

Abstract
Aging is driven by unavoidable entropic forces, physicochemical in nature, that damage the raw materials that constitute biological systems. Single cells experience and respond to stochastic physicochemical insults that occur either to the cells themselves or to their microenvironment, in a dynamic and reciprocal manner, leading to increased age-related cell-to-cell variation. We will discuss the biological mechanisms that integrate cell-to-cell variation across tissues resulting in stereotypical phenotypes of age.
Introduction
Aging - defined here as the time-correlated functional decline of biological systems - is a nearly universal phenomenon across somatic life. Virtually all known life, be it nematode, fish, or human, has an expiration date, which can vary greatly between even closely related species. Aging is perhaps the archetype of inevitably. At first, it might seem just another consequence of entropy: rooms get messy, appliances break, and the march of time grinds even mountains to dust. Increased entropy is inevitable (systems tend towards disorder), therefore biological systems are the embodiment of fighting entropy (negative entropy). Schrödinger described over 70 years ago how life consumes free energy, permitting living systems to locally reduce entropy and, at least in principle, avoid otherwise inevitable disorder. The biological manifestations of aging, and the apparent rates of aging, are heritable and reflect the variation and effectiveness with which different biological systems are negative entropy machines.
Aging results in a number of stereotypical states that are observable at the level of the organism. In humans, these states include wrinkles and spots in skin, graying hair, loss of muscle mass, general physiological and cognitive decline, and increased frailty. Stereotypical changes observed at the tissue level, such as changes in cellular composition (e.g. more adipose cells and less epithelia in breast or more granulocytes in blood) and changes in the extracellular matrix[1,2] likely underlie the more easily observed physiological changes. However, at the level of single cells, aging is associated with increased variation in molecular phenotypes. Thus, on the level of cells, the effects and phenotypes of aging are less predictable than in the tissues they comprise. Perhaps dysfunction of tissues with age results from the integration of many parts that are each broken in a different way. Here, we attempt to codify how cellular responses to entropic forces result in the stereotypical tissue-level changes that are characteristic of aging.
Entropic Drivers of Aging
The universality of degenerative aging implies that whatever processes drive aging should be universal as well. We suggest that the most upstream drivers of biological aging are physicochemical processes that are ubiquitous to life as we know it.
-
1)
Oxidation, especially by oxidizing by-products of aerobic metabolism, such as the hydroxyl radical[3].
-
2)
Ionizing radiation, both intrinsic, such as radioactive decay of cellular constituents, and extrinsic, such as radon gas or cosmic rays[4,5].
-
3)
Spontaneous (de)methylation, triggered by endogenous agents such as s-adenosylmethionine[6] or nitroso compounds[7]. In particular, spontaneous 5-methylcytosine methylation[8] and demethylation[9] are relevant to humans.
-
4)
The Amadori and Heyns reactions, causing glycative reactions between carbonyls and amines[10].
-
5)
An honorable mention to ultraviolet light, which causes thymidine dimerization and photolysis, especially in skin[11]. However, this is not truly universal, being avoided by marine life, troglodytes, and nocturnal animals.
-
6)
An honorable mention to peptide racemization, which sometimes changes protein structure[12]. This occurs slowly but measurably in humans[13], but functional consequences have not been demonstrated in long-lived proteins.
-
7)
An honorable mention to carbamylation, an adduct-forming reaction between proteins and urea derivatives[14]. However, the research connecting carbamylation and aging is still in a fledgling state.
These entropic processes act as primary drivers of aging in the absence of countervailing biological forces (Figure 1a). Irreversible processes like glycation and ionizing radiation should cause reaction products to accumulate with age. On the other hand, reversible processes like methylation should cause motion towards an equilibrium. In either case, these primary drivers damage the molecular constituents of cells, causing cellular effects that ultimately lead to aging-associated degenerate outcomes in the organism.
Figure 1-. Proposed causal chain of aging phenomena.
