

Abstract
Alcohol abuse causes brain damage and cognitive dysfunction. However, the underlying mechanisms remain elusive. Endoplasmic reticulum (ER) acts as machinery to ensure the proper folding of newly synthesized proteins. The perturbation of ER, i.e., ER stress, plays a pivotal role in some neurological disorders. Mammalian target of rapamycin (mTOR), a serine/threonine kinase, is involved in the regulation of ER stress. The current study sought to determine whether binge ethanol exposure induces ER stress in adult mouse brain and the role mTOR signaling during this process. Adult C57BL6 mice received binge ethanol exposure by daily gavage (5 g/kg, 25% ethanol w/v) for 1, 5 or 10 days. Binge ethanol exposure caused neurodegeneration and neuroinflammation after 5 days of exposure, and a concomitant increase of ER stress and inhibition of mTOR. However, ethanol exposure did not significantly alter spatial learning and memory, and spontaneous locomotor activity. Ethanol treatment induced ER stress and the death of cultured neuronal cells. Cotreatment with an ER stress inhibitor, sodium 4-phenylbutyrate (4-PBA) significantly diminished ethanol-induced ER stress and neuronal apoptosis, suggesting that ER stress contributes to ethanol-induced neurodegeneration. Furthermore, the blockage of mTOR activity by rapamycin increased ER stress in cultured neuronal cells; whereas the activation or inhibition of ER stress by tunicamycin or 4-PBA respectively had little effects on mTOR signaling. These results suggested that mTOR signaling is upstream of ER stress and may thereby mediate ethanol-induced ER stress.
Keywords: Alcohol use disorder, brain damage, endoplasmic reticulum stress, mTOR signaling, neurodegeneration, oxidative stress
Introduction
Human and animal studies suggest that excessive ethanol intake can lead to brain damage (Pfefferbaum et al., 1992; Ke et al., 2011). Binge alcohol drinking represents the most common pattern of drinking in 40–60% of alcohol abusers (Robin et al., 1998). Chronic binge drinking leads to the alteration of brain structure, neurological function impairment, and cognitive deficits in humans and rodents (Harding et al., 1997; Bowden et al., 2001; Nardelli et al., 2011; Fernandez et al., 2017). Ethanol-induced structural alteration varies across brain regions; cerebral cortex, cerebellum, and limbic system have the most neurodegeneration (Collins et al., 1996; Crews et al., 2000; Obernier et al., 2002; Crews and Nixon, 2009; Fernandez et al., 2017). However, the mechanisms underlying ethanol-induced neurodegeneration remain unclear.
It has been suggested that inflammation and oxidative stress play a pivotal role in ethanol-induced neurodegeneration (Lucas et al., 2006; Qin et al., 2008; Zou and Crews, 2010; Qin and Crews, 2012). Ethanol can increase proinflammatory cytokines in the central nervous system (CNS) through activating local neuroinflammation or systemic inflammation (Ferrier et al., 2006; Banks and Erickson, 2010; Leclercq et al., 2012). On the other hand, ethanol and its metabolite, acetaldehyde, can produce reactive oxygen species (ROS) and thereby cause oxidative stress in the brain (Guerri et al., 1994; Qin and Crews, 2012; Boyadjieva and Sarkar, 2013). There is considerable interaction between ROS and neuroinflammation (Kratsovnik et al., 2005; Morgan and Liu, 2011). This positive feedback loop between ROS and neuroinflammation may reinforce ethanol-induced neurodegeneration.
Endoplasmic reticulum (ER) is machinery that regulates protein folding and primes the degradation of misfolded proteins through proteasome, lysosome, and autophagy pathways (Kaushik and Cuervo, 2015). Protein folding in the ER is highly regulated, and only properly folded proteins can bypass quality control surveillance and shuttle to the Golgi compartment. In the ER, protein chaperones retain misfolded proteins through interactions and eventually target them for degradation either by the ubiquitin-proteasome system (UPS) or by the autophagy-lysosome system. A variety of conditions, such as pathogen infection, nutrient deprivation, inflammation, alterations in ER luminal Ca2+ or redox status, and toxic chemicals disrupt protein folding in the ER and cause the accumulation of unfolded proteins, which is referred to as ER stress (Ron, 2002). ER stress activates an unfolded protein response (UPR) to eliminate unfolded or misfolded proteins in the ER. The UPR is regulated through three ER- localized transmembrane proteins, Inositol-requiring kinase (IRE1), protein kinase RNA- like endoplasmic reticulum kinase (PERK), and activating transcription factor 6 (ATF6) (Marciniak and Ron, 2006). When ER stress is excessively activated, and the cellular function cannot be restored, it leads to cell death (Hetz and Saxena, 2017). mTOR is a member of a family of serine/threonine kinases. mTOR signaling interacts with ER stress on multiple levels (Appenzeller-Herzog and Hall, 2012). ER stress can act both downstream and upstream of mTOR signaling. The best-characterized ER stress pathway downstream of mTOR was Ire1α-JNK, in which activation of mTOR leads to ER stress-induced cellular apoptosis (Kato et al., 2012). ER stress also acts upstream of mTOR signaling in which ATF6 upregulates PI3K-Akt-mTOR axis (Kato et al., 2012).
Ethanol-induced ER stress is linked with multiple organ injuries. Ethanol feeding induces ER stress in pancreas; the enhancement of ER stress was found in acute pancreatitis (Kubisch et al., 2006; Pandol et al., 2010; Lugea et al., 2011). ER stress has been proposed as a key mechanism for alcohol-induced liver disease (ALD) (Ji et al., 2005; Ji and Kaplowitz, 2006; Dara et al., 2011; Ji et al., 2011). Recent studies also showed that ER stress is upregulated in the myocardium of alcohol-fed mice (Li and Ren, 2008; Li et al., 2009). We previously showed that ethanol induces ER stress in cultured neuronal cells and the developing brain (Chen et al., 2008; Ke et al., 2011). Here, we hypothesized that ER stress might also be involved in neuronal damage of alcoholics. In the present study, we sought to determine whether binge-like ethanol exposure induces ER stress in the adult mouse brain and whether mTOR signaling is involved in ethanol-induced ER stress.
Materials and Methods.
Reagents and antibodies
We purchased the reagents for the measurement of ethanol from Analox instruments (London, UK). 4-Hydroxynonenal (HNE) adduct assay was from Cell Biolabs, Inc. (San Diego, CA). Rapamycin was from EMD Millipore (Burlington, MA). ATF6 was from LifeSpan Biosciences (Seattle, WA). mTOR, p-mTOR, 4EBP1, p- 4EBP1, p70S6K, p-p70S6K, eukaryotic initiation factor 2α (eIF2α), p-eIF2α, Ki-67, p- PERK, cleaved caspase-3, and cleaved caspase-12 antibodies, were from Cell Signaling Technology (Danvers, MA). GRP78 antibody was from Novus Biologicals (Littleton, CO). X-box binding protein-1s (XBP1s) antibody was from BioLegend (San Diego, CA). Doublecortin (DCX) antibody was from Abcam (Cambridge, MA). CCAAT/- enhancer-binding protein homologous protein (CHOP) antibody was from Thermo Fisher Scientific (Rockford, IL). HRP-conjugated anti-rabbit and anti-mouse secondary antibodies were from GE Healthcare Life Sciences (Piscataway, NJ). Mounting media containing 4’, 6-diamidino-2-phenylindole (DAPI) was from Vector Laboratories (Burlingame, CA). Alexa-488 conjugated anti-rabbit and Alexa-594 conjugated anti mouse antibodies were from Life Technologies (Grand Island, NY). Ketamine/xylazine was from Butler Schein Animal Health (Dublin, OH). Other chemicals and reagents were purchased either from Sigma-Aldrich or Life Technologies (Frederick, MD).
