

Abstract
Site-selective protein modification based on covalent reactions of peptide tags and small molecules is a key capability for basic research as well as for the development of new therapeutic bioconjugates. Here, we describe the computation-guided rational design of a cysteine- and lysine-containing 11-residue peptide sequence that reacts with 2-cyanobenzothiazole (CBT) derivatives. Our data show that the cysteine residue reversibly reacts with the nitrile group on the CBT moiety to form an intermediate thioimidate, which undergoes irreversible SN transfer to the lysine residue, yielding an amidine-linked product. The concepts outlined herein lay a foundation for future development of peptide tags in the context of site-selective modification of lysine residues within engineered microenvironments.
Graphical Abstract
INTRODUCTION
Site-selective protein modification is an imsportant tool for imaging targets in vitro and in vivo, creating new therapeutics, and installing functionality on proteins to probe activity.1−4 However, the limited number of proteinogenic amino acids and their natural repetition in proteins limits them as targets for site-selective ligation. Easy access to different modifications, site selectivity, and the potential for a large substrate scope makes covalent modification of engineered peptide tags by small molecules an attractive and widely used platform for biotherapeutic and basic research.5 Much like an enzyme active site, these peptide tags are engineered to produce a microenvironment that enables preferential ligation to a peptide sequence of interest, thus allowing for selective and specific labeling of a protein. For application in biological systems, any reaction between the peptide tag and the small molecule should proceed in aqueous conditions, at physiological pH and temperature, and, if intended for in cellulo or in vivo studies, without deleterious effects.
Previously, Tsien and co-workers described a foundational approach for the rational design of a peptide sequence that can be covalently modified by a small molecule.6,7 In their sequence (Cys-Cys-Pro-Gly-Cys-Cys), four cysteine residues are preorganized into a secondary structure that rapidly reacts with FlAsH-EDT2, a fluorescein-based diarsenical probe. The engineered peptide tag consists entirely of proteinogenic amino acids, yet selective ligation is achievable—any off-target interactions with naturally occurring peptide sequences can be reversed with addition of ethanedithiol (EDT). As no unnatural amino acids or changes to the endogenous translational machinery are necessary, perturbations to the cell are minimized. The early success of this rationally designed peptide for protein imaging inspired an ongoing wave of research into novel peptide tags.
Recently, Pentelute and colleagues introduced the shortest small-molecule-binding peptide to date, the “π-clamp” (PheCys-Pro-Phe).8 Using a library selection method to identify peptides that are subject to arylation reactions in water, they found that this four-residue tag provided a favorable microenvironment for the cysteine residue to displace a fluorine substituent on a perfluoroaromatic probe via a nucleophilic aromatic substitution. As they had previously shown that endogenous cysteines do not react with perfluoroaryl moieties in water,9 the discovery of the π-clamp indicates the feasibility of designing other short natural amino acid sequences that elicit emergent functionality.
Both the FlAsH tag and the π-clamp exploit the tunability of cysteine’s reactivity when developing new peptide conjugation methods. In addition, cysteine is the most reactive of the 20 canonical amino acids at physiological pH and appears in proteins at a relatively low frequency, which makes it a prime target for ligation chemistry.2,3,5 However, cysteine plays fundamental structural and chemical roles in proteins, so modifying or removing endogenous cysteine residues from a protein of interest to accommodate an engineered peptide tag is not always feasible. Thus, engineered peptide tags that enable site-selective modification of different amino acid residues without requiring exogenous enzymes or translational machinery remain an unmet need.
Another amino acid that is nucleophilic under certain conditions is lysine. Cravatt and co-workers recently reported the results of a global profiling experiment on lysine reactivity in the human proteome.10 They found that of the more than 9000 lysines they quantified in the human proteome, a few hundred had heightened reactivity, and over 100 could be targeted by amine-reactive electrophilic fragments. As lysine is a common surface-exposed residue but non-nucleophilic at physiological pH due to protonation of the side chain amine, these data suggest peptide tags can form a microenvironment conducive to lysine modification. Further supporting this hypothesis is the recent work of Bernardes and co-workers, in which the siteselective modification of the single most reactive lysine on a given protein by sulfonyl acrylate reagents was reported.11 Moreover, the authors were able to computationally predict which lysine would be modified in each of the five proteins they analyzed.
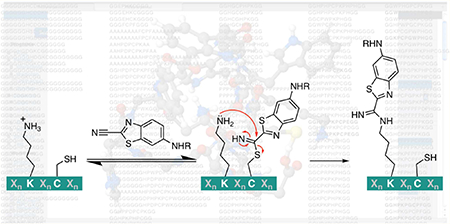
In designing a new small-molecule-binding peptide tag, we took inspiration from the enhanced amine reactivity seen in native chemical ligation (NCL) between an N-terminal cysteine and a C-terminal thioester (Figure 1A).12 During NCL, the nucleophilic N-terminal cysteine thiol undergoes reversible transthioesterification with the C-terminal thioester. Then, the acyl group is irreversibly transferred to the N-terminal amine, resulting in a new amide bond between the two original peptides. The transthioesterification step increases the effective concentration of the amine, greatly enhancing the rate of amide bond formation. Transglutaminases use a similar concept, forming a thioester bond between an enzyme cysteine and a substrate glutamine, which can then be attacked by a second peptide’s N-terminal amine or a lysine side chain to form an isopeptide bond.13,14 In the case of NCL, two distinct nucleophiles on the same residue are key to allowing amide bond formation to proceed at a reasonable rate. We hypothesized that an engineered peptide could position a cysteine residue and a lysine residue in such a way as to attain an analogous functionality to NCL, but without the requirement of an N-terminal cysteine.
Figure 1.

Chemoselective reactions involving aminothiols. (A) Mechanism of native chemical ligation. Transthioesterification occurs when an N-terminal cysteine on one peptide attacks a C-terminal thioester on another. An SN acyl transfer occurs, irreversibly forming a new amide bond. (B) 6-Hydroxy-2-cyanobenzothiazole and D-cysteine react to form firefly luciferin. The cysteine thiol attacks the nitrile group to form a thioimidate intermediate, and then the amino group attacks the thioimidate to form a tetrahedral intermediate. Finally, irreversible loss of ammonia drives the reaction to completion. (C) Proposed peptide (teal line) that uses cysteine and lysine residues to accomplish a reaction analogous to the native chemical ligation. The cysteine attacks the electrophile and then transfers it to the lysine residue in an irreversible reaction.