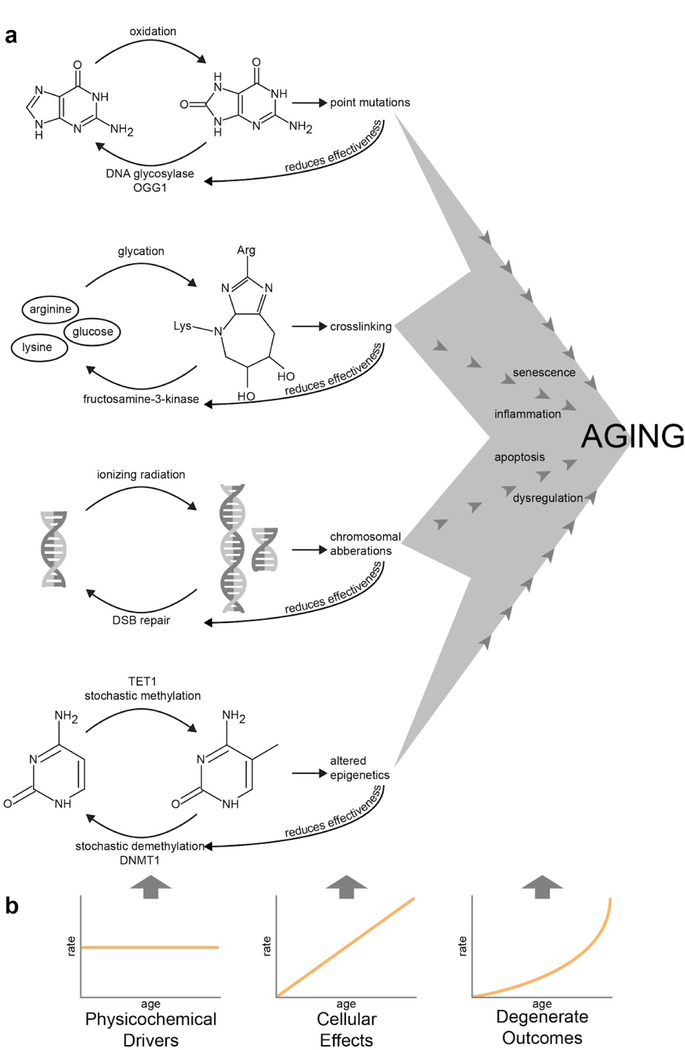
(a) A handful of distinct physicochemical drivers of aging, each antagonized by a biological repair mechanism, eventually leads to distinct cellular effects and shared degenerate outcomes. (b) Due to continually reduced effectiveness of the repair mechanisms, a constant level of physicochemical damage leads to an increasing quantity of cellular effects and an accelerating amount of degenerate outcomes.OGG1 repairs 8-oxoguanine[57], fructosamine 3-kinase deglycates proteins[17], double-strand break (DSB) repair involves various pathways[58], and TET1 and DNMT1 maintain methylation fidelity by acting on hemimethylated DNA[59].
Biological systems can, to varying degrees, repair the physicochemical insults of aging. DNA and a small subset of low-turnover proteins have half-lives longer than months[15,16] and are important substrates for physicochemical aging. The suite of DNA repair proteins is a subject of active investigation, and most common insults to DNA, such as 8-oxo-guanine formation or depurination, have known repair pathways. Similarly, damaged proteins are subject to autophagy and proteasomal degradation, and indirect evidence suggests that specific enzymes catalyze the repair of specific protein adducts[17]. Even if the forward rates of physicochemical damage are constant, variability in these reverse repair rates would be sufficient to cause variability in aging rates.
Assuming that repair mechanisms themselves also are subject to the physicochemical forces of aging, this model predicts that the apparent rate of aging will accelerate with age (Figure 1b). The ever-increasing gulf between the constant damaging forces and the diminishing repair forces would produce an acceleration analogous to the kinematic trajectory of a falling object. In reality, this is almost certainly complicated by various biological feedbacks. However, evidence in the literature is consistent with the linear accumulation of molecular defects such as oxidized DNA[18] and the superlinear accumulation of organismal damage, as seen in the seemingly exponential hazard function for human mortality[19].
The degenerative effects of aging do not appear to affect species at a multigenerational scale - only individuals age. Although degenerative aging of individual organisms is an all-but-universal observation in biology, species themselves do not degenerate - i.e., children are not degenerate relative to their parents. Various processes may contribute to this, including high activity of repair processes in the germ line. For instance, in C.elegans, oxidative damage to the germ line disappears during gamete maturation[20], apparently due to transiently elevated protein turnover. Furthermore, the mutation rate in human gametes appears to be two orders of magnitude lower than the mutation rate in human somatic cells[21]. This evidence supports the conclusion that, at least in specific cell lineages, organisms are able to negate the forward aging rate. In turn, this suggests variability in the aging rate across cell lineages within an individual.
Variability in Aging
One of the most striking aspects of aging is the wide range of variation observed. Across species, there are marked differences in aging rates even between closely related groups, e.g. compare the maximum lifespan of bowhead whales to minke whales (211 vs 50 years) or little brown bats to evening bats (34 vs 6 years)[22]. Within a species, differences in lifespan and healthspan also are apparent across populations. Regardless whether processes that drive aging are stochastic or deterministic, these processes converge into observable aging phenotypes that manifest in changes to tissue structure and function. Metastability is a physical concept, whereby an equilibrium state is stable for all intents and purposes, until acted upon by an external force. Aging may cause cells to transit across a continuum of metastable states, and these states may be maintained via epigenetic regulation[23]. In humans, aging is associated with changing DNA methylation states[24–26], and an ‘epigenetic clock’ requiring only 353 CpG sites has been shown to provide a robust estimate of age across many tissue types[27]. These marked phenotypic and epigenetic changes in aged tissues can be preserved in primary culture and shifted by altering the microenvironment[28,29], providing evidence that these states are likely metastable.
