and control groups (Fig. 4A), suggesting short-term treatment of NMN does not induce mitochondrial biogenesis or change mitochondrial respiration capacity. However, when using Palmitate as a long-chain fatty acid substrate, both basal and maximal OCR were significantly increased by NMN treatment (Fig. 4B), suggesting that NMN improved fatty acid oxidation (FAO). We further confirmed that such short-term NMN treatment (24 h) did not significantly induce the expression of FAO genes (i.e. PPARα, CD36, CPT1β, CPT2, LCAD, MCAD) or mitochondrial biogenesis genes (i.e. PGC-1α, PGC-1β, ERRα) (Fig. 4C), suggesting a posttranslational effect of NMN on FAO rather than transcriptional metabolic reprogramming.
Fig. 4.

NMN improves mitochondrial fatty acid oxidation.
(A, B) Mitochondrial respiration function in NRVM assessed by Seahorse Mito Stress test using (A) Pyruvate + Glucose and (B) Palmitate (BSA-conjugated palmitate) as substrates, respectively. OCR: oxygen consumption rate. Oligomycin: ATP synthase inhibitor. FCCP: mitochondrial uncoupler. Rot/AA: rotenone and antimycin A, specific inhibitors for ETC complex I and III respectively. Arrows indicate time for drug injection. Basal rate was calculated as OCR before addition of Oligomycin. Maximal rate was calculated as the highest OCR after addition of FCCP. NRVMs were treated with 2 mmol/L NMN for 24 h (NMN) or left untreated (Control). n = 3, *p < 0.05.
(C) The expression of FAO and mitochondrial biogenesis genes in NRVM treated with 2 mmol/L NMN for 24 h. n = 6, p ≥ 0.43.
(D) Mitochondrial protein acetylation and Sirt3 levels in NRVMs that were infected for 72 h with empty virus (Sh-EV) or virus expressing a ShRNA against rat KLF4 (Sh-KLF4).
(E) Mitochondrial respiration assessed using Pyruvate + Glucose and Palmitate (BSA-conjugated palmitate) as substrates, respectively. NMN groups were treated with 2 mmol/L NMN starting at 24 h post-infection and Seahorse assay was performed at 72 h post-infection. n = 3, *p < 0.05.
In another set of NRVM experiments, KLF4 was silenced using adenoviral shRNA to recapitulate the cardiac KLF4 deficiency. Similar to the CM-K4KO heart, loss of KLF4 in NRVM resulted in hyper-acetylation of mitochondrial protein (Fig. 4D). As expected, silencing KLF4 led to reduced mitochondrial respiration in NRVM (Fig. 4E, no NMN groups). Similar to studies on normal NRVM, NMN did not affect pyruvate oxidation but significantly improved FAO (Fig. 4E, NMN-treated groups).
Collectively, these NRVM-based mitochondrial respiration studies demonstrated that NMN treatment, even for a short-term, improved long-chain fatty acid oxidation. This effect of NMN can be achieved by either deacetylation of key FAO enzymes, such as LCAD (Figs. 1E & 3B) [22], or increase of NAD+ supply for the TCA cycle (Fig. 3A). Nevertheless, given that long-chain fatty acid is a major fuel of the heart, NMN can thus profoundly improve cardiac energetics and heart function.
3.5. NMN administration preserved mitochondrial ultrastructure and reduced cell death in the pressure overloaded hearts
Upon pressure overload, the CM-K4KO mice developed acute heart failure with severe disruption of mitochondrial homeostasis that manifested as dramatic alterations in mitochondrial ultrastructure, including degeneration, fragmentation, crista swelling, and heterogeneity (Fig. 5A). In contrast, no such change was observed in the MHC-Cre groups. Strikingly, NMN administration almost completely restored mitochondrial ultrastructure to the level comparable to the MHC-Cre group (Fig. 5B). These data were consistent with the cardiac function observed by echocardiography (Fig. 3C) and indicated that NMN successfully preserved mitochondrial homeostasis in the CM-K4KO hearts during pressure overload leading to improved cardiac function.
Fig. 5.