Animal model
Eight-week male C57BL/6 mice were from Jackson Laboratories (Bar Harbor, Maine). All study procedures are approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Kentucky and performed following regulations for the Care and Use of Laboratory Animals set forth by the National Institutes of Health (NIH) Guide. Twenty four mice were used for each time course and randomly assigned to two groups: water control group (n=12) and ethanol group (n=12). The mice received water or ethanol (5 g/kg, i.g., 25% ethanol w/v) intragastrically once daily for 1, 5 or 10 days with volume matched. At 1 hour after the last gavage, blood was extracted from the mouse tail veins, spun down, and the supernatant (serum) was aspirated directly to the measurement of blood ethanol concentration using an Analox AM 1 analyzer (Lunenburg, MA). At 6 hours after the last gavage, the mice were sacrificed and the brains were dissected for biochemical or histological analyses. For behavioral tests, twenty mice were gavaged with water (n=10) or ethanol (n=10) at the above dose for 10 consecutive days and then subjected to behavioral tests as described below.
Behavioral tests
Open Field Activity and Elevated Plus Maze are experimental tests used to assess general levels of locomotor activity and anxiety in rodents. Morris Water Maze is a behavioral procedure to assess spatial learning and memory in rodents. They have been widely used to study the effect of ethanol exposure on animal behavioral changes (Zhang et al., 2007; Ji et al., 2018). All behavioral tests were performed at the University of Kentucky Rodent Behavior Core.
Open Field Activity (OFA)
On the third day after the last gavage, all animals were brought from their housing room to the test room in clean holding cages for a ten-minute habituation period before testing. Open Field is a square chamber (50 × 50 × 40 cm) with opaque white walls and floors. Each animal was placed in the chamber and tested for 15 minutes using a computer-operated EthoVision XT system (Noldus, Wageningen, The Netherlands). The total distance traveled and time spent in the center area was calculated by tracking the animal’s midpoint relative to predetermined boundaries on the video images of each apparatus. The center was defined as a centered, square zone with a length and width half the length and width of the Open Field.
Elevated Plus Maze (EPM)
Following the OFA, the test animals were given a two-hour rest period before beginning the EPM test. EPM is made up of two open arms (50 × 10 cm) and two closed arms (50 × 10 × 40 cm) that are elevated above the floor. Each animal was placed in the intersection between open arms and closed arms and tested for a five-minute session using a computer-linked EthoVision XT system (Noldus, Wageningen, The Netherlands). The total distance traveled, and total entries to the open arms or closed arms were recorded and calculated by tracking the animal’s midpoint relative to predetermined boundaries on the video images of each apparatus.
Morris Water Maze (MWM)
On the fifth day after the last gavage, animals were transferred to the test room for a ten-minute habituation period before MWM testing. The MWM is a round plastic tub, about 107 cm in diameter, made opaque using white non-toxic water-based paint and then filled with water (25–26°C). We placed a hidden escape platform (15 × 15 cm) 1 cm under the surface of the water in a fixed location. Four visible extra-maze cues were posted on the walls around the maze. Each mouse was placed in the pool from different drop locations between trials and allowed to swim until they found the submerged platform. If they did not find the platform within 60 seconds, they were gently guided to the platform and remained on the platform for 10 seconds before being removed for a five-minute intertrial interval. Each animal had four trials for five consecutive days and the latency to reach the platform and distance traveled for each trial was recorded with Ethovision XT (Noldus Information Technology, The Netherlands).
Western blotting
Ethanol-induced neurodegeneration varies across brain regions; cerebellum, cortex and limbic system show the most sensitivity to ethanol neurotoxicity (Obernier et al., 2002; Crews and Nixon, 2009). We therefore examined the effect of ethanol on the cerebelum and cortex of mice. Proteins were extracted from cerebellum, cortex, and liver or cultured cells as previously described (Wang et al., 2015). Around 30–50 ug of extracted protein was used in Western blots to examine the levels of ER stress markers (GRP78, p-eIF2α, CHOP, XBP1s, and ATF6), mTOR signaling (p-mTOR, p-4EBP1, and p-p70S6K), and apoptotic markers (cleaved caspase-3 and caspase-12). The nitrocellulose membranes were first probed with specific primary antibodies overnight at 4°C. After washing with phosphate-buffered saline (PBS) containing 0.05% Tween-20 three times, the membranes were incubated with anti-rabbit or anti-mouse secondary antibodies (Horseradish peroxidase-conjugated) for 1 hour at room temperature. Protein-specific signals were then detected with enhanced chemiluminescence substrate (GE Healthcare, Chalfont, Buckinghamshire, UK) using a Chemi™Doc imaging system (Bio-Rad 215 Laboratories, Hercules, CA) and then quantified with the software of Image lab 5.2 (Bio-Rad Laboratories, Hercules, CA).
Fluoro-Jade C staining
Fluoro-Jade C is fluorescein-derived fluorochrome. Fluoro-Jade C staining is a commonly used method to label degenerating neurons due to its greatest signal to background ratio (Schmued et al., 2005). Brain tissues were fixed with 4% paraformaldehyde (PFA) overnight and then transferred to 30% sucrose to dehydrate. The brain tissues were sectioned sagittally at a thickness of 15 μm and then mounted onto superfrost/plus slides. After immunostained with NeuN (a marker for mature neurons) antibody, these sections were rehydrated in distilled water for 2 minutes and then transferred to a 0.06% potassium permanganate solution and incubated for 10 minutes. After rinsing in distilled water for 2 minutes, the sections were covered with Fluoro-Jade C working solution (0.0001% Fluoro-Jade C in 0.1% acetic acid) for 10 minutes. Next, the sections were rinsed in distilled water, dehydrated and covered with mounting and imaged with an inverted fluorescent microscope (IX81, Olympus).
Immunochemistry and immunofluorescence
We evaluated neurogenesis/proliferation level in the mouse brain by immunostaining of Ki-67 in subventricular zone (SVZ) and DCX in subgranular zone (SGZ). SVZ and SGZ are two regions in adult brain that have been demonstrated to give rise to neural stem cells or progenitor cells (Crews and Nixon, 2003). DCX is a microtubule-associated protein expressed in neuronal precursor cells and immature neurons. Ki-67 is used as a marker of proliferating cell in cancer biology and neuroscience. Mouse brains were dissected and processed for immunofluorescence (IF) or immunochemistry (IHC) staining as previously described (Ke et al., 2011; Chen et al., 2012). The brain sections were incubated with 0.3% H2O2 in methanol, permeabilized with 1% Triton X-100 and blocked with 1% BSA at room temperature. Doublecortin (DCX) in the SGZ of mouse brain was immunostained with rabbit polyclonal anti-DCX antibody (1:500). Ki-67 in the SVZ of mouse brain was immunostained with rabbit polyclonal anti-Ki-67 antibody (1:500). The sections were then incubated with anti-rabbit secondary antibody conjugated to Alexa Fluor 488 or biotin. The immunolabelings were visualized using nickel-enhanced 3,3’- Diaminobenzidine (DAB) or Alexa Fluor 488 (green) and imaged with an inverted fluorescent microscope (IX81, Olympus).
Protein carbonyl assay and HNE assays
Protein carbonyl assay is a colorimetric assay for the measurement of oxidized proteins, indicative of oxidative stress. Cerebellum, cortex or liver were homogenized in 1 ml of cold buffer containing 50 mM of phosphate, pH 6.7 and 1 mM EDTA with the final protein concentration at a range of 1–10 ug/ul. The homogenate was centrifuged, and the supernatant was collected for the assay. Briefly, 200 ul of the supernatant was transferred to two 2 ml tubes; one tube was the sample tube, and the other one was the control tube. 800 μl of 2,4-dinitrophenylhydrazine (DNPH) was added to the sample tube, and 800 ul of 2.5 M HCl was added to the control tube. Both tubes were vortexed every 15 minutes and remained at room temperature for one hour. Next, 1 ml of 20% trichloroacetic acid (TCA) was added to the sample tube and the control tube, and the mixtures remained on ice for five minutes. Proteins were precipitated by centrifuging, and the pellets were washed using ethanol/ethyl acetate mixture twice. The protein pellets were resuspended in 500 ul of guanidine hydrochloride. Then, 150 ul of protein was transferred from the sample tube and control tube to three wells in a 96-well plate, and the absorbance was measured at a wavelength of 360–385 nm using a plate reader. The corrected absorbance (CA) was calculated by subtracting the average absorbance of the control tube from the average absorbance of the sample tube. The final carbonyl content was determined using the following equation: Carbonyl content (mmol/mg)=[CA/(*11 mM−1)]*(500 μl/ 200 μl) /(protein mg/ml).