A number of studies have investigated the relationship between aging and gene expression variance. In one twin study, the concentrations of serum cytokines, chemokines, growth factors, and the frequencies of 95 different immune cell subsets showed that non-heritable influences dominated a majority of the observable variance, and that these measures became more variable with age[30]. Another twin study comprehensively measured gene expression in primary fat, skin, whole blood, and derived lymphoblastoid cell lines and found that over 36% of the tested genes showed age-associated expression in at least one tissue[31]. While additive genetic effects explained more of the variance in gene expression than age, this study concluded that gene expression variance changes with age. Measurements from bulk tissues are complicated by the multiple cell types present, a problem that is addressable by lineage-specific studies. In a study of mammary epithelia across individuals, aging manifested as a loss of lineage fidelity between the two principal epithelial lineages[29], such that the magnitude of difference in expression between the two lineages decreases with age. What stands out from this population-level transcriptomic profile of human mammary cells is that cellular aging is not stereotyped across individuals - loss of lineage fidelity occurs at different magnitudes and in different sets of genes.
With the advent of single-cell-RNA sequencing (sc-RNA-seq), cell-to-cell transcriptional heterogeneity (Figure 2a) has been shown in cell populations previously considered homogenous (e.g mouse bone marrow[32], mouse T-cells[33], and human myoblasts[34]). Several factors underlie this observed heterogeneity, including the sub-population structure of lineages, and stratification of expression based on spatial localization and/or the timing of regulatory events and cyclical processes – e.g. cell and hormonal cycles and circadian rhythm (Figure 2d, top panel). A number of sc-RNA-seq studies suggest that some of these underlying processes themselves are likely subject to age-dependent changes. Hematopoietic stem cells (HSCs) in young and old mice were found to have age-dependent gene expression variability associated with older HSCs traversing the G1 phase faster than young HSCs[35]. HSCs also show age-dependent shifts in balance away from self-renewal and towards differentiation[35]. Lineage bias of HSCs also were shown to change with age towards an increase in platelets, possibly explaining compositional shifts in hematopoietic lineages seen during aging[36]. The magnitude of response of the genes that are crucial for activation in naïve CD4 T-cells was found to be age-dependent, with reduced activation in older mice and increased cell-to-cell transcriptional variability with age[37]. In a human cohort whose ages ranged across six decades, pancreatic cells had increased transcriptional variation in the older subjects independent of cell composition, and these cells underwent fate drift where fractions of α- and β-cells with atypic hormone expression increased with age[38]. Increases in transcriptional variability (Figure 2b) and in compositional variability, either via shift in lineage bias or fate drift (Figure 2c), and the loss of stratification via aberrant timing and/or spatial localization of expression (Figure 2d, bottom panel) with age illustrate how increases in molecular noise can lead to aberrant phenotypes and function.
Figure 2-. Proposed etiology of aging-associated transcriptional variability.
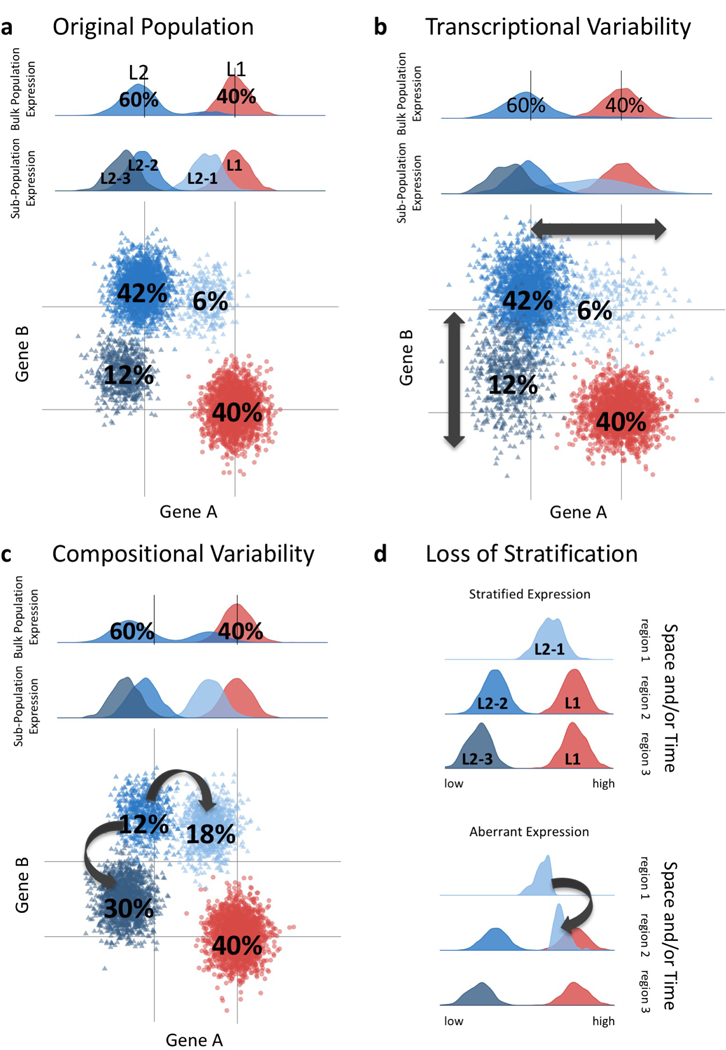
There are multiple routes to achieve age-associated variability among the cells that derive tissues. Hypothetical single-cell RNA sequencing shows heterogeneity of cell populations within a tissue. (a) Two hypothetical lineages each with a given frequency: L1=40% and L2=60% are depicted. At single cell resolution, one can observe multiple sub-populations of L2, e.g. L2–1 (6%), L2–2 (36%), and L2–3 (18%). Histograms of expression of Gene A of the bulk population and sub-populations shown in the top two panels. Dot-plot showing lineage-specific expression of Genes A and B are shown in the bottom panel. (b) Increased transcriptional variability with age has been reported. A hypothetical example in shown whereby expression of Genes A and B in the L2 sub-populations increases in variance, leading to an apparent loss of lineage-specific expression with age. (c) Changes with age in the composition of lineages that comprise a tissue has been reported. Change in sub-population frequency is shown with reduction of L2–2 (12%), and increases of L2–1 (18%) and L2–3 (30%) sub-populations with age. This could occur either via shift in lineage bias or by fate drift. (d) Loss of stratification with age is another hypothetical means of increasing transcriptional variability. Stratified expression of the original population in time and/or space is shown in the top panel. Loss of stratified expression with age is illustrated in bottom panel with half of L2–1 showing aberrant timing and/or spatial localization.
