

Abstract
The p53 tumor suppressor is a transcription factor (TF) that exerts antitumor functions through its ability to regulate the expression of multiple genes. Within the p53 protein resides a relatively short unstructured C-terminal domain (CTD) that remarkably participates in virtually every aspect of p53 performance as a TF. Because these aspects are often interdependent and it is not always possible to dissect them experimentally, there has been a great deal of controversy about the CTD. In this review we evaluate the significance and key features of this interesting region of p53 and its impact on the many aspects of p53 function in light of previous and more recent findings.
The p53 CTD: 25 Years and Counting
The p53 protein (see Glossary; also known as TP53), is a bona fide transcription factor (TF) (Box 1) that is conserved from mammals to Placosoans [1] and primarily exerts its ability to protect the genomes of the organisms it inhabits through its ability to regulate expression of multiple genes [2,3]. These target genes mediate numerous cellular outcomes including cell-cycle arrest, cell death, senescence, DNA repair, and several metabolic pathways [4,5]. In response to a variety of diverse stimuli, p53 undergoes (i) rapid and timely stabilization of its protein levels, (ii) efficient and specific binding to p53 cis elements within target promoter sequences, (iii) tissue-, time-, and stimulus-specific binding of numerous coactivators and modifiers, and (iv) dissociation from the DNA and ubiquitin-dependent degradation. The multidomain structure of the p53 protein (Figure 1) reflects the versatile organization of numerous transcriptional regulators in eukaryotes, which almost invariably possess regions necessary for transcriptional activation, DNA binding, and oligomerization [6–8]. In agreement with these canonical features p53 possesses a transcriptional activation region within its N terminus (NTD residues 1–61) that comprises two linked transactivation domains TAD I and II (NTD region; residues 20–40 and 40–61 respectively) that are N-terminal to a proline-rich domain (PR, residues 64–92) that can also regulate transcriptional choices [9]. This complex transactivation region within the N-terminal portion of p53 is adjacent to the large (~200 amino acid) central sequence specific ‘core’ DNA-binding domain (DBD; residues 96–293) [9]. The DBD is followed by a linker region (residues 300–323) and then the oligomerization domain (OD; residues 324–356). At the very end of the protein is a region that is somewhat unique to p53, namely the CTD (residues 364–393 [9]. Note that the above-mentioned residues refer to their locations within the human p53 protein. The individual domains of p53, as well as specific residues within them, have been the subject of many structural, biochemical, and in vivo studies that have generated an extensive list of their functions [2,9].
Box 1. Transcription Factors.
Transcription factors (TFs) are proteins that regulate the synthesis of an RNA copy of the genomic DNA template by DNA-dependent RNA polymerase (RNAP). Depending on the transcription stage TFs may be subdivided into three groups according to whether they regulate the initiation, elongation, or termination of transcription [6].
Some TFs regulate the initiation of transcription through direct binding to their corresponding cis elements (also known as binding sites or response elements) – specific short noncoding DNA sequences that mostly, but not always, reside within the promoters of genes [100]. These DNA sequence-dependent TFs have a modular organization and use their different structural modules (domains) for specific DNA binding (hence, DNA-binding domains, DBDs), transactivation (hence, TADs), and eitherhomo- or hetero-oligomerization (hence, ODs) [7]. The other domain swithin a given TF serve regulatory roles and may be responsible for TF stabilization and degradation, intracellular localization, allosteric regulation, etc.
TFs use their TAD (and other domains) for the recruitment of, and cooperative interaction with, other TFs and cofactors such as histone acetyltransferases (HAT), histone methylases, chromatin-remodeling complexes, etc. [6,100]. Collectively, they initiate and/or regulate the assembly of a promoter-specific transcriptionally-competent preinitiation complex (PIC). Deletions, mutations, or aberrant expression of the genes encoding TFs often result in deregulation of transcription of other genes, which subsequently leads to various cellular abnormalities and diseases [101].p53 isaclassic sequence-specific TF that directly controls the transcription of multiple genes involved in regulation of a diverse spectrum of intracellular processes ranging from metabolism to cell-cycle control and cell death, and is ultimately responsible for counteracting unrestrained cell proliferation and tumor development. Deletions and mutations of the TP53/Tp53 gene that result in either complete loss of p53 protein or expression of its transcriptionally-inactive counterparts are frequently found in numerous types of cancer. Those cancers that carry wild-type p53 very often restrain its activity by other means. Based on epidemiological studies with human cancer patients and genetic mouse models, p53 belongs to the diverse group of proteins known as tumor suppressors.
Figure 1. Domain Organization of p53 and Functions of the C-Terminal Domain.

The upper portion shows p53 domains boundaries (corresponds to human p53 major isoform ∝) together with their general structural classification. Abbreviations: CTD, C-terminal domain; DBD, DNA-binding domain; NTD, N-terminal transactivation domain; OD, oligomerization domain; PR, proline-rich domain. Solved structures of the DBD (PDB: 2AC0) and OD (PDB: 1PES) shown above the schematic p53 representation. The bottom portion of the figure shows the amino acid sequence of the human p53 CTD. Positively charged residues within this domain are in bold blue font. The functions of the CTD discussed in this review are listed.
The p53 CTD, the subject of this review, was first identified as such 25 years ago [10], about the time when the protein was recognized as a sequence-specific TF [11]. The original studies linked this domain with the ability of p53 to recognize various DNA substrates [3]. Remarkably, since then the relatively short unfolded CTD of p53 has been shown to participate in virtually every aspect of p53 performance as a TF, including DNA binding, cofactor recruitment, and protein stabilization. Because these aspects controlled by the CTD are very often interdependent and not always possible to dissect experimentally, there has been a great deal of controversy about this region of p53 [3,12]. In fact, after many years of intensive study, it is still very difficult to integrate all our findings on the CTD of p53 into a comprehensive unifying model.
In this review we evaluate the significance of the CTD and its impact on the many aspects of p53 function as a TF in light of previous and more recent findings. We also discuss the most important distinctive features of the CTD.
The CTD as an Example of an Intrinsically Disordered Region: Less Structure, More Functions
There are three well-defined intrinsically disordered regions (IDRs) within p53 (Box 2 and Figure 1); these are located within the NTD, in the linker region between the DBD and the OD, and in the CTD. Their IDR characteristics have been confirmed by experimental and computational structure-prediction studies [13–15]. A substitution frequency analysis focusing on the NTD and the CTD of p53 revealed substantial differences between these two IDRs in relative distribution of polar, charged, and hydrophobic residues [16]. The multifunctional nature of the CTD is likely dependent on its IDR, which possesses a unique amino acid composition that dictates both intrinsic flexibility and the potential presence of regions known as molecular recognition features (MoRFs) (Box 2) [17,18]. Multiple MoRFs within the CTD (as well as within the NTD) of p53 were predicted using cross-species alignment coupled with computational prediction algorithms [14]. The CTD of p53 has been shown experimentally to form helical structures when interacting with protein and DNA molecules [15,19]. Binding-associated structural changes within the CTD may be triggered by one or more post-translational modifications (PTMs) that are typical for IDRs in general [17] and for this region of p53 in particular [20–22].
Box 2. Intrinsically Disordered Regions (IDRs).
Human p53 is a 393 amino acid protein that has a multidomain organization (Figure 1). Transcriptionally-active p53 exists in an oligomeric form composed offour identical subunits (dimer of dimers, or homotetramer) [9]. From a structural point of view, the p53 monomer is made up of two distinct types of regions: those that demonstrate stable 3D shape and those that are unfolded under normal physiological conditions. The central DBD and the OD belong to the first group, while both the NTD and the CTD represent the second. The 30 amino acid polypeptide linker that connects the DBD with the OD is also disordered. Together, disordered regions constitute more than 40% of the entire p53 monomer [9].