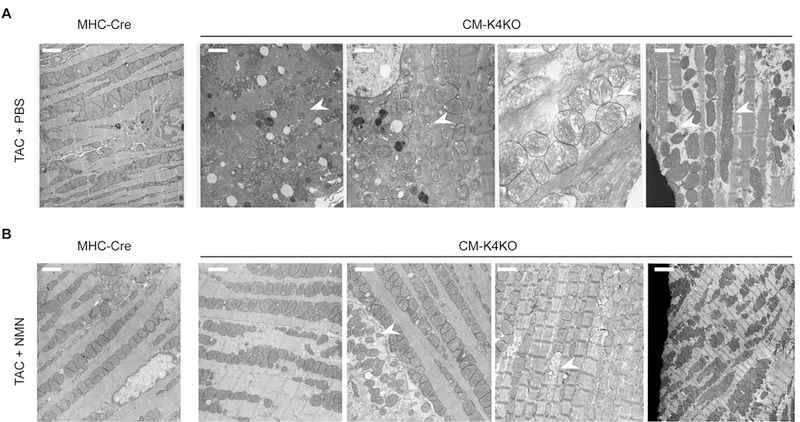
Administration of NMN protected cardiac mitochondria from TAC-induced damage.
(A) EM images from PBS (vehicle) treated hearts after 5 days of TAC.
(B) EM images from NMN (500 mg/kg/day) treated hearts after 5 days of TAC. Scale bar: 1 um. Arrows indicate damaged or abnormal mitochondria. Each image was from individual animal but different areas were chosen to display different phenotype. n = 3 in each group.
Mitochondrial damage is often associated with ROS stress and cell death. We found that TAC induced significant increase of ROS in the CM-K4KO myocardium and NMN treatment diminished such ROS burst (Fig. 6). Further, TAC induced significant cell death in the CM-K4KO hearts as > 10% nuclei showed TUNEL positive staining (Fig. 7). However, such TAC-induced cardiac cell death was reduced to < 5% in the CM-K4KO mice that received NMN treatment (Fig. 7). There was only minimal cell death detected in the MHC-Cre groups and it was similar before and after TAC. Collectively, these data suggested that short-term administration of NMN preserved cardiac mitochondrial function, reduced TAC-induced mitochondrial damage, reduced myocardial ROS and prevented cell death. The combined benefit of NMN-mediated effects at aforementioned multiple levels help maintain normal functions of cardiac mitochondria, cardiomyocytes and ultimately the heart.
Fig. 6.
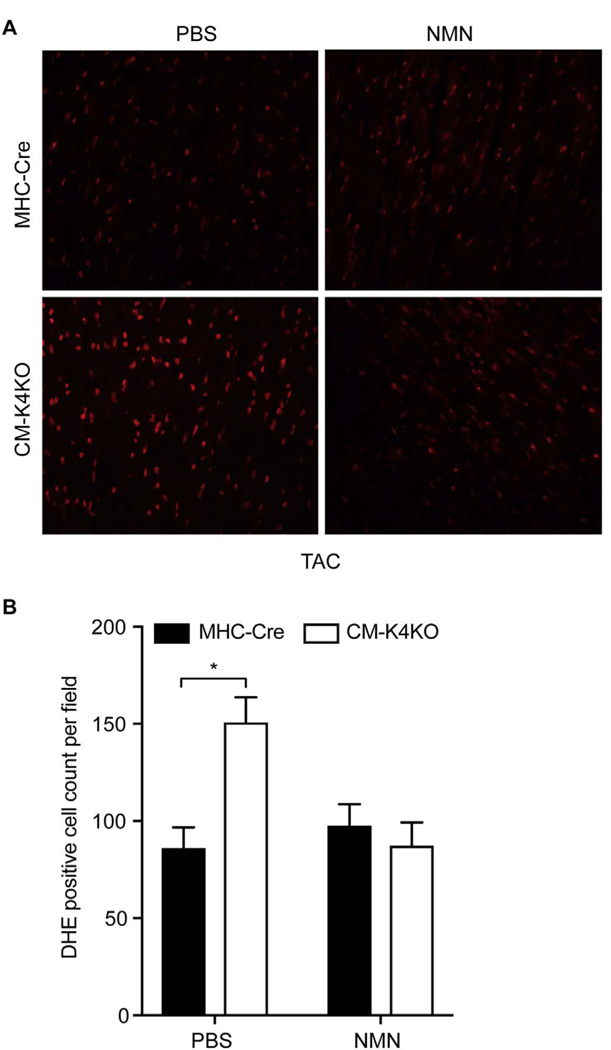
Administration of NMN reduced TAC-induced ROS generation in KLF4-deficient hearts.