Despite the cell-to-cell variability associated with aging, aging phenotypes at the macroscale are highly stereotyped. If robustness is an important characteristic of tissues[39], then healthy young cells should be able to compensate for perturbations and resist changes. Cells undergo cycles of proliferation and differentiation, and cells respond to cycles of systemic and local signaling that is dependent on each cell’s microenvironment. Therefore, cell-level gene-expression windows must be orchestrated among cells to produce dynamic and reciprocal population responses that allow proper tissue function. In coupled systems, as in gene networks, variability in expression could be viewed as evidence of a need for a compensatory mechanism (e.g. like shock absorbers for biological systems) that allows cell populations to dampen perturbations. Variability also can cause aberrant function when the optimal dynamic range of the system is exceeded, potentially manifesting as functional decline that is perceivable as aging at the tissue level.
Metastability of Aging Phenotypes
The inevitably of physicochemical damage does not imply the irreversibility of said damage. However, there are plausible mechanisms by which aging processes exhibit ratcheting, enabling metastability.
First, several types of physicochemical aging damage are of a two-step nature: a fast reversible process coupled to a slow irreversible process. In the case of glycation, this is fast imine formation coupled to slow glycosylamine formation coupled to slower Amadori rearrangements and permanent advanced glycation end-product formation[10]. Given an irreversible chemical reaction, accumulation of products is inevitable. Second, cell division can perpetuate the damage caused by DNA lesions, essentially making them permanent. Third, stable changes occur within tissues, including senescence and the stiffening of aged ECM. Fourth, there are epigenetic modifications that cause durable changes to cell states. Across a number of tissues, DNA methylation has been shown to change with age, although it is unclear by what mechanisms[29,40–42].
Conceptually, epigenetic changes can be divided into two classes. First, epigenetic drift, the gradual accumulation of DNA methylation changes that leads to increased diversity and variation in DNA methylation, corresponds with increased age-associated variation of transcription[31]. Second, epigenetic shifts are directional DNA methylation states at specific sites that seem to be stereotyped and age-specific across individuals. For example, methylation and chromatin accessibility of the genome organizer SATB1 is negatively correlated with age in T-cells[41], and genes used in autophagy are hypermethylated in aging macrophages[43]. Currently, the directness of the relationship between epigenetic marks and transcriptional outputs is still unclear. It is also unclear to what extent one is the cause and the other is the effect, or indeed whether their relationship is reciprocal. Epigenetics are not limited to DNA methylation and also include histone modification and higher-order changes to chromatin structure and nuclear architecture[44]. Indeed, nuclear-architecture-associated proteins such as CTCF likely reinforce these initial methylation changes[45].
The aging phenotype of a tissue results from many of these mechanisms of metastability working together. For example, physicochemical entropic drivers, like gamma radiation, will damage the raw materials used by cells, perhaps forcing cells to enter senescence. Senescent cells secrete molecules that cause chronic inflammatory states, which are a characteristic of aging (the so-called SASP)[46]. Mice engineered to kill senescent cells live longer and healthier lives compared to their wild-type cousins[47]. Muscle transplantation and parabiosis with young and old mice proved the existence of cell non-autonomous aging and provide excellent examples of aging as metastable states[48,49]. The SASP is an example of a cell non-autonomous means of spreading aging-states throughout a tissue. Other examples of cell non-autonomous aging include age-related NFkB signaling in muscle fibers controlling the activity of satellite cells[50], intestinal cells signalling to neurons to propagate the health and longevity benefits of diet restriction[51,52], and epithelial cells in breast communicating age-states through a cell-cell contact-dependent mechanism[29]. The multiple ratcheting mechanisms of aging that are at work in tissues suggest aging may be irreversible. However, the involvement of multiple epigenetic mechanisms suggests some effects of aging may be preventable or malleable.