Additionally, we were drawn to the reaction between 2-cyanobenzothiazole derivatives and 1,2-aminothiols. In the most widely used application, firefly luciferin is synthesized by reacting 6-hydroxy-2-cyanobenzothiazole with D-cysteine in the laboratory and in nature (Figure 1B).15,16 The cysteine thiol and the nitrile group react to form a thioimidate, which is then attacked by the lysine amine to form a tetrahedral intermediate. In contrast to NCL, in which the thiol “hands off” the acyl group to the amine, the tetrahedral intermediate eliminates a molecule of ammonia to reach the final product. Rao and co-workers found that the reaction of a 6-amino-2-cyanobenzothiazole (CBT) derivative with cysteine proceeds with a secondorder rate constant of 9 M−1 s−1 in vitro at room temperature and physiological pH.17 This feature as well as the reversibility of the reaction between CBT and endogenous thiols allowed them to develop a method to selectively label engineered N-terminal cysteines in vivo. Other groups have also devised clever ways to utilize this reaction.18−20 Most recently, Lin and co-workers reported a genetically encodable CBT derivative-binding peptide tag discovered through a phage display experiment.21 As with previous examples of engineered peptide tags, this too is a cysteine conjugation tag, and substituting adjacent residues with lysine did not appear to change the mode or mechanism of binding.
With both NCL and the CBT reaction in mind, we focused on developing a short, genetically encodable peptide tag that selectively and irreversibly ligates the nitrile group of a CBT derivative under physiological conditions. To do so, we designed in silico a peptide with a lysine and a cysteine residue oriented such that a selective reaction with the nitrile group of an N-acylated 6-amino-2-cyanobenzothiazole would occur (Figure 1C). Herein, we show that our first-generation engineered peptide creates a microenvironment for a “cysteine hand-off” mechanism analogous to NCL, resulting in an irreversible, covalent ligation.
RESULTS AND DISCUSSION
Initially, we considered selecting candidate peptide sequences using the phage display method reported previously Weiss, Wells, and co-workers.22−24 However, this method requires a disulfide bond in the peptide library to restrict the conformational space during the selection process, which complicates library generation and screening for our reaction of interest as our intended peptide would require a reduced cysteine for reaction with CBT. Considering the knowledge that phage display can lead to unanticipated results25 and the nontrivial nature of designing selection conditions that would favor a final product with the CBT analog attached to the lysine rather than the cysteine, we alternatively pursued a rational design in silico. This approach yielded several short peptide sequences (10 to 11 amino acids long) that efficiently bind cyanobenzothiazole derivatives under various conditions.
For the in silico approach, the online computational tool PEP-FOLD26,27 was used to model short peptides containing lysine, cysteine, histidine, aspartic or glutamic acid, and proline residues. Inspired by enzyme-resident catalytic triads, we hypothesized that histidine and aspartic acid would increase the nucleophilicity of cysteine and lysine residues if proline residues were incorporated to help lock the peptide into the desired conformation (Figure 2A). In addition, the protonated imidazole of the histidine residue could also catalyze the reaction between the cysteine thiol and the nitrile group of a CBT derivative.28,29 Of the 218 sequences modeled (Supplemental Table S1), 18 were selected for synthesis via solid-phase peptide synthesis (SPPS) and further analysis (Figure 2B,C). In Figure 2C, the two peptides on the left are representative of the best models: the lysine, histidine, aspartic acid, and cysteine residues are all in relatively close proximity. In particular, the histidine side chain appeared poised to help deprotonate the lysine residue. The two peptides on the right do not fit our criteria: either the histidine residue was not in a position to deprotonate the cysteine or the lysine residue or the cysteine and lysine residues appeared too far apart to feasibly work together in forming covalent bonds with a CBT derivative.
Figure 2.

Rational design of a peptide that covalently binds a CBT derivative. (A) Principles behind generating peptide sequences for computational modeling. Lysine and cysteine residues were interspersed with histidine and aspartic or glutamic acid residues to increase nucleophilicity. Prolines were added to lock the sequence (teal) into a favorable conformation for reaction with a CBT derivative. (B) Selection of sequences to test for reaction with a CBT derivative. 218 sequences were modeled using the program PEP-FOLD (see Supplemental Table S1 for a full list).26,27 Of these peptides, 18 that best fit our selection criteria were synthesized by solid-phase peptide synthesis (SPPS) and tested in a binding assay. (C) Examples demonstrating how peptides were selected for SPPS. The peptides on the left were two of the best models, whereas the two peptides on the right were not chosen for SPPS.
Based on MALDI-TOF MS, many of the peptides reacted with N-(2-(cyano-13C)-benzo[d]thiazol-6-yl)acetamide (CBTNAc, compound 1) in phosphate-buffered saline (PBS) at room temperature over 48 h even in the absence of reducing agents such as dithiothreitol (DTT) or tris(2-carboxyethyl)phosphine (TCEP) (assay workflow and representative data shown in Figure 3; data for all 18 peptides, peptides 4−21, shown in Supplemental Figure S1). Several but not all of the peptides also reacted with 2 equiv of the CBT derivative. We hypothesized that if only 1 equiv of CBTNAc was bound, it could be attached to either the lysine or cysteine residue, whereas if two were bound, they were attached to both the lysine and cysteine residues. In a follow-up experiment, free cysteine was added to the peptide−CBTNAc adducts to trap unbound CBTNAc as a luciferin analog. As the reaction of the peptide’s cysteine residue with the CBTNAc nitrile group is reversible, any CBTNAc bound to the cysteine and not transferred to the lysine residue should be in equilibrium with free peptide and CBTNAc. When free cysteine is added, it reacts irreversibly with unbound CBTNAc to form the luciferin analog. Depletion of the unbound CBTNAc in this manner will then drive the peptide−CBTNAc adduct toward free CBTNAc and an unmodified cysteine residue on the peptide by Le Chatelier̂ ‘s principle. Indeed, addition of free cysteine resulted in significant loss of CBTNAc from the peptides, with only 1 equiv remaining on some (Figure 3B and Supplemental Figure S1) and no modification remaining on others (Figure 3C and Supplemental Figure S1). Placing the cysteine and the lysine residues adjacent to one another resulted in minimal reaction with CBTNAc, but placing a proline between them was not in itself sufficient to ensure significant formation of a stable adduct (Supplemental Figure S1F,O), while placing additional residues between the cysteine and the lysine resulted in formation of just as much or more of the stable CBTNAc adduct as some peptides with cysteine and lysine close together (e.g., Supplemental Figure S1C,J,Q compared to S1F,O). Exchanging aspartic acid and glutamic acid altered the reactivity of a given peptide (Supplemental Figure 1A,E), as did replacing an aspartic or glutamic acid residue with or adding a residue such as histidine (Supplemental Figure S1H,O), tryptophan (Supplemental Figure S1L,M,N,P), or phenylalanine (Supplemental Figure S1Q,R).