(A) Representative images showing myocardial DHE staining.
(B) ROS level was calculated as DHE positive cells per field. TAC and NMN administration: 5 days. n = 3–5 animals in each group. *p < 0.05.
Fig. 7.
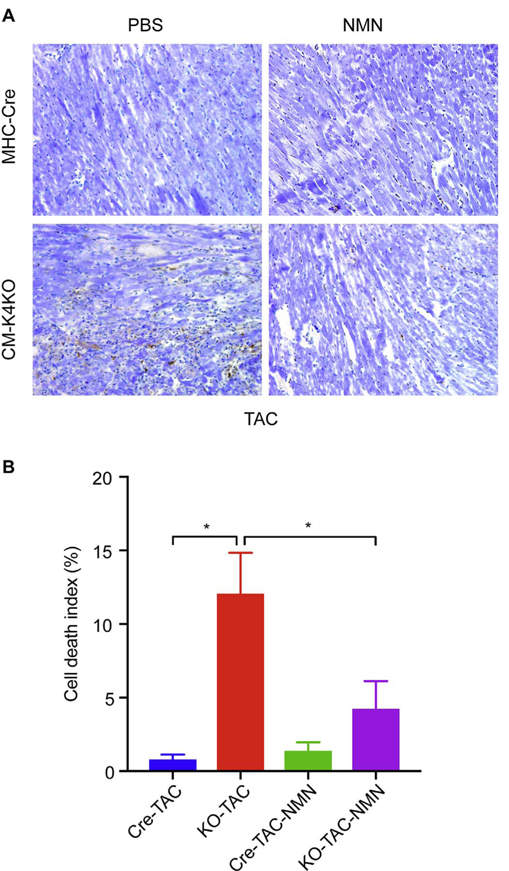
Administration of NMN reduced TAC-induced cell death in KLF4-deficient hearts.
(A) Representative TUNEL staining images from n = 3–5 animals in each group. TAC: 5 days. Apoptotic nuclei were stained in brown by DAB and normal nuclei were stained in light blue by Methyl green counter stain.
(B) Cell death index was calculated as percentage of brown nuclei in all nuclei. n = 3–5, *p < 0.05. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
4. Discussion
In this study, we demonstrate that cardiac KLF4-deficiency leads to hyperacetylation of mitochondrial proteins that predisposes the mitochondria and heart sensitive to stress. Short-term administration of NMN, a precursor of NAD+, preserved mitochondrial homeostasis and rescued heart function from pressure overload-induced heart failure. Our study thus identified protein hyperacetylation as a cause of mitochondrial dysfunction as well as a promising therapeutic target for heart failure. We demonstrate that administration of NMN (or other NAD+ repletion reagents) is capable of rescuing the heart through preservation of mitochondrial homeostasis.
Mitochondrial dysfunction has long been associated with heart failure. The investigation of mitochondrial function in human and animal models of heart failure shows a variety of mitochondrial alterations that point to defects at specific sites, i.e. the electron transport chain (ETC), the phosphorylation apparatus, or the supercomplexes assembly [27]. Although different etiology of heart failure leads to different presentation of mitochondrial dysfunction, in general severe heart failure is associated with reduced mitochondrial metabolic capacity and low ATP production [10]. Mitochondrial dysfunction is thought to be a result of accumulative injury from ROS due to its high-level oxidative metabolism. However, antioxidant therapy failed to be effective in clinical trials, suggesting novel therapies are required to target mitochondrial dysfunction.