IDRs represent a distinct class of polypeptides lacking 3D structural constraints and demonstrating broad conformational dynamics [17,18]. IDRs, highly abundant in all types of organisms, especially in eukaryotes [102], mediate diverse biochemical processes that range from cell signaling and protein modification and degradation to macromolecular interactions with other proteins or nucleic acids [17]. A computational analysis revealed that 82–94% of eukaryotic TFs possess long IDRs [103].
IDRs are known to differ in many respects from well-ordered polypeptide regions. They have different amino acid compositions that minimize structural restrictions, and use their hydrophobic residues not for stabilization of intramolecular structure but for intermolecular recognition [104]. They also provide notably larger relative intermolecular interfaces for interaction with potential partners [104].
The functional regions/elements mediating IDR intermolecular interactions are known as molecular recognition features (MoRFs). Two possible binding modes for MoRFs have been proposed: conformational selection and induced folding [17,105]. In the first mode, the conformation that ensures specific binding to a cofactor pre-exists within several other conformational ensembles characteristic to the IDR in question, and is then preferentially selected by the cofactor. The second mode implies that binding to a cofactor induces folding within MoRFs. The end-result of both modes is the emergence of relatively stable folding.
The presence of a relatively high number of positively charged residues, especially lysines, is a characteristic feature of the p53 CTD. Figure 2 shows the alignment of the CTDs originating from 32 species including mammals (19 entries), reptiles (4 entries), and fish (9 entries). The total number of lysines and arginines within the last 30 C-terminal amino acids is about 8–9 in mammals and varies significantly in more distant organisms, such as fish, from 3 to 12.
Figure 2. Alignment of the p53 C-Terminal Domains from 32 Species: 19 Mammals, Four Reptiles, and Nine Fishes.
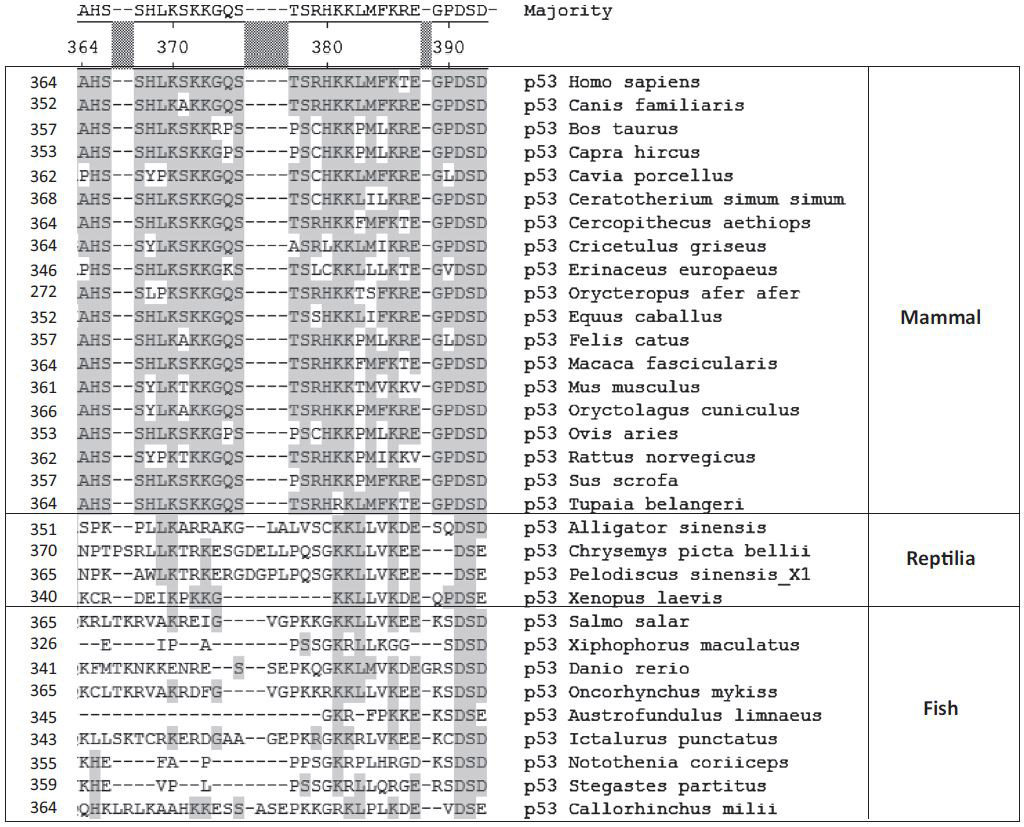
All p53 sequences are from www.ncbi.nlm.nih.gov/gene. The alignment was performed by Clustal W using MegAlign 5.03 software by DNASTAR Inc. The conserved amino acids are shaded.
In general, the amino acid sequences of IDRs are less conserved than those of well-structured globular domains and can tolerate a higher number of mutations without substantial loss of flexibility and function [18]. By contrast, predicted MoRF sequences within both terminal p53 domains appear to be more conserved than the sequences of other non-DBD regions [16]. Based on the alignment data, there are at least two highly conserved subregions within the CTD that can be traced down to cartilaginous fish (Figure 2). The first is K-K/R-Z (where Z is most frequently L, P, or another hydrophobic residue) at position 381–383 (all positions are given according to the human p53 sequence), where K381 and K382 are shown to be acetylated and ubiquitinated by either the p300 (EP300)/CBP (CREBBP) histone acetyltransferase (HATs) or the Mdm2 E3 ligase, respectively [23–25]. The second motif is D-S-D/E at 391–393, with S392 being a site of phosphorylation in response to UV irradiation and a substrate for several protein kinases [23]. In addition, K370 is conserved among the species shown in Figure 2, while K372 and K373 (a further site of acetylation by p300/CBP HAT [23,24]) are highly conserved largely in mammals. Therefore, from an evolutionary point of view, these conserved motifs within the CTD most likely reflect the preservation of vital contacts or functions of p53, and may be traced to specific related modulators or cofactors, such as HATs, protein kinases, E3 ligases, and other modifying activities. In fact, the genomes of the species used for the sequence conservation analysis in Figure 2 encode several notable modulators of p53, including p300 HAT, CK2, and Mdm2.
A future extensive analysis of p53 evolution together with a parallel analysis of the corresponding modulators of p53 activity (preferably, with known sites of interaction within p53) may be very helpful in reconstructing species-specific peculiarities in the p53 network.
It is worth mentioning that the other two members of the p53 family of proteins, p63 (TP63) and p73 (TP73), share not only overall domain organization with p53 but also many structural features. They possess natively disordered regions, although the degree of disorder across the p53 family varies significantly [26]. Computational disorder analysis identifies multiple IDRs within the longest isoforms of these proteins, TA p63∝ and TA p73∝, and predicts the presence of several corresponding MoRFs, some of which seem to coincide roughly with the related p53 MoRFs [16]. Nevertheless, the alignment of p53, p63, and p73 MoRF sequences shows little conservation, suggesting a diverse (although sometimes overlapping) set of interacting partners and different functions [16]. Notably, despite predicted IDRs within the C-terminal portions of both proteins, neither p63 nor p73 contain region(s) that could be considered to be functional homolog(s) of the CTD of p53. This is supported by related experimental findings – namely that Mdm2, an E3 ubiquitin ligase, is known to bind to p53 and p73 and inhibit their respective transcriptional activation functions [27]. The interaction between the NTD of Mdm2 and the TA of p63 is an order of magnitude weaker than that of Mdm2 with p53 or p73 TA [28], and data on Mdm2 binding to full-length p63 are controversial. Importantly, although Mdm2 also controls p53 half-life through ubiquitination of multiple lysine residues within its CTD, it has no effect on p73 (or p63) stability [27].