Conclusions
At the organismal level aging phenotypes appear stereotypical, but aging brings unpredictable and increasing variation at the single-cell level. Entropic forces act on the raw materials and building blocks of biological systems. However, biological systems are at odds with the notion of persistent and irreversible change because the molecular constituents are subject to continual renewal and repair. Cells manufacture the soluble factors, ECM scaffolds, and even the other cells that together comprise tissue microenvironments. They also produce the receptors required to interact with their microenvironment and the molecular machines needed to repair damage. Over time, as entropic forces alter the molecular constituents of cells, and cells in turn alter the microenvironment, cells exhibit a reciprocal response that alters the microenvironment further and changes how cells will respond to future perturbations (Figure 3). In a particular cell, if a physicochemical process alters a crucial repair or renewal mechanism, then the rate at which the cell resists the march of aging is forever altered. Thus, variation can arise in cells according to the distribution of damage to either the microenvironment or to the cells themselves.
Figure 3-. Entropic forces drive transitions through metastable states within an aging biological system.
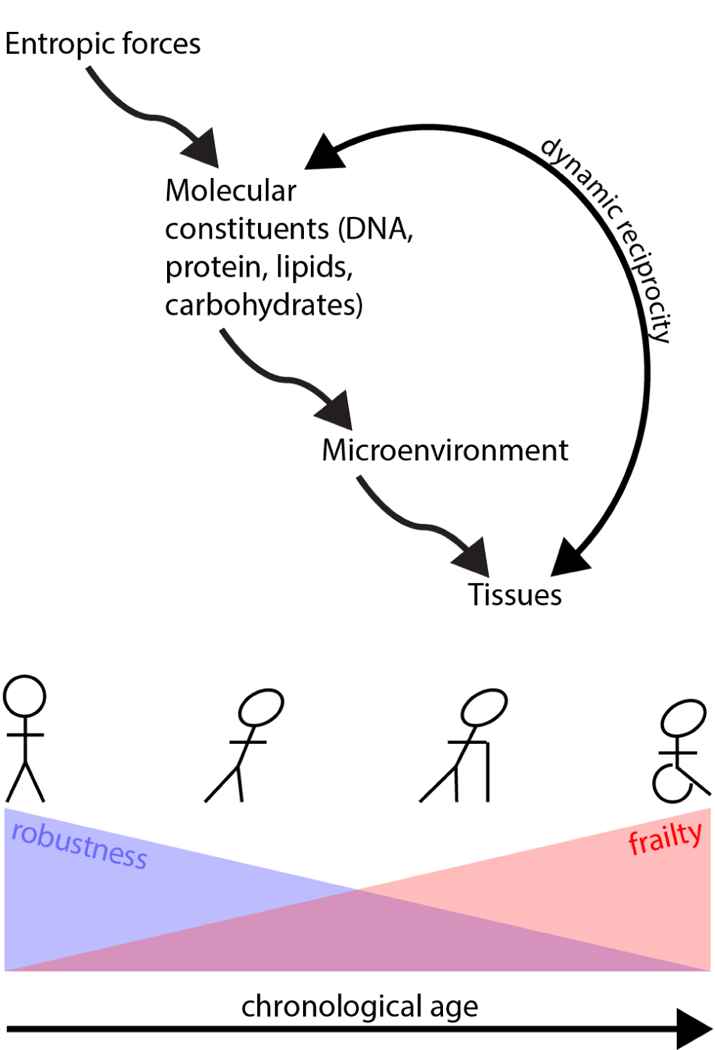
Over time, constant entropic forces act on molecular constituents, altering cells and extracellular matrix and ultimately evoking dynamic and reciprocal responses from the tissues. This, in turn, pushes cells through a series of metastable states of decreasing robustness and increasing frailty. This decreased robustness, combined with the stochastic nature of the entropic drivers, tends to increase cell-to-cell variability over time.
The rate of aging, defined both by the accumulation of molecular insults and the rate at which cellular function and physiological frailty accumulates, is non-linear, accelerating with age over time[53]. Thus, biological systems subjected to vigorously applied entropic forces should show accelerated aging. Indeed, children who receive myeloablative treatment prior to bone marrow transplant, or who receive chemotherapy, exhibit physiological ages that are many years beyond their peers[54–56]. Another way to explain the outpacing of molecular aging markers by physiological aging is that, for every direct molecular insult, there will be cascading interconnected consequences. The robustness of a biological system itself may decrease as well. Given the interconnectedness of cells within an organism, it is reasonable to conclude that advanced aging phenotypes, or state changes such as senescence, within a minority of cells may accelerate aging in the majority population. A growing body of evidence supports the hypothesis that molecular noise and heterogeneity of cells increases as an organism ages. Given this, outlier cells, especially cells, such as stem cells and fibroblasts, that exert an outsize effect on their surrounding tissue are especially likely candidates for aging acceleration.
Acknowledgements
We apologize to our colleagues whose important and relevant work we could not cite due to space limitations. We are grateful for support for our work on aging and breast cancer from the Era of Hope Scholar Award from the Congressionally Directed Medical Research Programs Breast Cancer Research Program (BC141351), the National Institute on Aging (R01AG040081), and the City of Hope Center for Cancer and Aging.