HNE adduct assay is for the measurement of lipid peroxidation, another index for oxidative stress. Protein lysates were prepared from samples (cerebellum, cortex, or liver). 50 μl of protein lysates or HNE-BSA standard were added to wells pre-coated with HNE conjugates. After a brief incubation, 50 μl of the diluted HNE antibody was added to the wells and incubated for 1 hour, followed by addition of an HRP conjugated secondary antibody. The wells were washed and then incubated with substrates for 20 minutes. The absorbance was read on a microplate reader using 450nm, and the content of HNE protein adducts was calculated by comparison with a predetermined HNE-BSA standard curve.
Cell culture and drug treatments
N2a cells are mouse neuroblastoma cell line that has been used to study neurite outgrowth, neurotoxicity, and Alzheimer’s disease (LePage et al., 2005). N2a cells were maintained in DMEM medium supplemented with 10% fetal bovine serum (FBS) and 1% streptomycin/penicillin. For ethanol treatment, N2a cells were serum starved overnight and then incubated with complete medium containing 0.2% or 0.4% (volume/volume) ethanol for 12 hours. Sodium phenylbutyrate (4-PBA) is a chemical chaperone used to block the induction of ER stress. We dissolved 4-PBA in water at a concentration of 20 mg/ml and then added it into the medium at a working concentration of 10 mM 2 hours before adding ethanol or tunicamycin. Tunicamycin is a conventional ER stress inducer by inhibiting N-linked glycosylation. We dissolved tunicamycin in dimethyl sulfoxide (DMSO) to a concentration of 10 mg/ml. In tunicamycin treatment, N2a cells were serum starved overnight and then treated with tunicamycin at a working concentration of 0.5 or 1 μg/ml for 12 hours. Rapamycin is an inhibitor for mTOR. Rapamycin was dissolved in DMSO at a concentration of 1 mM. For rapamycin treatment, N2a cells were serum starved overnight and then treated with rapamycin at a working concentration of 0, 0.5, 1, 2, 5 or 10 μM for 12 hours. The corresponding volume of DMSO was used as a control for the treatment of N2a cells.
RNA isolation and Real-time RT-PCR
Total RNA from the brain or liver was extracted using Trizol Reagent (Life Technologies) and treated with RNA-free DNAase I to remove remnant DNA as described previously (Kim et al., 2014). cDNA used for gene detection was synthesized as described previously using SuperScript III reverse transcriptase and random primers (Kim et al., 2014). We purchased the primers used for this study from Fisher Scientific and were as follows: Tnf-α, Mm099999068; IL1β, Mm00434228; IL-6, Mm00446190; MCP-1, Mm00441242; 18s rRNA, Mm03928990. The relative expression was normalized to an internal control (18s rRNA) using cycle time (Ct). The relative difference between control and treatment group was expressed and calculated as relative increases using 2-∆∆Ct and setting control as 1.
Statistics
The data are expressed as mean ± SEM, and statistical significance was determined using Student Unpaired t-test, one-way ANOVA or two-way ANOVA followed by Bonferroni Multiple Comparison test (GraphPad Prism version 7). A p value of less than 0.5 was considered statistically significant.
Results
Binge ethanol exposure induces neurodegeneration and neuroinflammation in mouse brain
Blood ethanol concentration (BEC) was measured on days 1, 5 and 10. The BEC was 351 ± 29 mg/dl on day 1, 374 ± 21 mg/dl on day 5, and 463 ± 25 mg/dl on day 10 in the ethanol-treated group. We first determined the effects of binge ethanol exposure on neurodegeneration. As shown in Fig. 1A-F, binge ethanol exposure for 1 day failed to induce the expression of cleaved caspase-3 and caspase-12. Cleaved caspase-3 is indicative of apoptosis, and cleaved caspase-12 suggests the activation of ER stress- specific apoptosis (Nakagawa et al., 2000). However, binge ethanol exposure for 5 or 10 days significantly increased the levels of cleaved caspase-3 and caspase-12 in the cerebellum and cerebral cortex. We confirmed ethanol-induced neurodegeneration using Fluoro-Jade C, a marker for degenerating neurons. As shown in Fig. 1G, ethanol exposure for 10 days increased the number of Fluoro-Jade C-positive cells in the cerebellum and cortex. The Fluoro-Jade C-labeled cells colocalized with NeuN, a marker of mature neurons, indicating neurodegeneration. Consistent with the findings in the brain, ethanol also induced apoptosis in the liver, the major organ for ethanol metabolism (Supplementary Fig. 2A-B). Ethanol also increased the expression of some proinflammatory cytokines and chemokines; binge ethanol exposure up-regulated IL-6 in the cerebellum and MCP-1 and IL1β in the cerebral cortex following 10 days of exposure (Fig. 1H).
Fig. 1. Binge ethanol exposure induces neurodegeneration in mouse brain.
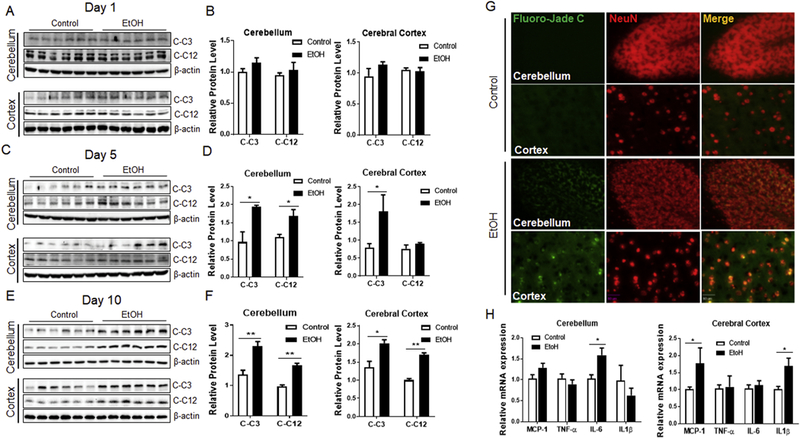
A, C, E. C57BL/6 mice received water (control) or ethanol intragastrically for 1, 5, or 10 days and brain tissues were dissected to assess the level of cleaved caspase-3 (C-C3) and cleaved caspase-12 (C-C12) by Western blotting. B, D, F. The protein levels of C-C3 and C-C12 were normalized to β-actin and expressed relative to control. Six animals (n=6) were used in each group to calculate the mean ± SEM. *P<0.05 or **P<0.01 vs. control. G. Representative images showing Fluoro-Jade C- (green) and NeuN- (red) positive cells in the brain of either water- or ethanol-treated mice for 10 days. H. Total RNA was prepared from the cerebellum or cerebral cortex of water- (control) or ethanol- treated mice for 10 days to assess the level of proinflammatory cytokines (MCP-1, TNF- α, IL-6, and IL1β) by RT-qPCR. The mRNA level of these cytokines was normalized to 18s rRNA and expressed relative to control. Three to six samples (n = 3–6) were used in each group to calculate the mean ± SEM. *P<0.05 vs. control. Scale bar: 20 μm.