Figure 3.

Assay for peptides that are covalently modified by CBTNAc (1) and representative results. (A) Overview of assay used to determine degree of irreversible covalent modification of candidate peptides. Candidate N-terminal acetylated, C-terminal amidated peptides were incubated with CBTNAc in PBS for 48 h at room temperature, and then an aliquot of the reaction mixture was analyzed by MALDI-TOF MS. The remaining solution was incubated for 48 h at room temperature with free cysteine, with or without DTT or TCEP reducing agent, and the reaction mixture was again analyzed by MALDI-TOF MS. (B) Representative MALDI-TOF MS spectra at each stage of the assay for a peptide with a lysine residue extensively modified by CBTNAc (peptide 4). The first spectrum (I) shows crude peptide alone, the second spectrum (II) was taken after incubation with CBTNAc and desalting, and the third spectrum (III) followed incubation with free cysteine. (C) Representative MALDI-TOF MS spectra at each stage of the assay for a peptide with a lysine residue not significantly modified by CBTNAc (peptide 5). The samples analyzed in each spectrum were prepared the same way as in B. Red boxes indicate the expected locations of masses corresponding to modified peptide.
Of the 18 peptides tested, we proceeded with characterization of the peptide modified to the greatest extent by CBTNAc, CBTag 1.0 (Ac-Gly-Gly-His-Pro-Asp-Pro-Cys-ProLys-Gly-Gly-NH2, 4). Performing the assay in the presence of 5 mM glutathione to mimic physiological thiol conditions still resulted in a significant amount of the post-cysteine treatment CBTag 1.0−CBTNAc adduct, though less of it than when conducting the assay in the absence of glutathione (Supplemental Figure S2), These results indicated that some competition between endogenous thiols and the CBTag 1.0 cysteine is probable, but, promisingly, a stable bond between CBTag 1.0 and CBTNAc can still form. To determine the necessity of the cysteine and the lysine residues, two modified peptides were synthesized: one with the lysine residue converted to arginine to preserve the positive charge while decreasing nucleophilicity further (22), and one with the cysteine residue converted to serine to decrease nucleophilicity (23). These derivatives were subjected to the conditions described in the previous paragraph. The lysine to arginine mutant reacted with 1 equiv of CBTNAc before cysteine treatment, likely due to the cysteine residue in the peptide (Supplemental Figure S3A). Treatment with free cysteine abrogated binding, resulting in free peptide, as expected. The cysteine to serine mutant did not bind any equivalents of CBTNAc, indicating that the cysteine residue is necessary for activity (Supplemental Figure S3B). Thus, both cysteine and lysine are necessary for irreversible covalent bond formation with CBTNAc. Combined with the earlier observation that not all of the synthesized peptides irreversibly reacted with CBTNAc, these data led us to propose that cysteine and lysine are necessary but not sufficient for CBTNAc binding, indicating that conformation of the peptide is important for the intended reactivity.
To further establish that the remaining equivalent of CBTNAc was not attached to the cysteine residue, the peptide−CBTNAc adduct was reacted with chloroacetate after treatment with free cysteine. Chloroacetate specifically reacts with thiols, not amines, and thus should only label the peptide−CBTNAc adduct if the CBTNAc is not bound to the cysteine residue (i.e., if the CBTNAc is bound to lysine). Indeed, a clean mass shift indicating reaction with chloroacetate was observed for the CBTag 1.0−CBTNAc adduct after cysteine treatment (Supplemental Figure S4A). Moreover, when CBTag 1.0 was incubated with CBTNAc but not treated with free cysteine, chloroacetate did not react with any peptide bound to 2 equiv of CBTNAc. If the CBTag 1.0 peptide was pretreated with chloroacetate, no binding of CBTNAc occurred, though this could be due to the addition of a negative charge causing a conformational shift (Supplemental Figure S4B,C). As a control, we confirmed that the cysteine to serine mutant did not react with chloroacetate, whereas the lysine to arginine mutant did (Supplemental Figure S4D).
Additionally, tandem mass spectrometry (MS/MS) on CBTag 1.0 following cysteine treatment confirmed that the remaining CBTNAc was indeed bound to lysine (Figure 4). Finally, 13C NMR spectra acquired after reaction between CBTNAc with a 13C-labeled nitrile group (13CBTNAc, compound 3) and CBTag 1.0 did not contain a peak corresponding to a stable tetrahedral intermediate but did contain peaks that match predicted values for the α-carbon of a thioimidate or amidine bond (CBTag 1.0−13CBTNAc amidine adduct 24, Supplemental Figure S5).
Figure 4.

Tandem mass spectrometry (MS/MS) on CBTag 1.0−CBTNAc demonstrates that modification is on the lysine residue. Top, predicted masses for MS/MS b and y product ions if the CBTNAc (purple star) is on the cysteine residue (left) or the lysine residue (right). Bottom, tandem MS/MS on the [M + CBTNAc + 2H]2+ ion shows product ions matching those predicted for lysine modification (green boxes).