References
- 1.Kishabongo AS, Katchunga P, Cikomola JC, De Somer FM, De Buyzere ML, Speeckaert MM, Delanghe JR: The presence of fructosamine in human aortic valves is associated with valve stiffness. J Clin Pathol 2016, 69:772–776. [DOI] [PubMed] [Google Scholar]
- 2.Shinno Y, Ishimoto T, Saito M, Uemura R, Arino M, Marumo K, Nakano T, Hayashi M: Comprehensive analyses of how tubule occlusion and advanced glycation end-products diminish strength of aged dentin. Sci Rep 2016, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cadet J, Davies KJA: Oxidative DNA damage & repair: An introduction. Free Radic Biol Med 2017, 107:2–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Richardson RB: Ionizing radiation and aging: rejuvenating an old idea. Aging 2009, 1:887–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wielopolski L, Asselin PK, Ramirez LK, Bauman WA, Bikit I: THE UBIQUITY OF BACKGROUND RADIATION AND THE CLINICAL UTILITY OF NATURALLY OCCURRING POTASSIUM-40 IN HUMAN BODY. In Gamma Rays: Technology, Applications and Health Implications Nova Science Publishers, Incorporated; 2012:345–359. [Google Scholar]
- 6.Barrows LR, Magee PN: Nonenzymatic methylation of DNA by S-adenosylmethionine in vitro. Carcinogenesis 1982, 3:349–351. [DOI] [PubMed] [Google Scholar]
- 7.Lawley PD: Methylation of DNA by N-methyl-N-nitrosourethane and N-methyl-N-nitroso-N′-nitroguanidine. Nature 1968, 218:580. [DOI] [PubMed] [Google Scholar]
- 8.Kasai H, Kawai K, Li Y-S: Free radical-mediated cytosine C-5 methylation triggers epigenetic changes during carcinogenesis. Biomol Concepts 2013, 4:213–220. [DOI] [PubMed] [Google Scholar]
- 9.Ehrlich M, Zhang XY, Inamdar NM: Spontaneous deamination of cytosine and 5-methylcytosine residues in DNA and replacement of 5-methylcytosine residues with cytosine residues. Mutat Res 1990, 238:277–286. [DOI] [PubMed] [Google Scholar]
- 10.Schalkwijk CG, Stehouwer CDA, van Hinsbergh VWM: Fructose-mediated non-enzymatic glycation: sweet coupling or bad modification. Diabetes Metab Res Rev 2004, 20:369–382. [DOI] [PubMed] [Google Scholar]
- 11.Christensen L, Suggs A, Baron E: Ultraviolet Photobiology in Dermatology. Adv Exp Med Biol 2017, 996:89–104. [DOI] [PubMed] [Google Scholar]
- 12.Sakaue H, Kinouchi T, Fujii N, Takata T, Fujii N: Isomeric Replacement of a Single Aspartic Acid Induces a Marked Change in Protein Function: The Example of Ribonuclease A. ACS Omega 2017, 2:260–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klumb K, Matzenauer C, Reckert A, Lehmann K, Ritz-Timme S: Age estimation based on aspartic acid racemization in human sclera. Int J Legal Med 2016, 130:207–211. [DOI] [PubMed] [Google Scholar]
- 14.Gorisse L, Pietrement C, Vuiblet V, Schmelzer CEH, Köhler M, Duca L, Debelle L, Fornès P, Jaisson S, Gillery P: Protein carbamylation is a hallmark of aging. Proc Natl Acad Sci U S A 2016, 113:1191–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Toyama BH, Hetzer MW: Protein homeostasis: live long, won’t prosper. Nat Rev Mol Cell Biol 2013, 14:55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lau E, Cao Q, Ng DCM, Bleakley BJ, Dincer TU, Bot BM, Wang D, Liem DA, Lam MPY, Ge J, et al. : A large dataset of protein dynamics in the mammalian heart proteome. Scientific Data 2016, 3:160015.This paper is special, quantifying the protein turnover rates for thousands of proteins in the mouse heart. This is important because the lowest turnover proteins are the likely candidates for aging-associated physicochemical damage.