Binge ethanol exposure induces endoplasmic reticulum stress in mouse brain
With this paradigm, we next sought to determine whether binge ethanol exposure induces ER stress. Consistent with the timeline of neurodegeneration, binge ethanol exposure for 1 day did not affect the levels of ER stress markers (Fig. 2A and B). However, ethanol exposure for 5 and 10 days significantly increased the levels of ER stress markers including GRP78, p-eIF2α, CHOP, XBP1s, and ATF6 both in the cerebellum and cerebral cortex (Fig. 2C-F). Ethanol also drastically increased the levels of ER stress markers in the liver (Supplementary Fig. 2A and B). Oxidative stress has been suggested as a potential mechanism for ethanol-induced neurodegeneration (Lucas et al., 2006; Block and Hong, 2007). Therefore, we examined the level of oxidative stress following binge ethanol exposure. While ethanol caused oxidative stress in the liver, manifested by an increased level of protein carbonyl and HNE adducts, it had little effect on that in the brain (Supplementary Fig. 1).
Fig. 2. Binge ethanol exposure induces endoplasmic reticulum stress in mouse brain.
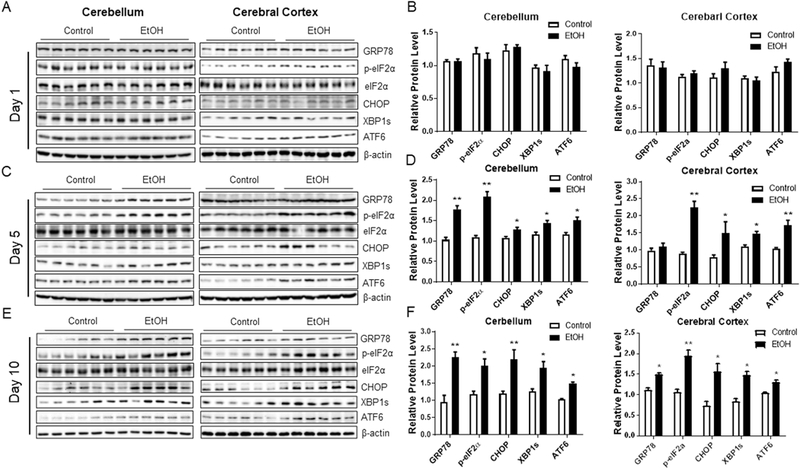
A, C, E. C57BL/6 mice were treated with water or ethanol as described in Figure 1 and brain tissues were dissected to assess the level of ER stress markers by Western blotting. B, D, F. The protein levels of ER stress markers were normalized to β-actin and expressed relative to control. Six animals (n = 6) were used in each group to calculate the mean ± SEM. *P<0.05 or **P<0.01 vs. control.
Binge ethanol exposure downregulates mTOR signaling in mouse brain
Since there is evidence of considerable interaction between mTOR signaling and ER stress (Appenzeller-Herzog and Hall, 2012), we assessed the effects of binge ethanol exposure on mTOR signaling in the brain. We found that binge ethanol exposure downregulated mTOR activity and its downstream signaling in the mouse brain, manifested by the decreased level of p-mTOR, and its target effectors, p-4EBP1 and p-p70S6K (Fig. 3C-F). Interestingly, the timeline of ethanol-induced downregulation of mTOR signaling paralleled to that of neurodegeneration and ER stress; that is, ethanol-induced downregulation of mTOR was only evident after 5 days of ethanol exposure.
Fig. 3. Binge ethanol exposure downregulates mTOR signaling in mouse brain.
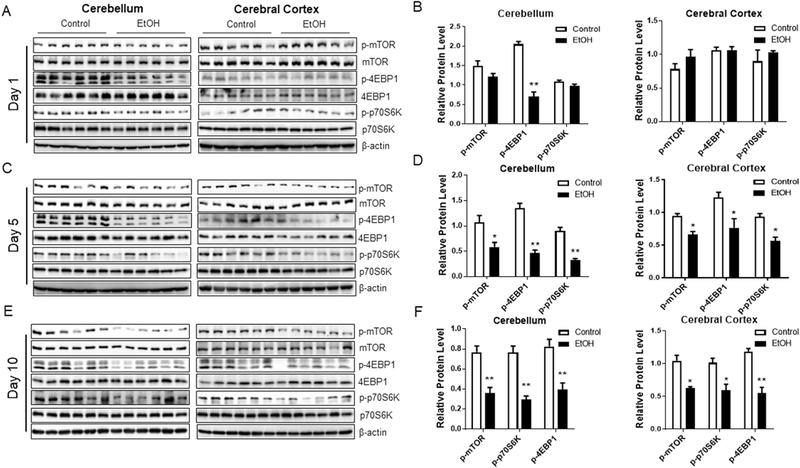
A, C, E. C57BL/6 mice were treated with water or ethanol as described in Figure 1 and brain tissues were dissected to assess the activity of mTOR signaling by Western blotting. B, D, F. The protein levels of p-mTOR and its downstream effectors were normalized to β-actin and expressed relative to control. Six animals (n = 6) were used in each group to calculate the mean ± SEM. *P<0.05 or **P<0.01 vs. control.
Inhibition of ER stress ameliorates ethanol-induced neurodegeneration
To determine whether ER stress contributes to ethanol-induced neurodegeneration, we performed in vitro experiments using a mouse neuroblastoma cell line (N2a). First, we found that ethanol at a concentration of 0.2% or 0.4% was able to increase the level of ER stress markers (GRP78, XBP1s, and ATF6) and cleaved caspase-3 in N2a cells (Fig. 4A, B, C, D, and F), confirming ethanol-induced ER stress. Consistent with the findings in vivo, ethanol also downregulated mTOR activity in N2a cells (Fig. 4A and E). Moreover, cotreatment with 4-PBA, a chemical chaperone used to inhibit ER stress, was able to alleviate the induction of ER stress and rescue neuronal apoptosis elicited by ethanol treatment (Fig. 4A, B, C, D, and F). These results suggest ER stress contributes to ethanol-induced neurodegeneration.
Fig. 4. Inhibition of ER stress reduces ethanol-induced neuronal death.
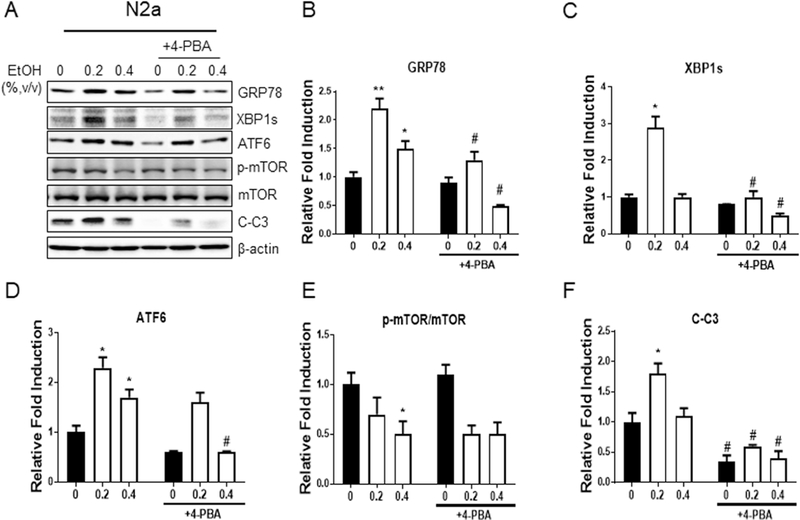
A. N2a cells were serum starved overnight, followed by pretreatment with or without 4-PBA (10 mM) for 2 hours before ethanol treatment (0.2% or 0.4%, v/v) for 12 hours. Cell lysates were collected to assess the levels of ER stress markers and cleaved caspase-3 (C-C3) by Western blotting. B-F. The protein levels of ER stress markers and C-C3 were normalized to β-actin and expressed relative to control (without treatment by ethanol and 4-PBA). Three independent experiments (n = 3) were performed to calculate the mean ± SEM. *P<0.05 or **P<0.01 vs. control. #P<0.05 vs. the corresponding points without 4-PBA treatment.