In order to quantitate relative amounts of starting materials, intermediates, and products over time, we developed a liquid chromatography/mass spectrometry (LC-MS) method to follow the course of the reaction. As expected, the lysine adduct, the cysteine adduct, and the doubly modified peptide were resolvable using our method, though higher concentrations of the peptide than were used in the MALDI-TOF assay were necessary in order to clearly observe a 210 nm signal suitable for integration (Supplemental Figures S6 and S7). In order to deconvolute the data, TCEP was included in the injected samples to remove peaks corresponding to disulfide dimers. Comparing peak areas in the chromatographs over time, we found that at the first time point (1.75−2.8 h), the majority of the CBTNAc attached to CBTag 1.0 at the final time point (40−45 h) was already covalently bound to either a cysteine or a lysine residue; however, only 7−10% of this peptide-bound CBTNAc population was attached to lysine, suggesting the thioimidate intermediate forms relatively quickly, but transfer to the lysine is slow (Supplemental Figure S8 and Tables S2−S4). By the end of the experiment, approximately 35−41% of peptide-bound CBTNAc was on a lysine residue whether the ratio of CBTag 1.0 to CBTNAc was 1:1 (22.5 mM each), 4:1 (22.5 and 5.63 mM), or 1:4 (5.63 and 22.5 mM), whereas the percentage of total CBTag 1.0 that had a lysine residue modified by CBTNAc ranged from 17 to 38% depending on the conditions (Supplemental Figure S8 and Tables S2−S4). The low variation in rate of formation of lysine-bound CBTNAc despite significant changes in concentration of the peptide also provide evidence that transfer of the CBTNAc is intramolecular, not intermolecular. These numbers matched the approximate yields of 16−36% we obtained when we isolated the amidine product after cysteine treatment. However, somewhat unexpectedly based on the MALDI-TOF MS results, conversion of the free peptide to peptide modified on the cysteine, the lysine, or both ranged from 38 to 80%—under the conditions of this assay, full conversion to a bound state was not observed even when CBTNAc was present in excess (Supplemental Figure S8 and Table S2−S4). We attribute this to reversion of some portion of the cysteine monoadduct to the free peptide and unreacted CBTNAc on passage through the column (no peaks corresponding to hydrolysis of CBTNAc were observed), unforeseen effects of including TCEP in the reaction, aggregation of the peptide or the CBTNAc at higher concentrations, or differences between running the reaction in a test tube and an LC-MS vial. Again, as a confirmation, the lysine to arginine mutant did form a peak corresponding to cysteine modification over time, and the cysteine to serine mutant spectrum did not change (Supplemental Figures S9−S11).
As an additional confirmation that transfer from cysteine to lysine is intramolecular and not intermolecular, a 1:1 mixture of CBTag 1.0 (K9R) (22) and CBTag 1.0 (C7S) (23) was subjected to our initial MALDI-TOF MS assay conditions (Supplemental Figure S12). As CBTag 1.0 (K9R) has a cysteine but not a lysine and reacts with 1 equiv of CBTNAc prior to cysteine treatment, and CBTag 1.0 (C7S) has a lysine but not a cysteine and does not react with CBTNAc at all, any formation of a CBTag (C7S)−CBTNAc adduct would have to be a result of intermolecular cysteine−lysine relay. Although a mass corresponding to formation of the CBTag 1.0 (K9R)−CBTNAc adduct was observed prior to cysteine treatment, no mass corresponding to the CBTag 1.0 (C7S)−CBTNAc adduct appeared. Moreover, treatment with cysteine reversed the CBTag 1.0 (K9R)− CBTNAc adduct, resulting in a MALDI-TOF MS spectrum that showed only the unreacted peptides. These data and the LC-MS data indicating that rate of formation of the CBTag 1.0−CBTNAc lysine adduct is independent of peptide concentration suggest an intramolecular cysteine−lysine transfer mechanism.
Together, our results support the hypothesis that the nucleophilic cysteine thiol rapidly reacts with the CBTNAc to form a thioimidate adduct, which is slowly transferred to the lysine, creating a stable amidine bond (Figure 5). To further rationalize why the tetrahedral intermediate proceeds to the amidine product instead of the product of the condensation reaction (as in luciferin), we used Maestro software to build both products and the integrated program MacroModel to search for the lowest energy conformations. The optimized conformation of the amidine product had a potential energy significantly lower than that of the product of the condensation reaction, even taking into account the released ammonia molecule (−488 and −456 kcal/mol, respectively; a difference of 22 kcal/mol) (Supplemental Figure S13). These results are not unexpected, as the entropic gain from the release of ammonia is unlikely to compensate for the entropic loss associated with the increased rigidity of an already large macrocycle, especially in the absence of conditions favoring the loss of ammonia. Additionally, the transition state for the amidine product is likely lower in energy relative to that of the condensation product. In both transition states, a partial positive charge builds up on a nitrogen atom, but in the transition state for the amidine product, the partial negative charge is on the cysteine sulfur atom, whereas in the transition state for the condensation product, the partial negative charge is on a nitrogen atom. Sulfur atoms are more polarizable than nitrogen atoms and are better able to stabilize a negative charge; thus, the amidine transition state is stabilized more than the condensation reaction transition state. In summary, both the transition state and the product energies favor the amidine product.
Figure 5.

Proposed mechanism of reaction between CBTag 1.0 and CBTNAc. First, the nucleophilic thiol on the cysteine residue reversibly forms a thioimidate bond with CBTNAc, likely catalyzed by the imidazole of the histidine residue.28,29 Then, the lysine amine attacks the thioimidate, resulting in a tetrahedral intermediate. Unlike the reaction to make firefly luciferin, a condensation step that releases ammonia does not proceed (red arrows). Instead, the end result is a stable amidine bond between the CBTNAc and the lysine residue in a “cysteine hand-off” mechanism analogous to that of NCL.
CONCLUSION
In conclusion, we have designed and characterized a peptide that forms an irreversible amidine bond between a uniquely reactive lysine residue and a CBT derivative. We have demonstrated that, similar to NCL, this mechanism occurs through transfer of the electrophilic group from a cysteine thiol to a lysine amine. These results are a promising first step toward a novel peptide tag for site-specific lysine functionalization, though significant optimization remains to be done before this system can be implemented as a practical tool. We are currently exploring mRNA display-based methods30 to select for an improved version of CBTag 1.0 as well as computational methods to engineer a peptide sequence that strongly favors the reactive conformation of the cysteine and lysine residues. Regardless, in using concepts that invoke themes from enzymology to bioorthogonal chemistry, we provide here a potential template for rational design of peptide tags with engineered “hot” lysines that can undergo site-selective covalent modification.