The Roles of the p53 CTD in DNA Binding
DNA interactions are fundamental for p53 transcriptional activity. Inactivation of p53 in various cancers very often results from missense mutations targeting the specific DBD of this factor [29]. p53-DNA interactions are also extremely complex and diverse because they are mediated by a combination of specific (via the DBD) and non-specific (via the CTD) contacts within the p53 homotetramer. The CTD contribution to non-sequence-specific DNA binding depends on electrostatic contacts between numerous positively charged lysine and arginine residues that reside within this domain and the phosphate backbone of DNA [15]. Reducing the number of lysine residues results in gradual abrogation of DNA binding to the CTD [15].
A crucial and functionally important outcome of the non-specific interactions of the CTD with DNA is the potential ability of p53 to use different modes for searching for cis elements within the complex genome of the eukaryotic cell. Specifically, the CTD has been shown to be indispensable for 1D p53 sliding (linear diffusion) along DNA [30–36]. The combination of 1D sliding with 3D hopping, which is provided by the DBD, may allow rapid and productive scanning of response elements (REs) by p53 following stress-induced protein stabilization [32].
Another essential feature of the CTD is its ability to mediate p53 DNA sequence-dependent core domain interactions with multiple non-linear DNA substrates, such as bent, supercoiled, nucleosomal, and stem-loop DNA [36–40]. Notably, all these forms of DNA are present in the cell and are important for numerous normal biological processes, including replication, nucleosome assembly and disassembly, and transcription [41–43]. Deletion or mutation of residues within the CTD have been shown to reduce sequence-specific p53 interactions with non-linear [30,38–40,44] and linear [40,44] DNA.
Results of studies on the chromatin state of p53 REs within genomic DNA, as well as biochemical studies on p53 interaction with mononucleosomal DNA, may be interpreted in favor of the importance of the CTD in those processes. First, Lidor et al. analyzed the distribution of ~2000 p53 REs within chromatin in unstressed versus stressed cells, and measured relative affinities to p53 [45]. Their results suggest that genomic regions bearing p53 REs have a relatively high nucleosomal content that is lost upon DNA damage and subsequent p53 activation. Second, Sammons et al. in a genome-wide analysis based on local chromatin context identified three distinct classes of p53 REs where, surprisingly, all three classes of REs (even easily accessible ones) showed significant mononucleosome enrichment, thereby questioning the existence of p53 REs in nucleosome-free DNA in unstressed cells [46]. Third, in vitro and in vivo binding of p53 to mononucleosomal REs within the promoter of the p21 (CDKN1A) gene has been demonstrated [47]. Fourth, binding of CTD-deleted, mutated, or p300/CBP-acetylated p53 to in vitro reconstituted mononucleosomes is significantly reduced compared to full-length p53 [40,44]. Finally, using cell-based assays where full-length or CTD-deleted p53 proteins were expressed at near physiological levels, Hamard et al. reconfirmed the requirement of the CTD for p53 binding to and transactivation of the promoters of several bona fide p53 targets, such as p21, Puma, and Mdm2 [48]. Importantly, they also showed that extremely high levels of CTD-deleted p53 transiently overexpressed under the strong human cytomegalovirus promoter may mask the detrimental effect of the CTD absence, emphasizing the need for more-physiological experimental conditions. Of note, gene-editing technologies such as transcription activator-like effector nucleases (TALENS) and clustered regularly-interspaced short palindromic repeats (CRISPR) [49,50] should provide an excellent alternative to experiments based on transient expression of genes. Taken together, the abovementioned findings are in agreement with discoveries made more than a decade ago by Espinosa and Emerson who originally pointed out the vital contributions of the CTD to the process of p53-dependent transactivation on chromatin DNA [38].
How does the CTD exercise its control over p53 sequence-specific DNA interactions? A biochemical analysis of the DNA-binding properties of a series of p53 variants with altered C termini (including a p300-acetylated version) provides a possible explanation [44]. This revealed the importance of the unmodified CTD lysine residues for binding to DNA, and suggested that the CTD controls the stability of a binary p53–DNA complex. At least in part, the CTD does this by promoting structural changes within the individual DBDs and increasing cooperative interactions between them when bound to cognate DNA. Correspondingly (and contrary to the results of an early biophysical analysis of full-length and CTD-deleted p53 [51]), new data obtained using molecular dynamics (MD) suggest that the CTD may promote conformational changes within the p53 tetramer through long-range interactions of the DBD of p53 when bound to cognate DNA [52].
Figure 1 schematically depicts the most ubiquitously expressed isoform of the p53 gene, known as p53∝. While this is also the most transcriptionally active one, at least eight other p53 isoforms have been discovered that are expressed at different levels in a tissue- and stimulus-specific manner [53]. The protein products of the two types of p53 isoforms, namely β and γ, lack the amino acids corresponding to the OD and the original CTD owing to alternative splicing of intron 9. Instead, they possess unique 10 (β) or 15 (γ) C-terminal amino acid sequences [53,54]. In principle, p53 isoforms could exert their biological activities in one of two ways: independently of the major p53 isoform or by modulating its activity. Currently, data on the roles of these isoforms remain scarce, although the most recent studies confirmed the abilities of p53β and p53γ to modulate the activity of the longest p53 isoform and regulate cellular response in a context-dependent manner [55]. Even so, the individual DNA-binding and transactivating properties of β and γ isoforms are greatly diminished compared to full-length p53 when analyzed in transient transfection or stable retroviral transduction experiments [53,56]. Future comprehensive research will be necessary to make accurate conclusions as to the functions of the p53 isoforms in general, and the C-terminally deleted types in particular, in normal and cancer cells.
Structural Studies on p53: An ENDless Story
Our current understanding of how cancer-derived mutations of p53 suppress its transcriptional activity has been illuminated by X-ray- and NMR-based studies showing how the DBD contacts cognate DNA sequences and how these mutant forms of p53 fail to do so [57–61]. Nevertheless, a detailed structural analysis of full-length p53 in complex with DNA is still beyond our grasp. The existence of long IDRs within p53 (more than 40% of the total length of the protein) poses a significant technical problem for structural studies on this TF, which has not been overcome. To date, all published high-resolution p53 structures lack information on the position of the CTD and its possible inter-domain contacts in the context of a p53 oligomer, owing to the physical absence of the disordered domains within the p53 constructs used for crystallization. Further, despite the use of full-length p53 constructs, data obtained with low-resolution electron microscopy, or with a combination of several biophysical techniques such as NMR spectroscopy and small-angle X-ray scattering, have revealed no structural information on either CTD within the p53 tetramer or the trajectory of the DNA molecule in the binary complex [62–64]. p53 is an extremely complex protein. While biochemical, structural, and MD studies suggest that interaction of p53 with DNA is accompanied by multiple conformational changes that take place within the individual domains [61], as well as within the tetramer of p53 [44,52], fitting these data into current structural models of the p53-DNA binary complex is daunting. Thus, obtaining molecular insight into the structural details of p53-DNA interactions calls for complex and nontrivial approaches. Recently, Wu et al. described a hybrid approach that combines atomic force microscopy (AFM) with electrostatic force microscopy (EFM) [65]. This method, called dual resonance-frequency enhanced EFM (DREEM), allows exquisitely sensitive resolution of the DNA within protein-DNA complexes and could be employed for studies of wild-type and domain-deficient p53 variants. In addition, there are examples where fruitful alternatives other than NMR, X-ray crystallography, or electron microscopy (EM) have been used to obtain accurate structure–function information about composite multidomain proteins. For example, crosslinkable derivatives of both nucleotides and amino acids have been successfully employed in studies elucidating binary and ternary complexes of bacterial DNA-dependent RNA polymerase (RNAP) with DNA [66] and elongation factors [67]. Depending on the chemical nature of the reagent (crosslinkable moiety, length of the spacer), it might be possible to study both intra- and intermolecular interactions within the p53 tetramer, alone or in complex with DNA. In fact, use of a UV-crosslinkable nucleotide derivative for studying position-specific p53-DNA interactions has been already successfully demonstrated [44]. Furthermore, site-specific p53 crosslinking derivatives can be subjected to subsequent mass spectrometry (MS) analysis of the protein [68]. As one can see, the arsenal of possible alternative methods that can be employed in p53 structure–function studies is far from being exhausted.