Inhibition of mTOR signaling contributes to the induction of ER stress
To delineate the crosstalk between mTOR signaling and ethanol-induced ER stress, we used pharmaceutical approaches to manipulate mTOR signaling and ER stress in N2a cells. The treatment of tunicamycin, an ER stress inducer by inhibiting N- glycosylation, caused ER stress while 4-PBA alleviated ER stress; however, they had no effects on mTOR signaling (Fig. 5A-E). Likewise, although 4-PBA could alleviate ethanol-induced ER stress, it could not reverse ethanol-mediated downregulation of mTOR signaling in N2a cells (Fig. 4A and E). Conversely, rapamycin, a mTOR kinase inhibitor, inhibited mTOR signaling and meanwhile induced ER stress (Fig. 5F-J). Collectively, these results suggest that mTOR signaling operates upstream of ethanol- induced ER stress and is involved in the regulation of ER stress in response to binge ethanol exposure.
Fig. 5. Inhibition of mTOR signaling induces ER stress.
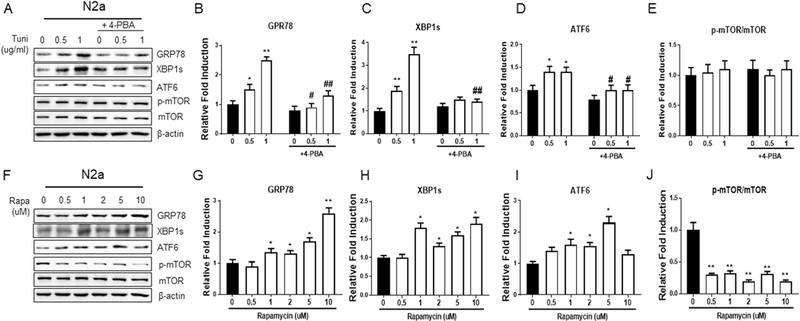
A. N2a cells were serum starved overnight, followed by pretreatment with or without 4-PBA (10 mM) for 2 hours before tunicamycin treatment (0.5 or 1 ug/ml) for 12 hours. Cell lysates were collected to assess the levels of ER stress markers and mTOR signaling. B-E. The protein levels of ER stress markers and p-mTOR/mTOR were normalized to β-actin and expressed relative to control (without treatment by tunicamycin and 4-PBA). Three independent experiments (n = 3) were performed to calculate the mean ± SEM. *P<0.05 or **P<0.01 vs. control. #P<0.05 vs. the corresponding points without 4-PBA treatment. F. N2a cells were serum starved overnight followed by treatment with rapamycin (0, 0.5, 1, 2, 5 or 10 uM) for 12 hours to assess the level of ER stress markers and mTOR signaling. G-J. Quantification was normalized to β-actin and expressed relative to control (without rapamycin treatment) and calculated as mean ± SEM for three independent experiments (n = 3). *P<0.05 or **P<0.01 vs. control.
Effects of binge ethanol exposure on animal behaviors
To determine whether the paradigm of binge ethanol exposure affects animal behaviors, we evaluated locomotor activity by Open Field Activity (OFA), anxiety-like behaviors by Elevated Plus Maze (EPM), and spatial-based learning and memory by Morris Water Maze (MWM) following ethanol exposure. For OFA, although the mice in the ethanol group showed a tendency of less activity, the total distance traveled was not significantly different between the control group and ethanol group (Fig. 6A, p=0.0697). Also during OFA, the mice in the ethanol group spent a similar period in the center compared with the mice in the control group (Fig. 6B, p=0.4717). For EPM, the ratio of open arm entries versus total entries and the total distance traveled between ethanol group and control group was also not significant (Fig. 6C, p=0.0545; Fig. 6D, p=0.0757). Lastly, the results from the MWM test showed there was no significant difference in latency and traveled distance between the control group and ethanol group (Fig. 6E, p=0.2816; Fig. 6F, p=0.3285). Altogether, these results suggested that this paradigm of binge ethanol exposure did not cause a significant change in locomotor activity, anxietylike behaviors, and spatial memory and learning.
Fig. 6. The effects of ethanol-induced neurodegeneration on animal behaviors.
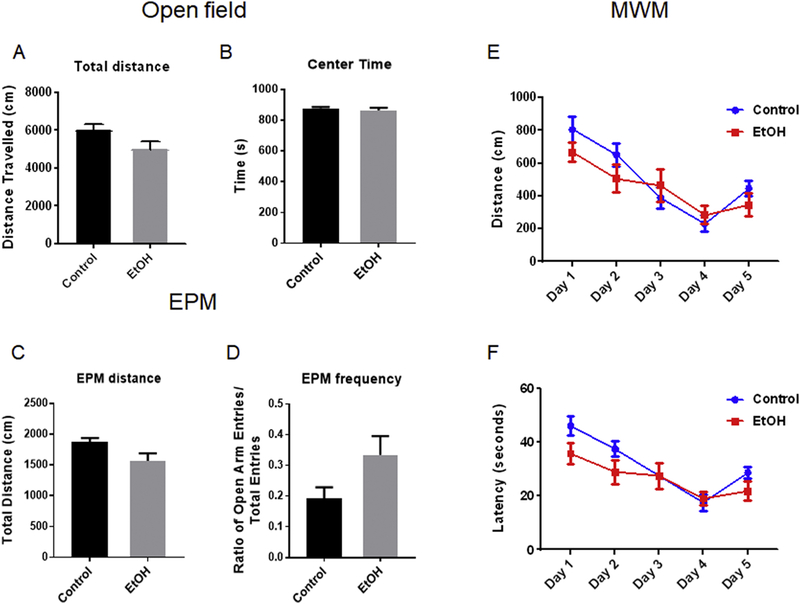
C57BL/6 mice received water (control) or ethanol intragastrically for 10 days. At 3 days after the last gavaging, animals were subjected to Open Field Activity (OFA) test (A-B) once. After 2 hours following OFA, animals were subjected to Elevated Plus Maze (EPM) test once (C-D). At 5 days after the last gavaging, animals were subjected to Morris Water Maze (MWM) test (E-F) for 5 consecutive days. Quantification of each parameter was calculated as the mean ± SEM of ten animals (n = 10) in each group. Unpaired student t-test was used to assess the difference between control group and ethanol group in OFA and EPM and two-way ANOVA was used in MWM to assess the difference between these two groups.
Discussion
We demonstrated that binge ethanol exposure caused duration-dependent neurodegeneration, neuroinflammation, and ER stress in the cerebellum and cortex of adult mouse brain. However, this paradigm of ethanol exposure did not cause significant alterations in behavioral outcomes which were tested by OFA, EPM, and MWM. In vitro studies suggested that ER stress contributes to the ethanol-induced death of neuronal cells. It appeared that mTOR signaling pathway was involved in ethanol-induced ER stress.
This paradigm of binge ethanol exposure produced high BECs (300–400 mg/dl). Although the National Institute on Alcohol Abuse and Alcoholism (NIAAA) defines binge drinking as a pattern of drinking leading to a BEC of 80 mg/dl, the actual amount of alcohol consumed in human alcoholics is much higher. BECs greater than 300 mg/dl are not uncommon for human alcoholics (Cartlidge and Redmond, 1990; Jones, 1999; van Hoof et al., 2011; Jones and Harding, 2013; Malejko et al., 2016). Some alcoholics with a BEC larger than 400 mg/dl are coherent and able to drive (Cartlidge and Redmond, 1990; Jones and Harding, 2013) and BECs greater than 500 mg/dl were reported in heavy drinkers (Jones, 1999; van Hoof et al., 2011; Malejko et al., 2016). The paradigms of binge ethanol exposure similar to ours have been used to investigate ethanol-induced neurodegeneration, glial activation, and neuroinflammation in the brain of adult and adolescent mice (Qin et al., 2008; Qin and Crews, 2012; Kane et al., 2014).