- 17.Delpierre G, Veiga-da-Cunha M, Vertommen D, Buysschaert M, Van Schaftingen E: Variability in erythrocyte fructosamine 3-kinase activity in humans correlates with polymorphisms in the FN3K gene and impacts on haemoglobin glycation at specific sites. Diabetes Metab 2006, 32:31–39. [DOI] [PubMed] [Google Scholar]
- 18.Kondo T, Ohshima T, Ishida Y: Age-dependent expression of 8-hydroxy-2’-deoxyguanosine in human pituitary gland. Histochem J 2001, 33:647–651. [DOI] [PubMed] [Google Scholar]
- 19.Weon BM, Je JH: Trends in scale and shape of survival curves. Sci Rep 2012, 2:504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bohnert KA, Kenyon C: A lysosomal switch triggers proteostasis renewal in the immortal C. elegans germ lineage. Nature 2017, 551:629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Milholland B, Dong X, Zhang L, Hao X, Suh Y, Vijg J: Differences between germline and somatic mutation rates in humans and mice. Nat Commun 2017, 8:15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Magalhães JP, Costa J: A database of vertebrate longevity records and their relation to other life-history traits. J Evol Biol 2009, 22:1770–1774. [DOI] [PubMed] [Google Scholar]
- 23.LaBarge MA, Mora-Blanco EL, Samson S, Miyano M: Breast Cancer beyond the Age of Mutation. Gerontology 2015, 62:434–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bocklandt S, Lin W, Sehl ME, Sánchez FJ, Sinsheimer JS, Horvath S, Vilain E: Epigenetic predictor of age. PLoS One 2011, 6:e14821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, Klotzle B, Bibikova M, Fan J-B, Gao Y, et al. : Genome-wide Methylation Profiles Reveal Quantitative Views of Human Aging Rates. Mol Cell 2013, 49:359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weidner CI, Lin Q, Koch CM, Eisele L, Beier F, Ziegler P, Bauerschlag DO, Jöckel K-H, Erbel R, Mühleisen TW, et al. : Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol 2014, 15:R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Horvath S: DNA methylation age of human tissues and cell types. Genome Biol 2013, 14:R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garbe JC, Pepin F, Pelissier FA, Sputova K, Fridriksdottir AJ, Guo DE, Villadsen R, Park M, Petersen OW, Borowsky AD, et al. : Accumulation of Multipotent Progenitors with a Basal Differentiation Bias during Aging of Human Mammary Epithelia. Cancer Res 2012, 72:3687–3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miyano M, Sayaman RW, Stoiber MH, Lin C-H, Stampfer MR, Brown JB, LaBarge MA: Age-related gene expression in luminal epithelial cells is driven by a microenvironment made from myoepithelial cells. Aging 2017, 9:2026–2051.This paper describes how gene expression in a tissue is driven by the microenvironment. It is of special interest because (1) it provides evidence for aging-associated loss of robustness and (2) it shows mechanisms by which aging cells may affect one another.
- 30.Brodin P, Jojic V, Gao T, Bhattacharya S, Angel CJL, Furman D, Shen-Orr S, Dekker CL, Swan GE, Butte AJ, et al. : Variation in the human immune system is largely driven by non-heritable influences. Cell 2015, 160:37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Viñuela A, Brown AA, Buil A, Tsai P-C, Davies MN, Bell JT, Dermitzakis ET, Spector TD, Small KS: Age-dependent changes in mean and variance of gene expression across tissues in a twin cohort. Hum Mol Genet 2017, doi:10.1093/hmg/ddx424. [DOI] [PMC free article] [PubMed]
- 32.Shalek AK, Satija R, Adiconis X, Gertner RS, Gaublomme JT, Raychowdhury R, Schwartz S, Yosef N, Malboeuf C, Lu D, et al. : Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature 2013, 498:236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Buettner F, Natarajan KN, Casale FP, Proserpio V, Scialdone A, Theis FJ, Teichmann SA, Marioni JC, Stegle O: Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells. Nat Biotechnol 2015, 33:155–160. [DOI] [PubMed] [Google Scholar]
- 34.Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL: The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 2014, 32:381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kowalczyk MS, Tirosh I, Heckl D, Rao TN, Dixit A, Haas BJ, Schneider RK, Wagers AJ, Ebert BL, Regev A: Single-cell RNA-seq reveals changes in cell cycle and differentiation programs upon aging of hematopoietic stem cells. Genome Res 2015, 25:1860–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grover A, Sanjuan-Pla A, Thongjuea S, Carrelha J, Giustacchini A, Gambardella A, Macaulay I, Mancini E, Luis TC, Mead A, et al. : Single-cell RNA sequencing reveals molecular and functional platelet bias of aged haematopoietic stem cells. Nat Commun 2016, 7:11075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martinez-Jimenez CP, Eling N, Chen H-C, Vallejos CA, Kolodziejczyk AA, Connor F, Stojic L, Rayner TF, Stubbington MJT, Teichmann SA, et al. : Aging increases cell-to-cell transcriptional variability upon immune stimulation. Science 2017, 355:1433–1436.This paper is of special interest, providing direct evidence for the aging-associated increase in transcriptional variability. This is important because, although aging-associated variability is predicted from first principles, this paper provides empirical verification.