EXPERIMENTAL SECTION
General.
Reactions were performed in flame- or oven-dried glassware under an inert nitrogen atmosphere unless otherwise noted. Anhydrous solvents were either purchased or obtained by passing solvent through an activated alumina column via a Pure Process Technology Glass Contour solvent purification system. All reagents and solvents were used as received unless otherwise noted. Water was passed through a Milli-Q filtration system prior to use. Where noted, samples were concentrated in vacuo at 40 °C using a BüCHI Rotavapor R-114 equipped with a BÜCHI B-480 heating bath and a Welch self-cleaning dry vacuum system (model 2025) or an IKA RV 10 basic rotary evaporator equipped with an IKA HB 10 basic heating bath and a Welch self-cleaning dry vacuum system (model 2025). If necessary, compounds were then further dried under high vacuum using an Edwards RV8 two-stage rotary vane pump or by lyophilization in a LABCONCO FreeZone 4.5Plus.
Thin layer chromatography was performed using SiliCycle SiliaPlate glass-backed silica gel plates containing a fluorescent indicator (Fisher Scientific 50964470). Plates were visualized using a UVGL-25 compact UV lamp, 254/365 nm, 4 W (P/N 95–0021-12). For flash column chromatography, the stationary phase was SiliCycle SiliaFlash P60 or Fisher silica gel sorbent (230−400 mesh, grade 60) silica gel. For purifications involving preparative or semipreparative reversed-phase high-performance liquid chromatography (RP-HPLC), the following conditions were used: the instrument consisted of an Agilent Technologies ProStar 325 UV−vis detector, two PrepStar solvent delivery modules, and a 440-LC fraction collector; the column was either a Varian Microsorb 100 Å C18, 8 μm, 21.4 × 250 mm Dynamax preparative column (R0080220C8) equipped with a Microsorb 100 Å C18, 8 μm guard column (R0080220G8) or an Agilent ZORBAX Eclipse XDB 80 Å C18, 5 μm, 9.4 × 250 mm semipreparative column (990967–202). Solvent A was 0.1% TFA in Milli-Q water; solvent B was 0.1% TFA in acetonitrile (MeCN). The UV−vis detector was used to monitor wavelengths at 210 and 254 nm. All pure compounds and stock solutions were stored at −20 °C.
LC-MS experiments were performed on an Agilent Technologies 1260 Infinity attached to a 6120 Quadrupole MS and a Peak Scientific NM32LA nitrogen generator. An Agilent InfinityLab Poroshell 120 EC-C18, 2.7 μm, 4.6 × 50 mm analytical LC column (part number 699975–902) was used. As with the HPLC, solvent A was 0.1% TFA in Milli-Q water; solvent B was 0.1% TFA in MeCN. Wavelengths of 210, 254, and 280 nm were monitored using the diode array detector. For ESI-MS, the 105−2000 m/z range was monitored in both positive and negative ion mode. Data were processed and analyzed using LC/MSD ChemStation (Agilent Technologies, Rev. B.04.03[16]). High-resolution mass spectrometry (HRMS) data were acquired by ESI-LC/MS on a Waters Acquity UPLC and Thermo Exactive Orbitrap mass spectrometer by Dr. Theresa McLaughlin at the Stanford University Mass Spectrometry facility.
MALDI-TOF MS spectra were obtained on either an Applied Biosystems Voyager-DE PRO BioSpectrometry Workstation using an Applied Biosystems Sample Plate SS, Numbers & Circles (P/N V700666) and the Applied Biosystems Voyager Instrument Control Panel v5.10 software (QB3/Chemistry Mass Spectrometry Facility, UC Berkeley) or a Bruker microFlex MALDI-TOF (S/N 256969.00028) using a microScout Target MSP 96 target polished steel BC plate (P/N 8280800) and the Bruker Daltonics FlexControl software (Vincent Coates Foundation Mass Spectrometry Laboratory, Stanford University). In all cases, the matrix was α-cyano-4-hydroxycinnamic acid dissolved in 50% MeCN in 0.1% TFA. Plates were spotted with a mixture of 0.5 μL of sample and 0.5 μL of matrix. For the Voyager, parameters were as follows: reflectron mode, positive ion detection; pulsed nitrogen laser (337 nm, 20 Hz); accelerating voltage 20 kV, grid voltage 94%, guide wire voltage 0.05%, delay time 270 ns; mass range 200−2500 Da, low mass gate 200 Da; spectrum acquisition−manual control, 1500 shots/spectrum; manual laser intensity ∼1500−2500. For the Bruker microFlex, the parameters were as follows: reflectron mode, positive ion detection; pulsed nitrogen laser (337 nm, 3 ns pulse width, pulse energy 150 μJ, 60 Hz); detector gain (reflector) 4.0×, 1810 V; sample rate 2.00 GS/s; laser power percentage was adjusted on a sample-by-sample basis to obtain an arbitrary intensity of 103−104 and a flat baseline; mass range 0−4000 Da; spectrum acquisition−random walk partial sample mode, results were the sum of at least 3 × 100 laser shots; calibration of the system was performed using the Bruker peptide calibration standard mixture (part number 8206195). Spectra were analyzed using the open-source software mMass (version 5.5.0)31−33 after data had been exported as an ASCII file from Voyager (Applied Biosystems) or FlexAnalysis (Bruker Daltonics). Nuclear magnetic resonance (NMR) spectra were obtained on a Bruker AVIII HD 400 MHz NMR spectrometer (UC Berkeley), a Varian Inova 500 (Stanford University), or an Agilent VNMRS 800 MHz NMR spectrometer (Stanford University). The 800 MHz NMR spectra were obtained by Dr. Corey Liu and Dr. Stephen Lynch of the Stanford University Department of Chemistry NMR Facility. Spectra were processed using MestReNova v10.0.2–15465 (Mestrelab Research S.L., 2015).
PEP-FOLD Screening.
PEP-FOLD26,27 was used for initial in silico screening of peptide sequences (Supplemental Table S1). MacPyMOL Molecular Graphics System v1.4 (Schrödinger, LLC, New York, NY, 2010) was used to visualize output.
Binding Assay (Adapted from Ref 22).