Recently, mitochondrial protein hyperacetylation has been recognized as a common mechanism underlying mitochondrial dysfunction in multiple organs including heart, liver, brain, kidney and muscle [28,29]. In general, hyperacetylation of mitochondrial proteins is believed to be detrimental to metabolism as demonstrated by numerous studies on the Sirt3-deficient mice [21–23,25,29–34] and recently by studies in clinical heart failure samples [12,28]. Conversely, deacetylation of mitochondrial proteins, i.e. by overexpression of Sirt3, has been shown to enhance metabolism [25,35]. Pharmacologically, administration of NAD+ precursors, such as NMN and nicotinamide riboside (NR), could normalize the NADH/NAD+ ratio, restore protein acetylation and boost metabolic function [36]. However, there are also studies suggested a correlation between increased acetylation and increased FAO enzyme activity [37,38]. Such discrepancy may be due to the different metabolic models used in different studies. In the studies that showed protein acetylation increases FAO, they were studying animals that are suffering obesity, diabetes or heart failure [38], conditions that are known to have profound alterations in metabolism, FAO in particular.
In present study, we demonstrated that NMN rescued TAC-induced mitochondrial dysfunction and cardiac failure in the KLF4-deficient heart. Our short-term administration of NMN did not affect the mitochondrial biogenesis or metabolic capacity but dramatically improved FAO, prevented mitochondrial damage and cell death. In studies that reported increased metabolic function by NAD+ precursors, the administration duration was often long-term (weeks or months) where some reprograming of cellular metabolic machinery including mitochondrial biogenesis could take place in multiple organs leading to the overall metabolic rate change of the whole organism [36,39]. However, here we showed that even short-term administration of NMN could be dramatically beneficial.
Obviously NMN can have impact at multiple levels as discussed below. We think the therapeutic effect from NMN administration is a combination of all.
First, NMN administration can potentially correct metabolic defects or enhance metabolism through repletion of intracellular NAD+ pools including the mitochondrial NAD+ pool. NAD+ is a critical coenzyme for mitochondrial ATP production. NAD+ gains two electrons at multiple steps of TCA cycle to form NADH, which is the major reducing equivalent driving ETC. As the TCA cycle and ETC require NAD+ and NADH, respectively, an optimal NAD+/NADH ratio is needed for efficient mitochondrial metabolism. Recent studies have demonstrated that NAD+ levels are rate-limiting for mitochondrial respiration [40]. As such, supplement of NMN can immediately boost mitochondrial function through increasing the NAD+/NADH ratio. Further, Sirt3-NAD+-dependent deacetylation of key FAO enzymes, such as LCAD, can improve mitochondrial metabolic function as well [22]. NAD+ also can activate Sirt1 leading to deacetylation and activation of PGC1α, the master regulator of metabolism and mitochondrial biogenesis [40]. Our mitochondrial respiration data from NRVM demonstrated significant improvement in FAO by NMN but not in pyruvate oxidation, suggesting short-term treatment of NMN might have a quick effect on NAD+/ NADH ratio and/or FAO enzymes deacetylation but not PGC1α-mediated mitochondrial biogenesis. Consistently, there was no significant changes in FAO or mitochondrial biogenesis genes during the short-term NMN treatment. Finally, before entering the TCA cycle all long-chain fatty acids are oxidized to Acetyl-CoA through β-oxidation that requires NAD+ as coenzyme. As such, FAO may require higher NAD+ concentration than pyruvate oxidation and NMN therefore can have more impact on FAO.