Multiple mechanisms have been proposed for ethanol-induced neurodegeneration including neuronal excitotoxicity, oxidative stress, and proinflammatory response (Lovinger, 1993; Zou and Crews, 2010). In our binge ethanol drinking mice, ethanol caused little oxidative stress in the brain, although it induced significant oxidative stress in the liver. Nevertheless, we found that IL-6 in the cerebellum and MCP-1 and IL1β in the cerebral cortex were induced by ethanol exposure, suggesting that proinflammation occurred in the mouse brain. These proinflammatory cytokines may at least partially contribute to ethanol-induced neurodegeneration. ER stress is the cellular machinery used to promote the proper folding and secretion of proteins and can initiate a proapoptotic programme when the folding challenge is overwhelming. We showed that binge ethanol exposure induced ER stress manifested by an increase of ER stress markers, such as GRP78, p-eIF2α, CHOP, ATF6, and XBP1s. Interestingly, the timeline of ethanol-induced ER stress correlated well with that of ethanol-induced neurodegeneration. Ethanol activated caspase-12 which is located in the ER and activated by ER stress (Nakagawa et al., 2000; Shiraishi et al., 2006). Cleaved caspase-12 may activate other caspases including caspase-3, and caspase-9 (Morishima et al., 2002; Kerbiriou et al., 2009). The activation of caspase-12 leads us to hypothesize that ER stress may be involved in ethanol-induced neurodegeneration. This hypothesis was supported by our in vitro study showing that 4-PBA, an ER stress inhibitor, significantly reduced ethanol-induced ER stress as well as the death of neuronal cells. Together, these data suggested that ER stress may at least partially mediate ethanol-induced neurodegeneration.
The effect of ethanol on ER stress in the mature brain is different from that in the developing brain. Our previous study indicated that ethanol rapidly triggered neurodegeneration and ER stress in the developing brain; ethanol-induced ER stress in the neonatal brain was evident after 4–8 hours of exposure, and a drastic caspase-3 activation and neurodegeneration were observed after 8 hours of exposure (Ke et al., 2011). In the adult brain, however, ethanol-induced neurodegeneration and ER stress were only observed after 5 days of exposure and to a much lesser extent. These results imply that the adult brain has a more mature unfolded protein response (UPR) system that can better cope with ER stress.
The underlying mechanisms for ethanol-induced ER stress are unclear. Acetaldehyde, a metabolite of ethanol can form adducts with proteins, nucleic acids, and lipids, which may affect protein folding (Ji, 2012). Ethanol was also reported to cause the alteration of Ca2+ homeostasis in the ER (Ji, 2012), which could lead to ER stress. Additionally, ethanol metabolism generates ROS, thereby disrupting the redox status in the ER that leads to ER stress (Ji, 2012). Neuroinflammation has also been shown to initiate ER stress in the brain (Lin et al., 2013; Clayton and Popko, 2016; Patel et al., 2017). mTOR belongs to the family of serine/threonine kinases. It exists in two complexes, mTORC1 and mTORC2 that have different subunit compositions and undertake distinct cellular tasks (Appenzeller-Herzog and Hall, 2012). It has been reported that ethanol downregulated mTOR signaling to promote autophagy in the developing brain, and cultured astrocytes and neurons (Luo, 2014; Pla et al., 2016). The current study showed that binge ethanol exposure downregulates mTOR signaling both in vivo and in vitro. However, we did not observe a significant increase of autophagy in the brain following ethanol exposure (data not shown). Instead, inhibition of mTOR signaling by rapamycin could induce ER stress in neuronal cells. While 4-PBA could diminish ethanol- or tunicamycin-induced ER stress, it could not reverse the ethanol- induced inhibition of mTOR signaling. This data suggested that mTOR signaling was upstream of ethanol-induced ER stress. Hence, it is likely that ER stress might act as a hub that converges signals from inflammatory response, oxidative stress and mTOR signaling.
While the effect of binge ethanol exposure on behavioral deficits in rats was well discussed in previous studies (Nixon and Crews, 2002; Nixon and Crews, 2004; Crews and Nixon, 2009; Morris et al., 2010), only a few studies focused on binge ethanol exposure-induced behavioral deficits in adult mice. Some studies showed that binge ethanol exposure led to behavioral deficits, such as impaired spatial learning and memory, particularly in adolescent rats (Thomas et al., 1996; Obernier et al., 2002). We did not observe significant behavioral alterations on our mice. There are several possibilities for this finding. First, it is possible that the extent of neurodegeneration under this paradigm is not adequate to incur the behavioral alterations. It may be necessary to extend the duration of ethanol exposure in future studies. Second, ethanol-induced alterations in behaviors may be reversible. The current behavioral tests were performed 3–5 days after ethanol exposure. Third, depending on the animal model and timing/dosage of ethanol exposure, ethanol could also induce neurogenesis during or after ethanol exposure (Aberg et al., 2005; Taffe et al., 2010). Our results showed binge ethanol exposure increases neurogenesis/proliferation in the subgranular zone (SGZ) and subventricular zone (SVZ) after 10 days of ethanol exposure (Supplementary Fig. 3A-C), manifested by the increase of doublecortin (DCX) and Ki-67. The newly developed neurons by neurogenesis may compensate for the neuronal deficit elicited by binge ethanol exposure.
In summary, binge ethanol exposure induces ER stress and neurodegeneration in adult mice. The induction of ER stress contributes to ethanol-induced neurodegeneration. Mechanistically, ethanol-induced ER stress is regulated at least partially by mTOR signaling. Nevertheless, it remains to be confirmed whether ER stress indeed contributes to ethanol-induced neurodegeneration in vivo. Additionally, the mechanisms for ethanol-induced ER stress have not been completely elucidated.
For example, the pathways/intermediates bridging mTOR signaling and ER stress is unknown. Also, does ethanol-induced neuroinflammation contributes to the induction of ER stress or vice versa? A better understanding of the role of ER stress in ethanol- induced neurodegeneration may offer novel approaches in the prevention and treatment of ethanol-associated neurotoxicity.
Supplementary Material
Binge ethanol exposure causes ER stress and neurodegeneration.
Blocking ER stress rescues neurons from ethanol neurotoxicity.
mTOR signaling is involved in ethanol-induced ER stress.
Acknowledgments
This study was supported by grants from the National Institute of Health (NIH) [AA017226 and AA015407] and NIH Training Grant [T32 DK007778]. It is also supported in part by the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development [Biomedical Laboratory Research and Development: Merit Review (BX001721)]. We would like to thank the University of Kentucky Rodent Behavior Core for their assistance with the behavioral tests.