Modifications of the CTD Elicit Alterations in p53 Stability, DNA Binding, and Cofactor Recruitment.
p53 is subjected to a plethora of PTMs which have been discussed and tabulated in excellent reviews [23,24,69–71]. The most extensively studied (and likely most abundant) of these are modifications of serine and threonine by phosphorylation, of lysine by acetylation and methylation, and ubiquitination and arginine methylation [23]. In addition, p53 lysines can be SUMOylated and NEDDylated and, even less well studied, O-glycosylation and GlcNAcylation of serine have also been reported [71]. An MS-based analysis of PTM distribution within p53 isolated from normal human fibroblasts that were infected with adenovirus deficient for p53 degradation identified 99 amino acid residues as targets for a staggering 222 modifications [72]. Unsurprisingly, given its numerous modifiable residues and accessibility, a considerable number of such modifications take place within the CTD. In fact, the same proteomics study showed that every serine and threonine residue within the last 30 C-terminal amino acids can be phosphorylated, and all the positively charged residues (6 lysines and arginine 379) may be modified in a wide variety of ways (Figure 3A) [72]. It is obvious that all these modifications (often mutually exclusive) cannot be present within the same molecule of p53, and different subpopulations of modified p53 must exist in myriad contexts and conditions [72]. Because this subject has been extensively covered in previous reviews, we focus here on three key outcomes of CTD modifications that are relevant to p53 as a regulator of transcription: protein stabilization or degradation, modulation of sequence specific DNA-binding properties of p53, and cofactor recruitment.
Figure 3. Post-Translational Modifications (PTMs) within the C-Terminal Domain (CTD) of p53.

(A) PTM sites within the CTD according to a comprehensive MS analysis [72]. Only the most-abundant modifications detected in [72] are indicated. (Below) Alignment of the p53 CTDs derived from the organisms most commonly used in cancer research and cancer models. (B) Representative examples of various structural shapes of the CTD (shown in dark blue) induced by intermolecular interactions with different cofactors (from left to right): S100B(ββ) (PDB: 1DT7), CBP bromodomain (PDB: 1JSP), and TTD 53BP1 (PDB: 2MWP and 4×34).
Efficient transcriptional responses depend on the intracellular levels of a given TF. p53 stability is primarily controlled by its key negative regulator, the E3 ligase Mdm2 [73,74]. Competitive binding to p53 and mutually exclusive modifications of its C terminus by either Mdm2 or the p300 HAT have suggested a possible regulatory switch that controls the stability of p53 through extensive ubiquitination (leading to degradation) versus acetylation (leading to stabilization) of corresponding lysines [24]. Although the high-affinity p53-Mdm2 interaction relies primarily on specific amino acids within the NTDs of both proteins [75], the impact of the CTD of p53 on this interaction is also significant because acetylation or especially deletion of the p53 C terminus destabilizes the p53–Mdm2 complex [76]. The CTD may contribute to p53 stability not only in a passive manner, by accepting modifications that either promote or prevent its degradation, but also as an active player providing an important secondary interaction site for proper Mdm2 binding.
Roles of the CTD and residues within it have been addressed using targeted mouse models (Table 1). While initial targeting of S389 or the six terminal lysines did not reveal dramatic phenotypes [77,78], mice with deletion of the last 31 or 24 C-terminal amino acids [79,80] were shown to be phenotypically distinct from mice where the p53 CTD lysines were substituted with arginines (so-called 6KR and 7KR mice, depending on the number of substituted lysines, see Table 1), thereby preserving charge but preventing all lysine modifications [78,81]. Both the Δ24 and Δ31 mouse models presented with anemia and bone marrow failure, suggesting crucial functions for the CTD of p53. Importantly, similar phenotypes have been observed in mice with reduced levels of expression of the p53 negative regulators, Mdm2 and Mdm4 [82–84]. Likewise the 7KR mouse shows this effect, but only after irradiation [85]. Furthermore, DCTD/DCTD p53 mice demonstrate partial stabilization of p53 in a tissue-dependent manner [79,80], while multiple lysine substitutions, preventing position-specific ubiquitination and acetylation but preserving the overall charge, led to no significant changes in the p53 half-life [78,81].The latter observation is in agreement with the finding that the identical substitutions within the CTD have no effect on the stability of the p53–Mdm2 complex in vitro [76]. Collectively, these data imply that (i) in the absence of the C-terminal lysines, Mdm2 may promote ubiquitination of other lysine residues within p53 [86], (ii) a fraction of p53 may be degraded via a non-ubiquitin-dependent pathway [87], (iii) as a secondary interaction site, the CTD may provide necessary stabilization of p53-Mdm2 interaction [76], and (iv) while deletion of the CTD may impact on overall p53 stability and activity, mutations that prevent modification of individual residues within the CTD are unlikely to have an obvious effect on the functions and Mdm2-mediated degradation of p53 in vivo, which is in agreement with the intrinsic properties of IDRs. Discrepancies between the dramatic changes in p53 stability associated with the status of specific lysine or serine CTD residues, as originally observed in the experiments with cancer cell lines, and the somewhat mild effects of their mutations in mouse models, are likely due to the use of overexpression assays in the former case.
Table 1.
Genetically Engineered Mouse Models with Targeted Mutation(s) of the C Terminus of p53
| Designation | Phenotype | Refs |
|---|---|---|
| S389A | Reduced apoptosis and impaired transcriptional responses to UV but not gamma radiation. | [77] |
| 6KRa | Normal p53 stabilization but impaired transcriptional response. | [78] |
| 7KRb | Normal p53 stabilization; anemia and hematopoiesis defects only after irradiation; enhanced transcriptional activity only after irradiation. | [81,85] |
| Δ24c | Decreased size and lifespan; anemia and ataxia, with defects in hematopoiesis and cerebellar development; altered transcriptional activity in target gene selective and tissue-specific manner. | [80] |
| Δ31d | Decreased size and lifespan; anemia and pulmonary fibrosis; increased p53 levels and hypertranscriptional activity in mouse embryo fibroblasts. | [79] |
K367R; K369R; K370R; K378R; K379R; K383R.
K367R; K369R; K370R; K378R; K379R; K383R; K384R.
Δ366–390.
Δ360–390.