N-terminal acetylated, C-terminal amidated peptides (36 μL of a 1 mM stock solution, pH adjusted to approximately 7) were incubated with CBTNAc (0.4 or 4 μL of a 100 mM stock solution in DMF) or DMF alone (negative control) in PBS for 48 h at room temperature. An aliquot (0.5 μL) of each reaction mixture was analyzed by MALDI-TOF MS either without prior purification or after desalting with a μ-C18 ZipTip.
Cysteine Competition Assay.
After MALDI-TOF MS analysis, half (20 μL) of the binding assay solution was incubated for 48 h at room temperature after an equal volume of cysteine (10 mM or 10 equiv relative to CBTNAc, unless cysteine would interfere with a later assay, in which case 1 mM or 1.1 equiv was used; pH adjusted to approximately 7) was added with or without DTT or TCEP reducing agent (10−15 mM or 10−15 equiv, unless the reducing agent would interfere with a later assay, in which case 1 mM or 1.1 equiv was used; pH adjusted to approximately 7). The remainder of the binding assay solution was incubated with an equal volume of PBS or an equal volume of DTT or TCEP reducing agent (10−15 mM or 10−15 equiv, unless the reducing agent would interfere with a later assay, in which case 1 mM or 1.1 equiv was used; pH adjusted to approximately 7). The reaction mixtures were again analyzed by MALDI-TOF MS either without prior purification or after desalting with a μ-C18 ZipTip.
Binding Assay in the Presence of Glutathione.
CBTag 1.0 (18 μL of a 1 mM stock solution in PBS) was incubated with reduced glutathione (0.2 μL of a 500 mM stock solution in PBS) and CBTNAc (2 μL of a 10 mM stock solution in DMF) or DMF alone (negative control) in PBS for 48 h at room temperature. After completion, an equal volume (20 μL) of a solution of cysteine and DTT (10 and 15 mM, respectively) in PBS was added, and the resulting solution was incubated a further 48 h at room temperature. An aliquot (0.5 μL) of each reaction mixture was analyzed by MALDI-TOF MS without prior purification.
Chloroacetate Labeling of Cysteine Residues.
Peptide solution (0.9 mM; 0.45 mM if after treatment with a slight excess of cysteine) was incubated with an equal volume of 30 or 500 mM chloroacetate (pH adjusted to approximately 7) overnight at room temperature. The reaction mixtures were analyzed by MALDI-TOF MS without prior purification.
Tandem LC-MS/MS.
After the cysteine competition assay (using 1.1 equiv of cysteine), samples were diluted 1:9 with PBS and then 1:4 with water to give final concentrations (where applicable) of 10 μM peptide, 10 μM CBTNAc, 10 μM cysteine, and 15 μM DTT. Dr. Theresa McLaughlin of the Stanford University Mass Spectrometry facility analyzed the samples by ESI-MS on an Agilent 1260 HPLC connected to a Bruker MicroTOF-QII mass spectrometer and equipped with an Agilent Pursuit 5 diphenyl 2.1 × 150 mm LC column held at 60 °C. The injection volume was 10 μL, the flow rate was 0.3 mL/min, and data were collected in autoMS2 mode with a mass range of 50−2000 Da. Solvent A was 0.1% formic acid (FA) in water, solvent B was 0.1% FA in MeCN, and the gradient was as follows: 5% B for 2 min, 5−95% B over 5 min, 95% B for 1 min, 95−5% B over 0.1 min, and 5% B for 6.9 min. Predicted fragment ions were calculated using the MS/MS fragment ion calculator in the Proteomics Toolkit provided by the Institute for Systems Biology (http://db.systemsbiology.net/proteomicsToolkit/FragIonServlet.html).
LC-MS Time-Course Experiment.
Samples were prepared as follows: In an LC-MS vial equipped with a 300 μL insert, a solution of 22.5 or 5.63 mM peptide (CBTag 1.0, CBTag 1.0 (K9R), or CBTag 1.0 (C7S); adjusted to pH 7 in PBS), 2 mM TCEP (adjusted to pH 7 in water), and 22.5 or 5.63 mM 13CBTNAc (3) was prepared in 5−6% DMF in PBS (total volume of 10 μL) and the starting time noted. Ratios of 1:1, 1:4, and 4:1 peptide/13CBTNAc were assessed. An LC-MS sequence was prepared such that each sample was injected five times over approximately 45 h, with an injection volume of 1 μL each time. The LC-MS gradient was 5% solvent B for 2 min, 5−75% solvent B over 15 min, 75−100% solvent B over 1 min, 100% solvent B for 6 min, 100−5% solvent B over 1 min, and 5% solvent B for 3 min, where solvent A was 0.1% TFA in water and solvent B was 0.1% TFA in acetonitrile. Identification of peaks was performed by comparison to the spectra of pure compounds, determining whether predicted masses were present and checking whether addition of an equal volume of 200 mM cysteine (adjusted to pH 7 in water) caused the disappearance of the peak over time. The relevant peaks in the 210 and 254 nm chromatographs for two replicates were integrated manually and used to calculate degree of 13CBTNAc modification over time. See Supporting Information Figures S6−S11 and Tables S2−S4 and LC-MS spectra and integration results section for details.
Computational Determination of Lowest Energy Conformer for Amidine and Condensation Reaction Products.
For conformational searching and molecular dynamics calculations, Maestro (version 10.7.015, MMshare version 3.5.015, Release 2016–3, Platform Darwin-x86_64, Schrödinger, LLC, New York, NY, 2016) and the integrated program Macromodel (Schrödinger, LLC, New York, NY, 2016) were used. In general, standard parameters were applied. For calculating potential energy, an OPLS3 force field with water as the solvent was used. Structures were minimized using the Polak-Ribiere conjugate gradient method until a gradient below the convergence threshold of 0.05 kJ/Å-mol was reached, with a maximum of 2500 iterations. For conformational searching, a mixed torsional/low-mode sampling method (multi-ligand) was used, with 1000 steps maximum, 100 steps per rotatable bond, and the energy window for saving structures set to 5.02 kcal/mol. For molecular dynamics simulations, a stochastic dynamics method was used, the simulation temperature was 300.0 K, the time step was 1.5 fs, the equilibration time was 1.0 ps, and the simulation time was 10 ns. Each structure was subjected to five rounds of conformational searching, with molecular dynamics simulated after rounds 2, 3, and 4. MacPyMOL Molecular Graphics System v1.4 (Schrödinger, LLC, New York, NY, 2010) was used to save images of the lowest energy conformer for the amidine product and the condensation reaction product (Supplemental Figure S13).