Second, NMN administration can protect cardiomyocytes from stress-induced cell death. We observed a strikingly high rate of cell death (> 10%) in CM-K4KO myocardium after stress, which could be a key contributor to acute heart failure. Such massive cell death, however, was significantly blocked by NMN administration (Fig. 7). It has been reported that, mitochondrial NAD+ levels, the highest among all intracellular NAD+ pools, dictate cell survival [41]. Depletion of mitochondrial NAD+ induces cell death and overexpression of NAMPT protects against genotoxic cell death. Of note, NAMPT-mediated protection requires Sirt3 and Sirt4, suggesting a mitochondrial acetylation-related mechanism [41]. In present study, we found reduction in Sirt3, NAMPT and NAD+ levels in CM-K4KO myocardium, all of which could contribute to cell death. Because of no detectable cell death at baseline in CM-K4KO hearts, it is likely that the CM-K4KO heart has low but above critical threshold level of NAD+ at baseline. However, upon TAC stress, NAD+ level declines and it may reach critical threshold level in CM-K4KO heart to trigger metabolic problems and cell death. Conversely, supplement of NMN help replete the NAD+ pools to maintain mitochondrial function and prevent cell death.
Third, NMN administration can protect mitochondrial homeostasis. The mitochondria in KLF4-deficient hearts suffered dramatic damage after TAC (Fig. 5). Consistently, there is more inflammation and ROS in the CM-K4KO myocardium as well (Figs. 3D & 6). NMN administration significantly rescued the mitochondrial ultrastructure, reduced ROS and inflammation in CM-K4KO myocardium. In part, NAD+-mediated improvement of mitochondrial metabolism could contribute here. Moreover, we found hyperacetylation of CypD and SOD2 in KLF4-deficient cardiac mitochondria (Fig. 1E). It has been shown that CypD hyperacetylation is associated with heart failure [12]. Acetylation of CypD promotes its binding to oligomycin-sensitive conferring protein (OSCP) and increases the sensitivity of mitochondrial permeability pore (mPTP) [12,42]. SOD2 is the most important antioxidant in the mitochondrial matrix, where it neutralizes the high toxic superoxide generated from ETC. Sirt3-mediated deacetylation activates SOD2, while acetylation of SOD2 impairs its function leading to ROS stress [21,43]. Both mPTP opening and ROS has been associated with cell death, inflammation and the development of heart failure [12,44]. Therefore, NMN can preserve mitochondrial homeostasis through stabilization of mPTP and reduction of ROS.
Finally, NMN can reduce myocardial inflammation. Damaged mitochondria can be very inflammatory, likely through ROS and its prokaryotic genomic DNA [45]. Inflammation is a vicious positive feedback cycle in which the initial injury in cardiomyocytes would induce infiltration of immune cells to further amplify myocardial inflammation. By protecting mitochondria from stress-induced damage (SOD2, mPTP, etc.), NMN thus can cut off the initial inflammatory signal, leading to reduced myocardial inflammation (Fig. 3D).
A surprising observation is that NMN did not exhibit detectable benefit in the Cre group. A simple explanation could be that these Cre mice are in fact perfectly healthy having normal NAD+ and protein acetylation levels at baseline. Although TAC does reduce cardiac NAD+ levels (Fig. 2C), it may never become rate-limiting in a normal mouse heart at the early stage of pressure overload hypertrophy (1–5 days in present study). Of note, this is quite different than chronic heart failure patients who have already reached late stage of the diseases. Reduced NAD+ levels and hyperacetylation of cardiac mitochondrial proteins have been observed in experimental heart failure models and clinical heart failure samples [12,28]. Further, normalization of NAD+ has been shown to be beneficial in mitochondrial mutant and WT mice that suffered heart failure [12].
In summary, we employed the cardiac KLF4-deficient mice as a pressure overload-induced acute heart failure model for a proof of principle study to demonstrate that administration of NMN (even short-term) could be an effective therapy for heart failure.
Acknowledgements
The authors thank Dr. Mukesh K. Jain for discussion and critical reading of the manuscript. We thank Jiayin Du for assistance in experiments.
Funding
This work was supported by American Heart Association National Scientist Development grant 12SDG12070077 (to X. Liao) and Postdoc Fellowship17POST33650110 (to Y. Shen), NIH grant K08HL123551 (to L. Zhang), and National Natural Science Foundation of China81400347 (to L. Zhou). This work was generously supported by Tom F. Peterson.
Footnotes
Disclosures
None.