The relevance of CTD modification status to the DNA-binding properties of p53 is a longstanding subject of inquiry [3]. p300/CBP-dependent acetylation of the C-terminal lysines [88] was considered to promote sequence-specific DNA binding of p53 via the central DBD, presumably by relieving a proposed inhibitory effect of the CTD [3]. The connection between the acetylation status of the CTD and the DNA-binding properties of p53 was originally supported by a set of in vitro experiments that employed the electrophoretic mobility shift assay (EMSA). The original interpretation was subsequently challenged by later studies that used multiple biochemical approaches [38,40,44,89]. Furthermore, chromatin immunoprecipitation (ChIP) experiments that utilized an acetylation-specific anti-p53 antibody [90] and sought to prove increased DNA-binding potential of the C-terminally acetylated p53 did not distinguish whether acetylation of CTD lysine(s) precedes sequence-specific DNA binding, or if p53 binding to DNA occurs before acetylation of the CTD. Findings that specific individual, as well as multiple, acetylation events within the CTD lead to destabilization of p53 in complex with its cognate DNA [40,44] actually suggest a destabilizing impact of CTD lysine acetylation on p53 sequence-specific DNA binding. In view of this, we can speculate that CTD hyperacetylation denotes the end-point of p53-dependent transactivation and signifies p53 dissociation from the promoter. Relevantly, a genome-wide study found that the number of sites bound by p53 induced with Nutlin-3 was sixfold greater than by p53 induced with doxorubicin (Dox) [91]. The latter is known to trigger multiple modifications within p53, including acetylation of the C-terminal lysines [92], whereas p53 is relatively undermodified in Nutlin-3 treated cells [93].p53 binding to its RE is followed by stimulus- and promoter-specific cofactor recruitment, a complex process that drives the formation of a transcriptionally competent pre-initiation complex (PIC). Although numerous key regulators bind to p53 through high-affinity contacts with its TAD (e.g., p300/CBP HAT, Mdm2, MdmX), the CTD provides a secondary interaction site for some of these and also interacts with many other cofactors [23,70]. The CTD may not be fully accessible until the formation of a binary complex between p53 and cognate DNA [94]. Stress-induced dissociation of Mdm2 followed by p53 binding to its cognate site may expose the CTD and make it available for both modifications and intermolecular interactions through its MoRFs. Many CTD-driven interactions are characterized by relatively low affinities, and can be tailored through cooperative binding with other promoter-specific factors as well as by individual PTMs within the CTD [20–22,95,96]. This ensures rapid and flexible control over the expression of numerous p53 transcriptional targets. Crosstalk with other specific or general TFs colocalized in the vicinity of the promoter may regulate the recruitment of various chromatin modifiers. Indeed, data from recent biochemical and structural studies have supported this scenario. For example a study using in vitro reconstituted p53-driven transcription on a chromatin template demonstrated the existence of a multistep cooperative process of cofactor (p300 HAT, and arginine methyltransferase CARM1) recruitment and subsequent chromatin modifications that were dependent on the CTD [97]. Further, Brd4, an epigenetic reader and a member of the BET family of transcriptional regulators, cooperates with p53 on particular p53-dependent promoters and physically interacts with it through the CTD [98]. PTMs of specific residues within the CTD alter the interacting regions and may direct binding to specific partners. For instance, an unmodified CTD-derived peptide (residues 367–388) binds to dimeric S100B(ββ) protein in a helical conformation [18] (Figure 3B). Acetylation of K382, but not K373, within a peptide spanning residues 367–386 of the CTD promotes binding to the bromodomain of CBP in vitro [20] (Figure 3B). The CTD peptide in this complex exists in a β-turn-like conformation. By contrast, in vitro interaction of the p53 CTD-derived peptide (residues 377–387) with the tandem Tudor domain (TTD) of 53BP1 has been shown to possess great plasticity and multiple binding modes depending on the PTM status of specific lysine residues within the CTD peptide [21,22]. A 12 amino acid peptide containing dimethylated K382 was found to bind to the TTD in a U-shape conformation, while an 11 amino acid peptide spanning residues 377–387 with acetylated K381 and dimethylated K382 appeared in the ∝-helical fold in the complex with the same TTD [22] (Figure 3B). The CTD has been implicated in secondary interactions with E3 ligase Mdm2 and, although it is unknown whether this interaction induces any specific structural change within the p53 CTD, acetylation of the C terminus was shown to destabilize the p53–Mdm2 complex [76]. It is very likely that we have only illuminated a small portion of the enormously complex and fluid interaction network that is dependent on the p53 CTD. Alternative approaches will be necessary to clarify the roles of individual cofactors recruited via the CTD and even the roles of individual CTD residues in this process.
Concluding Remarks: A Model for the Roles of the p53 CTD in the Complex Process of Transcriptional Activation by p53
As the saying goes, the devil is in the details, and, in the case of p53, it is easy to become lost in them. Even so, it is indisputable that the CTD controls and adjusts p53 transactivation potential at many different levels. We summarize here the steps in initiation of transcription on p53 target genes that are regulated by the p53 CTD: (i) p53 becomes stabilized in response to intrinsic or extrinsic signaling pathways; (ii) p53 searches for its sites on chromatin using a combination of 1D sliding (via the CTD) and 3D hopping (via the DBD) followed by binding to nucleosomal DNA; (iii) the CTD assists in stabilization of the sequence-specific p53-DNA complex; (iv) once the sequence-specific binary complex is formed, the CTD is then available for a dynamic range of modifications and cofactor interactions leading to the formation of the PIC and the initiation of transcription; (v) p53 ends up bound to a naked DNA fragment that has undergone significant conformational changes, as has the p53 tetramer itself. Even dissociation and promoter shut-off seem to partially depend on the CTD because acetylation of K373 and K382 is required for binding of TAF1 and subsequent phosphorylation of threonine at position 55 that leads to p53 dissociation from the promoter [99]. Although there is ample evidence for the role of the CTD in each of the above steps, this does not preclude the possibility that there may be additional aspects of its function in p53 biology that remain to be discovered (see Outstanding Questions).
Outstanding Questions.
Can the differences within the CTD of p53 in different vertebrate species be linked to the evolution of specific factors that bind to and regulate p53?
What is the exact position of the CTD within the p53 tetramer? How does it change between three different p53 states: (i) unbound, or bound to (ii) specific or (iii) non-specific DNA? In addition, what are the structural consequences of deletion of the CTD? Such information will provide further insight into how the p53 DBD binds specifically to DNA.
Can we dissect the timing (pre- vs post-DNA binding) of PTM of the CTD upon stress signaling to p53?
How does p53 dissociate from its cognate site in vivo, and what is the role of the CTD (and the corresponding PTMs) in this process? At present, we have little knowledge of this step in the process of p53-dependent transactivation.
How does the CTD function to regulate p53 in different tissues? Mouse models have revealed profound differences in the requirement for the CTD to regulate different p53 target genes [80]. Is this due to tissue-specific chromatin organization and/or availability of particular CTD-binding cofactors, and if so what are they? Currently, this area of p53 research is underdeveloped and requires our utmost attention because it may provide us with direct answers to many longstanding questions in p53 biology.
Supplementary Material
Trends.
p53 is a TF that exerts its antitumor activity predominantly through the transcriptional regulation of multiple target genes.
Rapid identification of cognate p53 sites within the chromatin context of the eukaryotic cell, stable sequence-specific DNA binding, and cofactor recruitment are vital for an accurate and efficient transcriptional outcome mediated by p53 in response to a variety of stimuli of exogenous and endogenous nature.
The intrinsically disordered CTD participates in all aspects of p53 functioning as a TF.
Lack of structural constraints, conformational dynamics, and a unique amino acid composition are important for functions of the CTD within p53.
Despite the current lack of structural information on the CTD within p53, the CTD is an integral part of the tetramer and participates in its binding-induced structural changes.