- 38.Enge M, Arda HE, Mignardi M, Beausang J, Bottino R, Kim SK, Quake SR: Single-Cell Analysis of Human Pancreas Reveals Transcriptional Signatures of Aging and Somatic Mutation Patterns. Cell 2017, 171:321–330. e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koltover VK: Mathematical Theory of Reliability and Aging: Teaching Comes from Kiev. In 2016 Second International Symposium on Stochastic Models in Reliability Engineering, Life Science and Operations Management (SMRLO) 2016:386–392. [Google Scholar]
- 40.Zykovich A, Hubbard A, Flynn JM, Tarnopolsky M, Fraga MF, Kerksick C, Ogborn D, MacNeil L, Mooney SD, Melov S: Genome-wide DNA methylation changes with age in disease-free human skeletal muscle. Aging Cell 2014, 13:360–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tserel L, Kolde R, Limbach M, Tretyakov K, Kasela S, Kisand K, Saare M, Vilo J, Metspalu A, Milani L, et al. : Age-related profiling of DNA methylation in CD8+ T cells reveals changes in immune response and transcriptional regulator genes. Sci Rep 2015, 5:13107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hernandez DG, Nalls MA, Gibbs JR, Arepalli S, van der Brug M, Chong S, Moore M, Longo DL, Cookson MR, Traynor BJ, et al. : Distinct DNA methylation changes highly correlated with chronological age in the human brain. Hum Mol Genet 2011, 20:1164–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khalil H, Tazi M, Caution K, Ahmed A, Kanneganti A, Assani K, Kopp B, Marsh C, Dakhlallah D, Amer AO: Aging is associated with hypermethylation of autophagy genes in macrophages. Epigenetics 2016, 11:381–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ucar D, Márquez EJ, Chung C-H, Marches R, Rossi RJ, Uyar A, Wu T-C, George J, Stitzel ML, Palucka AK, et al. : The chromatin accessibility signature of human immune aging stems from CD8+ T cells. J Exp Med 2017, 214:3123–3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McClay JL, Aberg KA, Clark SL, Nerella S, Kumar G, Xie LY, Hudson AD, Harada A, Hultman CM, Magnusson PKE, et al. : A methylome-wide study of aging using massively parallel sequencing of the methyl-CpG-enriched genomic fraction from blood in over 700 subjects. Hum Mol Genet 2014, 23:1175–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Coppé J-P, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, Nelson PS, Desprez P-Y, Campisi J: Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 2008, 6:2853–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A, et al. : Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016, 530:184–189.This paper is of special interest, demonstrating that the induced destruction of senescent cells improved the lifespan of mice. This is key evidence that aging-associated cell subpopulations can have outsize effects on the whole organism.
- 48.Carlson BM, Faulkner JA: Muscle transplantation between young and old rats: age of host determines recovery. Am J Physiol 1989, 256:C1262–6. [DOI] [PubMed] [Google Scholar]
- 49.Conboy IM, Conboy MJ, Wagers AJ, Girma ER, Weissman IL, Rando TA: Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature 2005, 433:760–764. [DOI] [PubMed] [Google Scholar]
- 50.Oh J, Sinha I, Tan KY, Rosner B, Dreyfuss JM, Gjata O, Tran P, Shoelson SE, Wagers AJ: Age-associated NF-κB signaling in myofibers alters the satellite cell niche and re-strains muscle stem cell function. Aging 2016, 8:2871–2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Minnerly J, Zhang J, Parker T, Kaul T, Jia K: The cell non-autonomous function of ATG-18 is essential for neuroendocrine regulation of Caenorhabditis elegans lifespan. PLoS Genet 2017, 13:e1006764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gelino S, Chang JT, Kumsta C, She X, Davis A, Nguyen C, Panowski S, Hansen M: Intestinal Autophagy Improves Healthspan and Longevity in C. elegans during Dietary Restriction. PLoS Genet 2016, 12:e1006135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Finch CE, Crimmins EM: Constant molecular aging rates vs. the exponential acceleration of mortality. Proc Natl Acad Sci U S A 2016, 113:1121–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hurria A, Jones L, Muss HB: Cancer Treatment as an Accelerated Aging Process: Assessment, Biomarkers, and Interventions. Am Soc Clin Oncol Educ Book 2016, 35:e516–22.This paper is of special interest, reviewing the relationship between cancer treatments and accelerated aging. This is key evidence connecting physiochemical insults and the aging process.
- 55.Ariffin H, Azanan MS, Abd Ghafar SS, Oh L, Lau KH, Thirunavakarasu T, Sedan A, Ibrahim K, Chan A, Chin TF, et al. : Young adult survivors of childhood acute lymphoblastic leukemia show evidence of chronic inflammation and cellular aging. Cancer 2017, 123:4207–4214. [DOI] [PubMed] [Google Scholar]
- 56.Arora M, Sun C-L, Ness KK, Teh JB, Wu J, Francisco L, Armenian SH, Schad A, Namdar G, Bosworth A, et al. : Physiologic Frailty in Nonelderly Hematopoietic Cell Transplantation Patients: Results From the Bone Marrow Transplant Survivor Study. JAMA Oncol 2016, 2:1277–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mikkelsen L, Bialkowski K, Risom L, Løhr M, Loft S, Møller P: Aging and defense against generation of 8-oxo-7,8-dihydro-2′-deoxyguanosine in DNA. Free Radical Biology and Medicine 2009, 47:608–615. [DOI] [PubMed] [Google Scholar]
- 58.Maier P, Hartmann L, Wenz F, Herskind C: Cellular Pathways in Response to Ionizing Radiation and Their Targetability for Tumor Radiosensitization. Int J Mol Sci 2016, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tricarico R, Bellacosa A: Active DNA Demethylation in Development, Human Disease, and Cancer. In DNA Replication, Recombination, and Repair Springer, Tokyo; 2016:517–548. [Google Scholar]