Abbreviation
- ATF6
Activating transcription factor 6
- IRE1α
Inositol-requiring kinase
- PERK
Protein kinase RNA-like endoplasmic reticulum kinase
- CHOP
CCAAT/-enhancer-binding protein homologous protein
- eIF2α
Eukaryotic initiation factor 2α
- XBPIs
X-box binding protein-ls
- mTOR
Mammalian target of rapamycin
- p70S6K
Ribosomal protein S6 Kinase beta-1
- 4EBP1
Eukaryotic translation initiation factor 4E-binding protein 1
- DAPI
4’, 6-diamidino-2-phenylindole
- 4-PBA
Sodium phenylbutyrate
- DNPH
2,4-dinitrophenylhydrazine
- HNE
4-Hydroxynonenal
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
All authors have read and approved the manuscript. We do not have conflict of interest. This manuscript has not been and will not be submitted or published in other scientific journals in whole or in part while it is under the consideration of Toxicol Appl Pharmacol
References
- Aberg E, Hofstetter CP, Olson L, Brene S, 2005. Moderate ethanol consumption increases hippocampal cell proliferation and neurogenesis in the adult mouse. Int J Neuropsychopharmacol 8, 557–567. [DOI] [PubMed] [Google Scholar]
- Appenzeller-Herzog C, Hall MN, 2012. Bidirectional crosstalk between endoplasmic reticulum stress and mTOR signaling. Trends Cell Biol 22, 274–282. [DOI] [PubMed] [Google Scholar]
- Banks WA, Erickson MA, 2010. The blood-brain barrier and immune function and dysfunction. Neurobiol Dis 37, 26–32. [DOI] [PubMed] [Google Scholar]
- Block ML, Hong JS, 2007. Chronic microglial activation and progressive dopaminergic neurotoxicity. Biochem Soc Trans 35, 1127–1132. [DOI] [PubMed] [Google Scholar]
- Bowden SC, Crews FT, Bates ME, Fals-Stewart W, Ambrose ML, 2001. Neurotoxicity and neurocognitive impairments with alcohol and drug-use disorders: potential roles in addiction and recovery. Alcoholism, clinical and experimental research 25, 317–321. [PubMed] [Google Scholar]
- Boyadjieva NI, Sarkar DK, 2013. Microglia play a role in ethanol-induced oxidative stress and apoptosis in developing hypothalamic neurons. Alcoholism, clinical and experimental research 37, 252–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartlidge D, Redmond AD, 1990. Alcohol and conscious level. Biomed Pharmacother 44, 205–208. [DOI] [PubMed] [Google Scholar]
- Chen G, Ke Z, Xu M, Liao M, Wang X, Qi Y, Zhang T, Frank JA, Bower KA, Shi X, Luo J, 2012. Autophagy is a protective response to ethanol neurotoxicity. Autophagy 8, 1577–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Ma C, Bower KA, Shi X, Ke Z, Luo J, 2008. Ethanol promotes endoplasmic reticulum stress-induced neuronal death: involvement of oxidative stress. J Neurosci Res 86, 937–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton BLL, Popko B, 2016. Endoplasmic reticulum stress and the unfolded protein response in disorders of myelinating glia. Brain Res 1648, 594–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins MA, Corso TD, Neafsey EJ, 1996. Neuronal degeneration in rat cerebrocortical and olfactory regions during subchronic “binge” intoxication with ethanol: possible explanation for olfactory deficits in alcoholics. Alcoholism, clinical and experimental research 20, 284–292. [DOI] [PubMed] [Google Scholar]
- Crews FT, Braun CJ, Hoplight B, Switzer RC 3rd, Knapp DJ, 2000. Binge ethanol consumption causes differential brain damage in young adolescent rats compared with adult rats. Alcoholism, clinical and experimental research 24, 1712–1723. [PubMed] [Google Scholar]
- Crews FT, Nixon K, 2003. Alcohol, neural stem cells, and adult neurogenesis. Alcohol research & health: the journal of the National Institute on Alcohol Abuse and Alcoholism 27, 197–204. [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Nixon K, 2009. Mechanisms of neurodegeneration and regeneration in alcoholism. Alcohol and Alcoholism 44, 115–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dara L, Ji C, Kaplowitz N, 2011. The contribution of endoplasmic reticulum stress to liver diseases. Hepatology 53, 1752–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez GM, Lew BJ, Vedder LC, Savage LM, 2017. Chronic intermittent ethanol exposure leads to alterations in brain-derived neurotrophic factor within the frontal cortex and impaired behavioral flexibility in both adolescent and adult rats. Neuroscience 348, 324–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrier L, Berard F, Debrauwer L, Chabo C, Langella P, Bueno L, Fioramonti J, 2006. Impairment of the intestinal barrier by ethanol involves enteric microflora and mast cell activation in rodents. The American journal of pathology 168, 1148–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerri C, Montoliu C, Renau-Piqueras J, 1994. Involvement of free radical mechanism in the toxic effects of alcohol: implications for fetal alcohol syndrome. Advances in experimental medicine and biology 366, 291–305. [DOI] [PubMed] [Google Scholar]
- Harding AJ, Wong A, Svoboda M, Kril JJ, Halliday GM, 1997. Chronic alcohol consumption does not cause hippocampal neuron loss in humans. Hippocampus 7, 78–87. [DOI] [PubMed] [Google Scholar]
- Hetz C, Saxena S, 2017. ER stress and the unfolded protein response in neurodegeneration. Nat Rev Neurol 13, 477–491. [DOI] [PubMed] [Google Scholar]
- Ji C, 2012. Mechanisms of alcohol-induced endoplasmic reticulum stress and organ injuries. Biochem Res Int 2012, 216450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji C, Kaplowitz N, 2006. ER stress: can the liver cope? Journal of Hepatology 45, 321–333. [DOI] [PubMed] [Google Scholar]
- Ji C, Kaplowitz N, Lau MY, Kao E, Petrovic LM, Lee AS, 2011. Liver-specific loss of glucose-regulated protein 78 perturbs the unfolded protein response and exacerbates a spectrum of liver diseases in mice. Hepatology 54, 229–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji C, Mehrian-Shai R, Chan C, Hsu YH, Kaplowitz N, 2005. Role of CHOP in hepatic apoptosis in the murine model of intragastric ethanol feeding. Alcoholism, clinical and experimental research 29, 1496–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Z, Yuan L, Lu X, Ding H, Luo J, Ke ZJ, 2018. Binge Alcohol Exposure Causes Neurobehavioral Deficits and GSK3beta Activation in the Hippocampus of Adolescent Rats. Sci Rep 8, 3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones AW, 1999. The drunkest drinking driver in Sweden: blood alcohol concentration 0.545% w/v. J Stud Alcohol 60, 400–406. [DOI] [PubMed] [Google Scholar]
- Jones AW, Harding P, 2013. Driving under the influence with blood alcohol concentrations over 0.4 g%. Forensic Sci Int 231, 349–353. [DOI] [PubMed] [Google Scholar]
- Kane CJ, Phelan KD, Douglas JC, Wagoner G, Johnson JW, Xu J, Phelan PS, Drew PD, 2014. Effects of ethanol on immune response in the brain: region-specific changes in adolescent versus adult mice. Alcoholism, clinical and experimental research 38, 384–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Nakajima S, Saito Y, Takahashi S, Katoh R, Kitamura M, 2012. mTORC1 serves ER stress-triggered apoptosis via selective activation of the IRE1-JNK pathway. Cell Death Differ 19, 310–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik S, Cuervo AM, 2015. Proteostasis and aging. Nature medicine 21, 1406–1415. [DOI] [PubMed] [Google Scholar]
- Ke Z, Wang X, Liu Y, Fan Z, Chen G, Xu M, Bower KA, Frank JA, Li M, Fang S, Shi X, Luo J, 2011. Ethanol induces endoplasmic reticulum stress in the developing brain. Alcoholism, clinical and experimental research 35, 1574–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerbiriou M, Teng L, Benz N, Trouve P, Ferec C, 2009. The calpain, caspase 12, caspase 3 cascade leading to apoptosis is altered in F508del-CFTR expressing cells. PloS one 4, e8436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim IM, Wang Y, Park KM, Tang Y, Teoh JP, Vinson J, Traynham CJ, Pironti G, Mao L, Su H, Johnson JA, Koch WJ, Rockman HA, 2014. beta-arrestin1-biased beta1-adrenergic receptor signaling regulates microRNA processing. Circulation research 114, 833–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratsovnik E, Bromberg Y, Sperling O, Zoref-Shani E, 2005. Oxidative stress activates transcription factor NF-kB-mediated protective signaling in primary rat neuronal cultures. Journal of molecular neuroscience: MN 26, 27–32. [DOI] [PubMed] [Google Scholar]