Glossary
- Atomic force microscopy (AFM)
a type of scanning force microscopy technique capable of force measurement, imaging at high resolution, and manipulation
- Chromatin Immunoprecipitation (ChIP)
a molecular biology technique that is used for the analysis of protein interactions with specific genomic loci in the cell. It relies on two different techniques: protein immunoprecipitation and PCR amplification
- Cis elements
also known as response elements (RE) or binding sites (BS), cis elements are specific short noncoding DNA sequences that mostly reside within the promoters of genes recognized by sequence-specific TFs
- Clustered regularly-interspaced short palindromic repeats (CRISPR)
regions of prokaryotic DNA containing short repetitions of interspaced sequences
- Crosslinkable derivatives
chemically modified active derivatives of nucleic acid or protein that are capable of binding covalently to a targeted molecule. Crosslinking can be induced by manipulating the reaction conditions (e.g., by irradiating the sample with UV light of a specific wavelength)
- DNA-dependent RNA polymerase (RNAP)
an enzyme responsible for transcription. Depending on the organism, its complexity may vary from that of a single subunit (e.g., in some viruses) to a multisubunit complex (bacteria and eukaryotes). Eukaryotes have several types of multisubunit RNAPs that are responsible for the transcription of specific classes of RNA molecules
- Doxorubicin (Dox)
an anticancer (‘antineoplastic’ or ‘cytotoxic’) chemotherapy drug. Dox interacts with DNA by intercalation, which leads to inhibition of the enzyme topoisomerase II and, eventually, to the inhibition of DNA replication and cell death. p53 is activated in response to Dox treatment
- Electrophoretic mobility shift assay (EMSA)
a relatively simple and rapid non-equilibrium method for detecting intermolecular interactions, for example between a DNA-binding protein and cognate DNA. The method utilizes electrophoresis in (typically) polyacrylamide gels under native conditions to separate molecular complexes from the individual non-interacting components
- Electrostatic force microscopy (EFM)
an AFM technique employing a conducting cantilever and substrate. It enables the detection/imaging of electrical properties at high resolution
- Intrinsically disordered regions (IDRs)
a distinct class of polypeptides lacking 3D structural constraints and demonstrating broad conformational dynamics
- Isoform
an alternatively expressed mRNA from the same gene locus that may differ in transcription start-site (TSS), 5′- or 3′-untranslated regions, and protein-coding region sequences. Proteins translated from mRNA isoforms may have differences in their amino acid sequence, and hence in their functional properties
- Long-range interactions
a type of interaction between distant regions (domains) of a protein that plays an important role in maintaining the native structure of a protein
- Mdm2
this gene encodes the E3 ligase Mdm2. This gene is a transcriptional target of p53
- Mdm2 E3 ligase
an E3 Ligase that belongs to the RING family of proteins. Mdm2 inhibits p53 transcriptional activity and targets it for degradation
- Molecular dynamics (MD) simulations
a technique for computer simulation of complex systems, modeled at the atomic level
- Molecular recognition features (MoRFs)
the functional regions/elements mediating IDR intermolecular interactions
- Nucleosomal
the state of DNA when it is associated with core histones, primarily the core histone octamer
- Nutlin-3
a low molecular weight compound that specifically inhibits the interaction between Mdm2 and tumor-suppressor p53. Treatment with Nutlin-3 results in rapid stabilization of p53 and its derepression
- p21 (CDKN1A)
this gene encodes a potent cyclin-dependent kinase inhibitor, p21, and is a transcriptional target of p53
- p53 (TP53)
a tumor-suppressor protein that responds to diverse cellular stresses to regulate the expression of target genes, thereby inducing cell-cycle arrest, apoptosis, senescence, DNA repair, or changes in metabolism
- p63 (TP63) and p73 (TP73)
two other evolutionarily conserved members of the p53 family of proteins; these TFs share some structural features and functions with p53 but also display unique structural parts (e.g., SAM domain) and biological activities (e.g., regulate normal development)
- p300 (EP300)/CBP (CREBBP) histone acetyltransferases
two closely related histone acetyltransferases (HATs); common and essential activators of transcription and p53 binding partners
- Placozoans
a genetically diverse group of invertebrate organisms with simple structure
- Post-translational modifications (PTMs)
enzyme-dependent covalent attachment of chemical groups (e.g., phosphoryl, acetyl, methyl, and others) to specific amino acids within the polypeptide chain of a protein
- Preinitiation complex (PIC)
a multiprotein complex that is required for binding of RNAP to the promoter of genes in eukaryotes and archaea. The PIC includes multiple proteins of general and of promoter-specific nature
- Promoter
a region at the beginning of a gene that recruits RNAP and promoter-specific transcription factors for transcription initiation
- Puma (BBC3)
this gene encodes a protein that induces cell death (apoptosis) and is a transcriptional target of p53
- Stem-loop DNA
a non-linear form of DNA generated when two regions of the same strand, usually complementary in nucleotide sequence when read in opposite directions, anneal. Such interaction forms a double helix ending in an unpaired loop
- Supercoiled
the state of DNA that corresponds to its over- or underwinding
- Transcription activator-like effector nucleases (TALENS)
fusions of transcription activator-like (TAL) proteins and a FokI nuclease. Their nuclease specificity can be modified in a wide range such that they will bind to and cleave only the desired sites within genomic DNA
- Ubiquitin-dependent degradation
an intracellular protein-degradation pathway that requires conjugation of the target protein to ubiquitin before degradation by the proteasome
Footnotes
Supplemental Information
Supplemental information associated with this article can be found online athttp://dx.doi.org/10.1016/j.tibs.2016.08.011.
References
- 1.Belyi VA et al. (2010)The origins and evolution of the p53 family of genes. Cold Spring Harb. Perspect Biol 2, a001198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Riley T et al. (2008) Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell. Biol 9, 402–412 [DOI] [PubMed] [Google Scholar]
- 3.Laptenko O and Prives C (2006) Transcriptional regulation by p53: one protein, many possibilities. Cell Death Differ. 13,951–961 [DOI] [PubMed] [Google Scholar]
- 4.Levine AJ (1997) p53, the cellular gatekeeper for growth and division. Cell 88, 323–331 [DOI] [PubMed] [Google Scholar]
- 5.Vousden KH and Prives C (2009) Blinded by the light: the growing complexity of p53. Cell 137, 413–431 [DOI] [PubMed] [Google Scholar]
- 6.Lee TI and Young RA (2000) Transcription of eukaryotic protein-coding genes. Annu. Rev. Genet 34, 77–137 [DOI] [PubMed] [Google Scholar]
- 7.Ptashne M and Gann A (2001) Genes & Signals, Cold Spring Harbor Laboratory [Google Scholar]
- 8.Matthews JM (ed.) (2012) Protein Dimerization and Oligomerization in Biology (Advances in Experimental Medicine and Biology, Vol. 747), Springer Science+Business Media [Google Scholar]
- 9.Joerger AC and Fersht AR (2008) Structural biology of the tumor suppressor p53. Annu. Rev. Biochem 77, 557–582 [DOI] [PubMed] [Google Scholar]
- 10.Foord OS et al. (1991) A DNA binding domain is contained in the C-terminus of wild type p53 protein. Nucleic Acids Res. 19, 5191–5198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vogelstein B and Kinzler KW (1992) p53 function and dysfunction. Cell 70, 523–526 [DOI] [PubMed] [Google Scholar]
- 12.Ahn J and Prives C (2001) The C-terminus of p53: the more you learn the less you know. Nat. Struct. Biol 8, 730–732 [DOI] [PubMed] [Google Scholar]
- 13.Bell S et al. (2002) p53 contains large unstructured regions in its native state. J. Mol. Biol 322, 917–927 [DOI] [PubMed] [Google Scholar]