Reference
- [1].Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC, Myocardial fatty acid metabolism in health and disease, Physiol. Rev 90 (1) (2010) 207–258. [DOI] [PubMed] [Google Scholar]
- [2].Huss JM, Kelly DP, Mitochondrial energy metabolism in heart failure: a question of balance, J. Clin. Invest 115 (3) (2005) 547–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Riehle C, Abel ED, PGC-1 proteins and heart failure, Trends Cardiovasc. Med 22 (4) (2012) 98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Schilling J, Kelly DP, The PGC-1 cascade as a therapeutic target for heart failure, J. Mol. Cell. Cardiol 51 (4) (2011) 578–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Aubert G, Vega RB, Kelly DP, Perturbations in the gene regulatory pathways controlling mitochondrial energy production in the failing heart, Biochim. Biophys. Acta (BBA) 1833 (4) (April 2013) 840–847 (Molecular Cell Research). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].den Boer ME, Dionisi-Vici C, Chakrapani A, van Thuijl AO, Wanders RJ, Wijburg FA, Mitochondrial trifunctional protein deficiency: a severe fatty acid oxidation disorder with cardiac and neurologic involvement, J. Pediatr 142 (6) (2003) 684–689. [DOI] [PubMed] [Google Scholar]
- [7].Scarpulla RC, Transcriptional paradigms in mammalian mitochondrial biogenesis and function, Physiol. Rev 88 (2) (2008) 611–638. [DOI] [PubMed] [Google Scholar]
- [8].Hock MB, Kralli A, Transcriptional control of mitochondrial biogenesis and function, Annu. Rev. Physiol 71 (2009) 177–203. [DOI] [PubMed] [Google Scholar]
- [9].Heggermont WA, Papageorgiou AP, Heymans S, van Bilsen M, Metabolic support for the heart: complementary therapy for heart failure? Eur. J. Heart Fail 18 (12) (2016) 1420–1429. [DOI] [PubMed] [Google Scholar]
- [10].Neubauer S, The failing heart–an engine out of fuel, N. Engl. J. Med 356 (11) (2007) 1140–1151. [DOI] [PubMed] [Google Scholar]
- [11].Lesnefsky EJ, Moghaddas S, Tandler B, Kerner J, Hoppel CL, Mitochondrial dysfunction in cardiac disease: ischemia–reperfusion, aging, and heart failure, J. Mol. Cell. Cardiol 33 (6) (2001) 1065–1089. [DOI] [PubMed] [Google Scholar]
- [12].Lee CF, Chavez JD, Garcia-Menendez L, Choi Y, Roe ND, Chiao YA, Edgar JS, Goo YA, Goodlett DR, Bruce JE, Tian R, Normalization of NAD + redox balance as a therapy for heart failure, Circulation 134 (12) (2016) 883–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kris-Etherton PM, Lichtenstein AH, Howard BV, Steinberg D, Witztum JL, Nutrition Committee of the American Heart Association Council on nutrition, metabolism, antioxidant vitamin supplements and cardiovascular disease, Circulation 110 (5) (2004) 637–641. [DOI] [PubMed] [Google Scholar]
- [14].Yang W, Nagasawa K, Munch C, Xu Y, Satterstrom K, Jeong S, Hayes SD, Jedrychowski MP, Vyas FS, Zaganjor E, Guarani V, Ringel AE, Gygi SP, Harper JW, Haigis MC, Mitochondrial sirtuin network reveals dynamic sirt3-dependent deacetylation in response to membrane depolarization, Cell 167 (4) (2016) 985–1000 (e21). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Atkins GB, Jain MK, Role of Kruppel-like transcription factors in endothelial biology, Circ. Res 100 (12) (2007) 1686–1695. [DOI] [PubMed] [Google Scholar]