Synthesis.
N-(2-Cyanobenzo[d]thiazol-6-yl)acetamide (CBTNAc, 1).
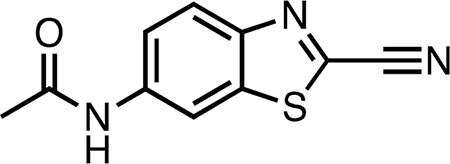
Synthetic procedures were adapted from ref 22; NMR spectra (methanol-d4 and chloroform-d) were reported in refs 22 and 29. 6-Amino-2-cyanobenzothiazole (12 mg, 0.069 mmol) was suspended in anhydrous DCM (0.60 mL). Pyridine (3 drops) and acetyl chloride (0.020 mL, 0.28 mmol, 4.8 equiv) were added, and the reaction was allowed to stir 1 h at room temperature. The resulting clear light yelloworange solution was concentrated in vacuo and then resuspended in ethyl acetate (5.0 mL) and aqueous saturated sodium bicarbonate (5.0 mL). The ethyl acetate layer was separated and concentrated in vacuo to a yellow-white solid. The crude product was purified by silica gel chromatography (1:5 ethyl acetate/hexanes to 100% ethyl acetate). By TLC (1:1 ethyl acetate/hexanes), the product was visible under shortwave UV light as a dark bright blue spot at Rf 0.15. Productcontaining fractions were combined and concentrated in vacuo and then lyophilized to yield 7.87 mg (52.5%) of an off-white powder. LC-MS indicated δ96% purity. 1H NMR (500 MHz, acetone-d6) δ 9.68 (br s, 1H), 8.81 (d, J = 2.0 Hz, 1H), 8.12 (d, J = 9.0 Hz, 1H), 7.72 (dd, J = 9.0, 2.0 Hz, 1H), 2.15 (s, 3H). 13C{1H} NMR (126 MHz, acetone-d6) δ 169.6, 149.0, 141.2, 137.9, 135.6, 125.8, 121.3, 114.1, 111.4, 24.4. HRMS (ESI) m/z: [M + H]+ calcd for C10H8N3OS 218.0383; found 218.0381.
N-(2-Chlorobenzo[d]thiazol-6-yl)acetamide (ClBTNAc, 2).
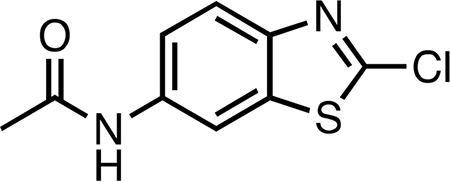
Synthetic procedures were adapted from ref 33. 6-Amino-2chlorobenzothiazole (0.66 g, 3.57 mmol) was dissolved in acetic anhydride (6.5 mL, 68.8 mmol, 19.2 equiv). Pyridine (1.5 mL, 19.3 mmol, 5.2 equiv) was added dropwise, and then the temperature of the light yellow-brown solution was slowly increased to boiling point (approximately 140 °C) in an oil bath. After being heated for 5 min, the dark orange-brown solution was cooled, poured into water (20 mL), and extracted three times with 20 mL aliquots of ethyl acetate. The combined bright yellow ethyl acetate layers were washed twice with 20 mL aliquots of brine, dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo to an orange-brown oil. The crude product was purified by silica gel chromatography, eluting with 2.5 column volumes of 40% ethyl acetate in hexanes then 5 column volumes of 50% ethyl acetate in hexanes. By TLC (1:1 ethyl acetate/hexanes) visualized with shortwave UV, three major spots eluted. By NMR peaks and mass spectrometry, these were identified as starting material (Rf 0.82), product (Rf 0.52), and starting material with the chlorine atom replaced by a hydrogen atom (Rf 0.32). Removal of the solvent in vacuo followed by drying under high vacuum overnight yielded recovered starting material as a pale orange-yellow powder (0.186 g, ∼28.2% unreacted) and product as a pale yellow-tan powder (0.364 g, 45% yield, 76.8% yield based on recovered starting material). LC-MS indicated 96−98% purity. 1H NMR (400 MHz, methanol-d4) δ 8.32 (d, J = 2.2 Hz, 1H), 7.75 (d, J = 8.8 Hz, 1H), 7.45 (dd, J = 8.8, 2.2 Hz, 1H), 2.15 (s, 3H). 13C{1H} NMR (101 MHz, methanol-d4) δ 171.7, 153.3, 148.3, 138.3, 137.9, 123.4, 120.6, 112.9, 23.9. HRMS (ESI) m/z: [M + H]+ calcd for C9H8ClN2OS 227.0040; found 227.0038.
N-(2-(Cyano-13C)-benzo[d]thiazol-6-yl)acetamide (13CBTNAc, 3).