- 14.Cheng Y et al. (2007) Mining alpha-helix-forming molecular recognition features with cross species sequence alignments. Biochemistry 46, 13468–13477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Friedler A et al. (2005) Modulation of binding of DNA to the C-terminal domain of p53 by acetylation. Structure 13, 629–636 [DOI] [PubMed] [Google Scholar]
- 16.Xue B et al. (2013) Intrinsically disordered regions of p53 family are highly diversified in evolution. Biochim. Biophys. Acta 1834, 725–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tompa P et al. (2015) Intrinsically disordered proteins: emerging interaction specialists. Curr. Opin. Struct. Biol 35, 49–59 [DOI] [PubMed] [Google Scholar]
- 18.Brown CJ et al. (2011) Evolution and disorder. Curr. Opin. Struct. Biol 21, 441–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rustandi RR et al. (2000) Structure of the negative regulatory domain of p53 bound to S100B(betabeta). Nat. Struct. Biol 7, 570–574 [DOI] [PubMed] [Google Scholar]
- 20.Mujtaba S et al. (2004) Structural mechanism of the bromodo-main of the coactivator CBP in p53 transcriptional activation. Mol. Cell 13, 251–263 [DOI] [PubMed] [Google Scholar]
- 21.Tong Q et al. (2015) Structural plasticity of methyllysine recognition by the tandem tudor domain of 53BP1. Structure 23, 312–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tong Q et al. (2015) An acetyl-methyl switch drives a conformational change in p53. Structure 23, 322–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boehme KA and Blattner C (2009) Regulation of p53 – insights into a complex process. Crit. Rev. Biochem. Mol. Biol 44, 367–392 [DOI] [PubMed] [Google Scholar]
- 24.Brooks CL and Gu W (2011) The impact of acetylation and deacetylation on the p53 pathway. Protein Cell 2, 456–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brooks CL and Gu W (2011) p53 regulation by ubiquitin. FEBS Lett. 585, 2803–2809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dos Santos HG et al. (2016) Functional diversification after gene duplication: paralog specific regions of structural disorder and phosphorylation in p53, p63, and p73. PLoS ONE 11, e0151961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maisse C et al. (2003) p73 and p63 protein stability: the way to regulate function? Biochem. Pharmacol. 66, 1555–1561 [DOI] [PubMed] [Google Scholar]
- 28.Zdzalik M et al. (2010) Interaction of regulators Mdm2 and Mdmx with transcription factors p53, p63 and p73. Cell Cycle 9, 4584–4591 [DOI] [PubMed] [Google Scholar]
- 29.Olivier M et al. (2010) TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol 2, a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McKinney K et al. (2004) p53 linear diffusion along DNA requires its C terminus. Mol. Cell 16, 413–424 [DOI] [PubMed] [Google Scholar]
- 31.Tafvizi A et al. (2008)Tumor suppressor p53 slides on DNA with low friction and high stability. Biophys. J 95, L01–L03 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tafvizi A et al. (2011) A single-molecule characterization of p53 search on DNA. Proc. Natl. Acad. Sci. U.S.A 108, 563–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khazanov N and Levy Y (2011) Sliding of p53 along DNA can be modulated by its oligomeric state and by cross-talks between its constituent domains. J. Mol. Biol 408, 335–355 [DOI] [PubMed] [Google Scholar]
- 34.Vuzman D and Levy Y (2012) Intrinsically disordered regions as affinity tuners in protein–DNA interactions. Mol. Biosyst 8,47–57 [DOI] [PubMed] [Google Scholar]
- 35.Murata A et al. (2015) One-dimensional sliding of p53 along DNA is accelerated in the presence ofCa2+ or Mg2+ at millimolar concentrations. J. Mol. Biol 427, 2663–2678 [DOI] [PubMed] [Google Scholar]
- 36.McKinney K and Prives C (2002) Efficient specific DNA binding by p53 requires both its central and C-terminal domains as revealed by studies with high-mobility group 1 protein. Mol. Cell. Biol 22, 6797–6808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gohler T et al. (2002) Specific interaction of p53 with target binding sites is determined by DNA conformation and is regulated by the C-terminal domain. J. Biol. Chem 277, 41192–41203 [DOI] [PubMed] [Google Scholar]
- 38.Espinosa JM and Emerson BM (2001) Transcriptional regulation by p53 through intrinsic DNA/chromatin binding and site-directed cofactor recruitment. Mol. Cell 8, 57–69 [DOI] [PubMed] [Google Scholar]
- 39.Palecek E et al. (2004) Enhancement of p53 sequence-specific binding by DNA supercoiling. Oncogene 23, 2119–2127 [DOI] [PubMed] [Google Scholar]
- 40.Kim H et al. (2012) p53 requires an intact C-terminal domain for DNA binding and transactivation. J. Mol. Biol 415, 843–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gilbert N and Allan J (2014) Supercoiling in DNA and chromatin. Curr. Opin. Genet. Dev 25, 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brázda V et al. (2011) Cruciform structures are a common DNA feature important for regulating biological processes. BMC Mol. Biol 5, 12–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alvarez M et al. (2003) Context-dependent transcription: all politics is local. Gene 313, 43–57 [DOI] [PubMed] [Google Scholar]
- 44.Laptenko O et al. (2015) The p53C terminus controls site-specific DNA binding and promotes structural changes within the central DNA binding domain. Mol. Cell 57, 1034–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lidor NE et al. (2010) p53 binds preferentially to genomic regions with high DNA-encoded nucleosome occupancy. Genome Res. 20, 1361–1368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sammons MA et al. (2015)TP53 engagement with the genome occurs in distinct local chromatin environments via pioneer factor activity. Genome Res. 25, 179–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Laptenko O et al. (2011)p53 binding to nucleosomes within the p21 promoter in vivo leads to nucleosome loss and transcriptional activation. Proc. Natl. Acad. Sci. U.S.A 108,10385–10390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hamard PJ et al. (2012) p53 basic C terminus regulates p53 functions through DNA binding modulation of subset of target genes. J. Biol. Chem 287, 22397–22407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mougiakos I et al. (2016)Next generation prokaryotic engineering: the CRISPR-Cas toolkit. Trends Biotechnol. 34, 575–587 [DOI] [PubMed] [Google Scholar]
- 50.Boettcher M and McManus MT (2015)Choosing the righttool for the job: RNAi, TALEN, or CRISPR. Mol. Cell 58, 575–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ayed A et al. (2001) Latent and active p53 are identical in conformation. Nat. Struct. Biol 8, 756–760 [DOI] [PubMed] [Google Scholar]
- 52.D’Abramo M et al. (2016)The p53 tetramer shows an induced-fit interaction of the C-terminal domain with the DNA-binding domain. Oncogene 35, 3272–3281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bourdon JC et al. (2005) p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 19, 2122–2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marcel V et al. (2011) Biological functions of p53 isoforms through evolution: lessons from animal and cellular models. Cell Death Differ. 18, 1815–1824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marcel V et al. (2014) Modulation of p53β and p53γexpression by regulating the alternative splicing of TP53 gene modifies cellular response. Cell Death Differ. 21, 1377–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Silden E et al. (2013) Expression of TP53 isoforms p53β or p53γ enhances chemosensitivity in TP53null cell lines. PLoS ONE 8, e56276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cho Y et al. (1994) Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science 265, 346–355 [DOI] [PubMed] [Google Scholar]
- 58.Rippin TM et al. (2002) Recognition of DNA by p53 core domain and location of intermolecular contacts of cooperative binding. J. Mol. Biol 319, 351–358 [DOI] [PubMed] [Google Scholar]
- 59.Kitayner M et al. (2006) Structural basis of DNA recognition by p53 tetramers. Mol. Cell 22, 741−753 [DOI] [PubMed] [Google Scholar]