- [16].Haldar SM, Ibrahim OA, Jain MK, Kruppel-like Factors (KLFs) in muscle biology, J. Mol. Cell. Cardiol 43 (1) (2007) 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].McConnell BB, Yang VW, Mammalian Kruppel-like factors in health and diseases, Physiol. Rev 90 (4) (2010) 1337–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Liao X, Haldar SM, Lu Y, Jeyaraj D, Paruchuri K, Nahori M, Cui Y, Kaestner KH, Jain MK, Kruppel-like factor 4 regulates pressure-induced cardiac hypertrophy, J. Mol. Cell. Cardiol 49 (2) (2010) 334–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Haldar SM, Lu Y, Jeyaraj D, Kawanami D, Cui Y, Eapen SJ, Hao C, Li Y, Doughman YQ, Watanabe M, Shimizu K, Kuivaniemi H, Sadoshima J, Margulies KB, Cappola TP, Jain MK, Klf15 deficiency is a molecular link between heart failure and aortic aneurysm formation, Sci. Transl. Med 2 (26) (2010) 26ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Liao X, Zhang R, Lu Y, Prosdocimo DA, Sangwung P, Zhang L, Zhou G, Anand P, Lai L, Leone TC, Fujioka H, Ye F, Rosca MG, Hoppel CL, Schulze PC, Abel ED, Stamler JS, Kelly DP, Jain MK, Kruppel-like factor 4 is critical for transcriptional control of cardiac mitochondrial homeostasis, J. Clin. Invest 125 (9) (2015) 3461–3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Dikalova AE, Itani HA, Nazarewicz RR, McMaster WG, Flynn CR, Uzhachenko RV, Fessel JP, Gamboa JL, Harrison DG, Dikalov SI, Sirt3 impairment and SOD2 hyperacetylation in vascular oxidative stress and hypertension, Circ. Res 121 (5) (August 18, 2017) 564–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, Stevens RD, Li Y, Saha AK, Ruderman NB, Bain JR, Newgard CB, Farese RV Jr., Alt FW, Kahn CR, Verdin E, SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation, Nature 464 (7285) (2010) 121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bharathi SS, Zhang Y, Mohsen AW, Uppala R, Balasubramani M, Schreiber E, Uechi G, Beck ME, Rardin MJ, Vockley J, Verdin E, Gibson BW, Hirschey MD, Goetzman ES, Sirtuin 3 (SIRT3) protein regulates long-chain acyl-CoA dehydrogenase by deacetylating conserved lysines near the active site, J. Biol. Chem 288 (47) (2013) 33837–33847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].He W, Newman JC, Wang MZ, Ho L, Verdin E, Mitochondrial sirtuins: regulators of protein acylation and metabolism, Trends Endocrinol. Metab 23 (9) (2012) 467–476. [DOI] [PubMed] [Google Scholar]
- [25].Hirschey MD, Shimazu T, Jing E, Grueter CA, Collins AM, Aouizerat B, Stancakova A, Goetzman E, Lam MM, Schwer B, Stevens RD, Muehlbauer MJ, Kakar S, Bass NM, Kuusisto J, Laakso M, Alt FW, Newgard CB, Farese RV Jr., Kahn CR, Verdin E, SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome, Mol. Cell 44 (2) (2011) 177–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hershberger KA, Martin AS, Hirschey MD, Role of NAD + and mitochondrial sirtuins in cardiac and renal diseases, Nat. Rev. Nephrol 13 (4) (2017) 213–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Rosca MG, Vazquez EJ, Kerner J, Parland W, Chandler MP, Stanley W, Sabbah HN, Hoppel CL, Cardiac mitochondria in heart failure: decrease in re-spirasomes and oxidative phosphorylation, Cardiovasc. Res 80 (1) (2008) 30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Horton JL, Martin OJ, Lai L, Riley NM, Richards AL, Vega RB, Leone TC, Pagliarini DJ, Muoio DM, Bedi KC Jr., Margulies KB, Coon JJ, Kelly DP, Mitochondrial protein hyperacetylation in the failing heart, JCI Insight 2 (1) (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Dittenhafer-Reed KE, Richards AL, Fan J, Smallegan MJ, Fotuhi Siahpirani A, Kemmerer ZA, Prolla TA, Roy S, Coon JJ, Denu JM, SIRT3 mediates multi-tissue coupling for metabolic fuel switching, Cell Metab 21 (4) (2015) 637–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Shimazu T, Hirschey MD, Hua L, Dittenhafer-Reed KE, Schwer B, Lombard DB, Li Y, Bunkenborg J, Alt FW, Denu JM, Jacobson MP, Verdin E, SIRT3 deacetylates mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 and regulates ketone body production, Cell Metab 12 (6) (2010) 654–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hallows WC, Yu W, Smith BC, Devries MK, Ellinger JJ, Someya S, Shortreed MR, Prolla T, Markley JL, Smith LM, Zhao S, Guan KL, Denu JM, Sirt3 promotes the urea cycle and fatty acid oxidation during dietary restriction, Mol. Cell 41 (2) (2011) 139–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hirschey MD, Shimazu T, Huang JY, Schwer B, Verdin E, SIRT3 regulates mitochondrial protein acetylation and intermediary metabolism, Cold Spring Harb. Symp. Quant. Biol 76 (2011) 267–277. [DOI] [PubMed] [Google Scholar]