- Kubisch CH, Sans MD, Arumugam T, Ernst SA, Williams JA, Logsdon CD, 2006. Early activation of endoplasmic reticulum stress is associated with arginine-induced acute pancreatitis. American journal of physiology. Gastrointestinal and liver physiology 291, G238–245. [DOI] [PubMed] [Google Scholar]
- Leclercq S, Cani PD, Neyrinck AM, Starkel P, Jamar F, Mikolajczak M, Delzenne NM, de Timary P, 2012. Role of intestinal permeability and inflammation in the biological and behavioral control of alcohol-dependent subjects. Brain Behav Immun 26, 911–918. [DOI] [PubMed] [Google Scholar]
- LePage KT, Dickey RW, Gerwick WH, Jester EL, Murray TF, 2005. On the use of neuro-2a neuroblastoma cells versus intact neurons in primary culture for neurotoxicity studies. Crit Rev Neurobiol 17, 27–50. [DOI] [PubMed] [Google Scholar]
- Li SY, Gilbert SA, Li Q, Ren J, 2009. Aldehyde dehydrogenase-2 (ALDH2) ameliorates chronic alcohol ingestion-induced myocardial insulin resistance and endoplasmic reticulum stress. J Mol Cell Cardiol 47, 247–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SY, Ren J, 2008. Cardiac overexpression of alcohol dehydrogenase exacerbates chronic ethanol ingestion-induced myocardial dysfunction and hypertrophy: role of insulin signaling and ER stress. J Mol Cell Cardiol 44, 992–1001. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Lin W, Lin Y, Li J, Fenstermaker AG, Way SW, Clayton B, Jamison S, Harding HP, Ron D, Popko B, 2013. Oligodendrocyte-specific activation of PERK signaling protects mice against experimental autoimmune encephalomyelitis. The Journal of neuroscience: the official journal of the Society for Neuroscience 33, 5980–5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM, 1993. Excitotoxicity and alcohol-related brain damage. Alcoholism, clinical and experimental research 17, 19–27. [DOI] [PubMed] [Google Scholar]
- Lucas SM, Rothwell NJ, Gibson RM, 2006. The role of inflammation in CNS injury and disease. British journal of pharmacology 147 Suppl 1, S232–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugea A, Tischler D, Nguyen J, Gong J, Gukovsky I, French SW, Gorelick FS, Pandol SJ, 2011. Adaptive unfolded protein response attenuates alcohol- induced pancreatic damage. Gastroenterology 140, 987–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, 2014. Autophagy and ethanol neurotoxicity. Autophagy 10, 2099–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malejko K, Graf H, Gahr M, 2016. Survival of Very High Blood Alcohol Concentration Without Consequential Damage in a Patient Without a Previous Substance Use Disorder. J Forensic Sci 61, 1155–1157. [DOI] [PubMed] [Google Scholar]
- Marciniak SJ, Ron D, 2006. Endoplasmic reticulum stress signaling in disease. Physiological reviews 86, 1133–1149. [DOI] [PubMed] [Google Scholar]
- Morgan MJ, Liu ZG, 2011. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res 21, 103–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishima N, Nakanishi K, Takenouchi H, Shibata T, Yasuhiko Y, 2002. An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. The Journal of biological chemistry 277, 34287–34294. [DOI] [PubMed] [Google Scholar]
- Morris SA, Eaves DW, Smith AR, Nixon K, 2010. Alcohol inhibition of neurogenesis: a mechanism of hippocampal neurodegeneration in an adolescent alcohol abuse model. Hippocampus 20, 596–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J, 2000. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 403, 98–103. [DOI] [PubMed] [Google Scholar]
- Nardelli A, Lebel C, Rasmussen C, Andrew G, Beaulieu C, 2011. Extensive deep gray matter volume reductions in children and adolescents with fetal alcohol spectrum disorders. Alcoholism, clinical and experimental research 35, 1404–1417. [DOI] [PubMed] [Google Scholar]
- Nixon K, Crews FT, 2002. Binge ethanol exposure decreases neurogenesis in adult rat hippocampus. Journal of neurochemistry 83, 1087–1093. [DOI] [PubMed] [Google Scholar]
- Nixon K, Crews FT, 2004. Temporally specific burst in cell proliferation increases hippocampal neurogenesis in protracted abstinence from alcohol. The Journal of neuroscience: the official journal of the Society for Neuroscience 24, 9714–9722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obernier JA, White AM, Swartzwelder HS, Crews FT, 2002. Cognitive deficits and CNS damage after a 4-day binge ethanol exposure in rats. Pharmacol Biochem Behav 72, 521–532. [DOI] [PubMed] [Google Scholar]
- Pandol SJ, Gorelick FS, Gerloff A, Lugea A, 2010. Alcohol abuse, endoplasmic reticulum stress and pancreatitis. Dig Dis 28, 776–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S, Sharma D, Kalia K, Tiwari V, 2017. Crosstalk between endoplasmic reticulum stress and oxidative stress in schizophrenia: The dawn of new therapeutic approaches. Neurosci Biobehav Rev. [DOI] [PubMed] [Google Scholar]
- Pfefferbaum A, Lim KO, Zipursky RB, Mathalon DH, Rosenbloom MJ, Lane B, Ha CN, Sullivan EV, 1992. Brain gray and white matter volume loss accelerates with aging in chronic alcoholics: a quantitative MRI study. Alcoholism, clinical and experimental research 16, 1078–1089. [DOI] [PubMed] [Google Scholar]
- Pla A, Pascual M, Guerri C, 2016. Autophagy Constitutes a Protective Mechanism against Ethanol Toxicity in Mouse Astrocytes and Neurons. PloS one 11, e0153097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Crews FT, 2012. NADPH oxidase and reactive oxygen species contribute to alcohol-induced microglial activation and neurodegeneration. J Neuroinflammation 9, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, He J, Hanes RN, Pluzarev O, Hong JS, Crews FT, 2008. Increased systemic and brain cytokine production and neuroinflammation by endotoxin following ethanol treatment. J Neuroinflammation 5, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin RW, Long JC, Rasmussen JK, Albaugh B, Goldman D, 1998. Relationship of binge drinking to alcohol dependence, other psychiatric disorders, and behavioral problems in an American Indian tribe. Alcoholism, clinical and experimental research 22, 518–523. [PubMed] [Google Scholar]
- Ron D, 2002. Translational control in the endoplasmic reticulum stress response. The Journal of clinical investigation 110, 1383–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmued LC, Stowers CC, Scallet AC, Xu L, 2005. Fluoro-Jade C results in ultra high resolution and contrast labeling of degenerating neurons. Brain Res 1035, 24–31. [DOI] [PubMed] [Google Scholar]
- Shiraishi H, Okamoto H, Yoshimura A, Yoshida H, 2006. ER stress-induced apoptosis and caspase-12 activation occurs downstream of mitochondrial apoptosis involving Apaf-1. Journal of cell science 119, 3958–3966. [DOI] [PubMed] [Google Scholar]
- Taffe MA, Kotzebue RW, Crean RD, Crawford EF, Edwards S, Mandyam CD, 2010. Long-lasting reduction in hippocampal neurogenesis by alcohol consumption in adolescent nonhuman primates. Proceedings of the National Academy of Sciences of the United States of America 107, 11104–11109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JD, Wasserman EA, West JR, Goodlett CR, 1996. Behavioral deficits induced by binge-like exposure to alcohol in neonatal rats: importance of developmental timing and number of episodes. Dev Psychobiol 29, 433–452. [DOI] [PubMed] [Google Scholar]
- Van Hoof JJ, Van Der Lely N, Bouthoorn SH, Van Dalen WE, Pereira RR, 2011. Adolescent alcohol intoxication in the Dutch hospital departments of pediatrics: a 2-year comparison study. J Adolesc Health 48, 212–214. [DOI] [PubMed] [Google Scholar]
- Wang H, Wang X, Ke ZJ, Comer AL, Xu M, Frank JA, Zhang Z, Shi X, Luo J, 2015. Tunicamycin-induced unfolded protein response in the developing mouse brain. Toxicol Appl Pharmacol 283, 157–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Morse AC, Koob GF, Schulteis G, 2007. Dose- and time-dependent expression of anxiety-like behavior in the elevated plus-maze during withdrawal from acute and repeated intermittent ethanol intoxication in rats. Alcoholism, clinical and experimental research 31, 1811–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J, Crews F, 2010. Induction of innate immune gene expression cascades in brain slice cultures by ethanol: key role of NF-kappaB and proinflammatory cytokines. Alcoholism, clinical and experimental research 34, 777–789. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.