- 60.Cañadillas JM et al. (2006) Solution structure of p53 core domain: structural basis for its instability. Proc. Natl. Acad. Sci. U.S.A 103, 2109–2114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Petty TJ et al. (2011)An induced fit mechanism regulates p53 DNA binding kinetics to confer sequence specificity. EMBO J. 30, 2167–2176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Okorokov AL et al. (2006) The structure of p53 tumor suppressor protein reveals the basis for its functional plasticity. EMBO J. 25, 5191–5200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wells M et al. (2008)Structure of tumor suppressor p53 and its intrinsically disordered N-terminal transactivation domain. Proc. Natl. Acad. Sci. U.S.A 105, 5762–5767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Melero R et al. (2011) Electron microscopy studies on the quaternary structure of p53 reveal different binding modes for p53 tetramers in complex with DNA. Proc. Natl. Acad. Sci. U.S.A 108, 557–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu D et al. (2016) Visualizing the path of DNA through proteins using DREEM imaging. Mol. Cell 61, 315–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nudler E et al. (1998) Spatial organization of transcription elongation complex in Escherichia coli. Science 281, 424–428 [DOI] [PubMed] [Google Scholar]
- 67.Parshin A et al. (2015) DksA regulates RNA polymerase in Escherichia coli through a network of interactions in the secondary channel that includes Sequence Insertion 1. Proc. Natl. Acad. Sci. U.S.A 112, E6862–E6871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Arlt C et al. (2015) Structure of full-length p53 tumor suppressor probed by chemical cross-linking and mass spectrometry. Proteomics 15, 2746–2755 [DOI] [PubMed] [Google Scholar]
- 69.Gu B and Zhu WG (2012) Surf the post-translational modification network of p53 regulation. Int. J. Biol. Sci 8, 672–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Meek DW and Anderson CW (2009) Posttranslational modification of p53: cooperative integrators of function. Cold Spring Harb. Perspect. Biol 1, a000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kruse JP and Gu W (2008) SnapShot: p53 posttranslational modifications. Cell 133, 930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.DeHart CJ et al. (2014) Extensive post-translational modification of active and inactivated forms of endogenous p53. Mol. Cell Proteomics 13, 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Toledo F and Wahl GM (2006) Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat. Rev. Cancer 6, 909–923 [DOI] [PubMed] [Google Scholar]
- 74.Pant V and Lozano G (2014) Limiting the power of p53 through the ubiquitin proteasome pathway. Genes Dev. 28, 1739–1751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kussie PH et al. (1996) Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 274, 948–953 [DOI] [PubMed] [Google Scholar]
- 76.Poyurovsky MV et al. (2010)The C terminus of p53 binds the N-terminal domain of MDM2. Nat. Struct. Mol. Biol 17, 982–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bruins W et al. (2004) Increased sensitivity to UV radiation in mice with a p53 point mutation at Ser389. Mol. Cell Biol 24, 8884–8894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Feng L et al. (2005) Functional analysis of the roles of post-translational modifications at the p53C terminus in regulating p53 stability and activity. Mol. Cell Biol 25, 5389–5395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Simeonova I et al. (2013) Mutant mice lacking the p53 C-terminal domain model telomere syndromes. Cell Rep. 3, 2046–2058 [DOI] [PubMed] [Google Scholar]
- 80.Hamard PJ et al. (2013) The C terminus of p53 regulates gene expression by multiple mechanisms in a target- and tissue-specific manner in vivo. Genes Dev. 27, 1868–1885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Krummel KA et al. (2005) The C-terminal lysines fine-tune P53 stress responses in a mouse model but are not required for stability control or transactivation. Proc. Natl. Acad. Sci. U.S.A 102, 10188–10193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liu G et al. (2007) The p53-Mdm2 network in progenitor cell expansion during mouse postnatal development. J. Pathol 213, 360–368 [DOI] [PubMed] [Google Scholar]
- 83.Abbas HA et al. (2010) Mdm2 is required for survival of hemato-poietic stem cells/progenitors via dampening of ROS-induced p53 activity. Cell Stem Cell 7, 606–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mendrysa SM et al. (2003) Mdm2 is critical for inhibition of p53 during lymphopoiesis and the response to ionizing irradiation. Mol. Cell Biol 23, 462–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang YV et al. (2011) Fine-tuning p53 activity through C-terminal modification significantly contributes to HSC homeostasis and mouse radiosensitivity. Genes Dev. 25, 1426–1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chan WM et al. (2006) Ubiquitination of p53 at multiple sites in the DNA-binding domain. Mol. Cancer Res 4, 15–25 [DOI] [PubMed] [Google Scholar]
- 87.Tsvetkov P et al. (2010) Ubiquitin-independent p53 proteasomal degradation. Cell Death Differ. 17, 103–108 [DOI] [PubMed] [Google Scholar]
- 88.Gu W and Roeder RG (1997) Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 90, 595–606 [DOI] [PubMed] [Google Scholar]
- 89.Anderson ME et al. (1997) Reciprocal interference between the sequence-specific core and nonspecific C-terminal DNA binding domains of p53: implications for regulation. Mol. Cell Biol 17, 6255–6264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Luo J et al. (2004) Acetylation of p53 augments its site-specific DNA binding both in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A 101,2259–2264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Menendez D et al. (2013) Diverse stresses dramatically alter genome-wide p53 binding and transactivation landscape in human cancer cells. Nucleic Acids Res. 41, 7286–7301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ito A et al. (2001) p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J. 20, 1331–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Thompson T et al. (2004) Phosphorylation of p53 on key serines is dispensable for transcriptional activation and apoptosis. J. Biol Chem 279, 53015–53022 [DOI] [PubMed] [Google Scholar]
- 94.Cesková P et al. (2006) On the mechanism of sequence-specific DNA- dependent acetylation of p53: the acetylation motif is exposed upon DNA binding. J. Mol. Biol 357, 442–456 [DOI] [PubMed] [Google Scholar]
- 95.Rajagopalan S et al. (2010) Mechanistic differences in the transcriptional activation of p53 by 14-3-3 isoforms. Nucleic Acids Res. 38, 893–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.van Dieck J et al. (2009) Posttranslational modifications affect the interaction of S100 proteins with tumor suppressor p53. J. Mol. Biol 394, 922–930 [DOI] [PubMed] [Google Scholar]
- 97.An W et al. (2004) Ordered cooperative functions of PRMT1, p300, and CARM1 in transcriptional activation by p53. Cell 117, 735–748 [DOI] [PubMed] [Google Scholar]
- 98.Wu SY et al. (2013) Phospho switch triggers Brd4 chromatin binding and activator recruitment for gene-specific targeting. Mol. Cell 49, 843–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wu Y et al. (2014) Phosphorylation of p53 by TAF1 inactivates p53- dependent transcription in the DNA damage response. Mol. Cell 53, 63–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Todeschini AL et al. (2014) Transcription factors: specific DNA binding and specific gene regulation. Trends Genet. 30,211–219 [DOI] [PubMed] [Google Scholar]
- 101.Lee TI and Young RA (2013)Transcriptional regulation and its misregulation in disease. Cell 152, 1237–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dunker AK et al. (2000) Intrinsic protein disorder in complete genomes. Genome Inform. Ser. Workshop Genome Inform 11, 161–171 [PubMed] [Google Scholar]
- 103.Liu J et al. (2006) Intrinsic disorder in transcription factors. Biochemistry 45, 6873–6888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mészáros B et al. (2007) Molecular principles of the interactions of disordered proteins. J. Mol. Biol 372, 549–561 [DOI] [PubMed] [Google Scholar]
- 105.Vacic V et al. (2007) Characterization of molecular recognition features, MoRFs, and their binding partners. J. Proteome Res 6, 2351−2366 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.