- [33].Jing E, Emanuelli B, Hirschey MD, Boucher J, Lee KY, Lombard D, Verdin EM, Kahn CR, Sirtuin-3 (Sirt3) regulates skeletal muscle metabolism and insulin sig-naling via altered mitochondrial oxidation and reactive oxygen species production, Proc. Natl. Acad. Sci. U. S. A 108 (35) (2011) 14608–14613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Yu W, Dittenhafer-Reed KE, Denu JM, SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status, J. Biol. Chem 287 (17) (2012) 14078–14086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lin L, Chen K, Abdel Khalek W, Ward JL, Yang H 3rd, Chabi B, Wrutniak-Cabello C, Tong Q, Regulation of skeletal muscle oxidative capacity and muscle mass by SIRT3, PLoS One 9 (1) (2014) e85636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Canto C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, Fernandez-Marcos PJ, Yamamoto H, Andreux PA, Cettour-Rose P, Gademann K, Rinsch C, Schoonjans K, Sauve AA, Auwerx J, The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity, Cell Metab 15 (6) (2012) 838–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Alrob OA, Sankaralingam S, Ma C, Wagg CS, Fillmore N, Jaswal JS, Sack MN, Lehner R, Gupta MP, Michelakis ED, Padwal RS, Johnstone DE, Sharma AM, Lopaschuk GD, Obesity-induced lysine acetylation increases cardiac fatty acid oxidation and impairs insulin signalling, Cardiovasc. Res 103 (4) (2014) 485–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Fukushima A, Lopaschuk GD, Acetylation control of cardiac fatty acid beta-oxidation and energy metabolism in obesity, diabetes, and heart failure, Biochim. Biophys. Acta 1862 (12) (2016) 2211–2220. [DOI] [PubMed] [Google Scholar]
- [39].Mills KF, Yoshida S, Stein LR, Grozio A, Kubota S, Sasaki Y, Redpath P, Migaud ME, Apte RS, Uchida K, Yoshino J, Imai SI, Long-term administration of nicotinamide mononucleotide mitigates age-associated physiological decline in mice, Cell Metab 24 (6) (2016) 795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Bai P, Canto C, Oudart H, Brunyanszki A, Cen Y, Thomas C, Yamamoto H, Huber A, Kiss B, Houtkooper RH, Schoonjans K, Schreiber V, Sauve AA, Menissier-de Murcia J, Auwerx J, PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation, Cell Metab 13 (4) (2011) 461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, Lamming DW, Souza-Pinto NC, Bohr VA, Rosenzweig A, de Cabo R, Sauve AA, Sinclair DA, Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival, Cell 130 (6) (2007) 1095–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Bernardi P, Di Lisa F, The mitochondrial permeability transition pore: molecular nature and role as a target in cardioprotection, J. Mol. Cell. Cardiol 78 (2015) 100–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Chen Y, Zhang J, Lin Y, Lei Q, Guan KL, Zhao S, Xiong Y, Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS, EMBO Rep 12 (6) (2011) 534–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Giordano FJ, Oxygen, oxidative stress, hypoxia, and heart failure, J. Clin. Invest 115 (3) (2005) 500–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida K, Akira S, Yamamoto A, Komuro I, Otsu K, Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure, Nature 485 (7397) (2012) 251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]