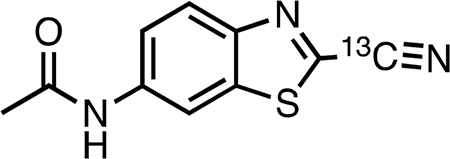
Synthetic procedures were adapted from ref 34; NMR spectra (chloroform-d) of the compound (not enriched for 13C) were reported in refs 22 and 35. [CAUTION: Potassium cyanide is extremely hazardous; compounds dissolved in DMSO are more easily absorbed by skin. Use appropriate safety measures.] Potassium cyanide-13C purchased from Cambridge Isotope Laboratories (48.3 mg, 0.71 mmol, 1.19 equiv) in anhydrous DMSO (12.5 mL) was heated at 130 °C in an oil bath until it was mostly dissolved into a clear solution (15 min). ClBTNAc (2) (139.4 mg, 0.615 mmol) was added, and the resulting dark orange reaction mixure was stirred for 2 h at 130 °C, becoming bright red over time. The solution was allowed to stand at room temperature overnight, turning a deep red-orange on cooling. The next day, the reaction mixture was poured into a separatory funnel, and 75 mL of 0.2 M monopotassium phosphate (KH2PO4, pH 4−5) was added. This aqueous solution was extracted once with 125 mL of diethyl ether and twice with 50 mL aliquots of diethyl ether. The combined neon yellow ether extracts were washed twice with 25 mL aliquots of water and then dried over anhydrous sodium sulfate. The solution was filtered, silica gel (1.1 g) was added, and the solvent was removed in vacuo. The dry residue was added to a column of silica gel (100 mL) prepacked with petroleum ether, and the product was eluted using 0−50% ethyl acetate in petroleum ether followed by 50−100% ethyl acetate in hexanes. Pure fractions were identified by TLC (1:1 ethyl acetate/hexanes), where the product was visible as a dark blue spot (Rf 0.26) under short- and long-wave UV light. The impurity (Rf 0.31) eluted just before the product. Concentration of the product-containing fractions in vacuo followed by drying under high vacuum overnight yielded 74 mg (55.1%) of product as a white to pale yellow solid. Product was ˷97% pure by LC-MS. 1H NMR (500 MHz, acetone-d6) δ 9.68 (br s, 1H), 8.81 (d, J = 2.1 Hz, 1H), 8.12 (d, J = 9.0 Hz, 1H), 7.72 (dd, J = 8.9, 2.1 Hz, 1H), 2.16 (s, 3H). 13C{1H} NMR (126 MHz, acetone-d6) δ 169.6, 149.0, 141.1, 137.9, 135.1, 125.8, 121.3, 114.1, 111.4, 24.4. HRMS (ESI) m/z: [M + H]+ calcd for C913CH8N3OS 219.0416; found 219.0414.
General Peptide Synthesis Conditions.
Peptides were made by solid-phase peptide synthesis either manually in a peptide synthesis vessel or using an AAPPTec Apex 396 automated peptide synthesizer equipped with a 96-well reactor. Solvents were HPLC grade or peptide synthesis grade; neither solvents nor reagents were anhydrous. Rink amide AM resin (100−200 mesh, 0.41 mmol/g loading) from EMD Millipore (855130) was used as the solid phase unless otherwise noted. All coupling, deprotection, and cleavage steps were performed at ambient temperature.
General Procedure for Manual Synthesis.
A 50 mL peptide synthesis vessel (VWR 80071–382) was equipped with a house nitrogen and a vacuum line. Rink amide AM resin (0.05 g, 0.02 mmol) was added and allowed to swell for 10 min in either dichloromethane (DCM) or N,N-dimethylformamide (DMF) without nitrogen agitation. The resin was drained and dried with several cycles of vacuum and nitrogen, and then the resin Fmoc protecting group was cleaved with 20% piperidine in DMF or N-methyl-2-pyrrolidone (NMP) (5−10 mL) for 15−20 min with nitrogen agitation, rinsing the sides every 5−10 min to ensure all beads were deprotected. Beads were drained and dried with several rounds of vacuum and nitrogen, then a Kaiser test36 was performed on a small amount of resin to ascertain degree of completion. If deprotection was complete, the first amino acid was coupled with monomer (0.2 mmol, 10 equiv), coupling reagent (0.2 mmol, 10 equiv of HATU for Fmoc-His(Trt) coupling, COMU for all others), and N,N-diisopropylethylamine (DIPEA; 15 equiv for Fmoc-Cys(Trt) coupling, 20 equiv for all others) in NMP (1.5 mL) or 1:1 DCM/NMP (1.5 mL; Fmoc-Cys(Trt) coupling only) with nitrogen agitation. Directly before coupling, the DIPEA was added to the monomer/coupling reagent solution and the vial gently inverted for 30−60 s before addition to resin. Again, sides of the vessel were rinsed with minimal DCM or DMF 5−10 min into the reaction. After 15−30 min, the resin was drained and washed thoroughly with DCM, then the first coupling reaction was repeated. The Kaiser test was used to confirm completion, then any remaining free amines on the resin were capped with a solution of 10% acetic anhydride and 5% DIPEA in DMF or NMP, agitating with nitrogen for 15−20 min. After the resin was drained and washed, the Fmoc group on the first amino acid was removed with 20% piperidine in DMF or NMP as noted previously. Subsequent couplings were performed only once unless the amino acid was β-branched or the Kaiser test did not show completion. After the coupling and deprotection of the final amino acid, the N-terminus was acetylated using a solution of 10% acetic anhydride and 5% DIPEA in DMF or NMP, agitating with nitrogen for 15−20 min. In preparation for deprotection and cleavage, the resin was thoroughly washed with DCM and left under vacuum until dry (15−30 min). A clean round-bottom flask was attached to the synthesis vessel, and the cleavage cocktail Reagent L (for 0.02 mmol resin: 0.96 g of dithiothreitol, 0.04 mL of triisopropylsilane, 0.1 mL of ddH2O, and trifluoroacetic acid (TFA) to a final volume of 2 mL) was added to cleave and deprotect the peptide. The resin was allowed to sit 1.5 h without nitrogen agitation; the mixture was manually stirred every 30 min with a glass stir rod. After draining the cleavage solution into the clean round-bottom flask, the solvent was removed in vacuo, and the resulting viscous cloudy pale yellow to pale white solution was pipetted into ice-cold ether (45 mL). The flask was rinsed with minimal TFA, and the resulting solution was also added to the ether. The suspension was centrifuged in a Sorvall Legend RT at 3700g and 4 °C for 8−10 min. The ether was decanted carefully, and the remaining pale yellow to off-white precipitate was washed twice more with ice-cold ether (45 mL). After the final centrifugation, the ether was carefully decanted and the precipitate allowed to dry 15−30 min in a hood to remove any remaining ether. Finally, the precipitate was resuspended in minimal ddH2O and lyophilized in a preweighed vial using a LABCONCO FreeZone 4.5Plus. Crude peptide was stored at −20 °C until purification.
General Procedure for Automated Peptide Synthesis.
Rink amide AM resin (0.05 g, 0.02 mmol) was added to a well in the AAPPTec Apex 396 automated peptide synthesizer. The instrument was programmed to complete the following steps: NMP (1 mL) was added, and the resin was mixed for 3 min at 600 rpm. After the solvent was drained, this step was repeated. To deprotect the first Fmoc group, 20% piperidine in DMF (1 mL) was added and the resin mixed for 3 min at 600 rpm. After draining, another aliquot of 20% piperidine in DMF (1 mL) was added, and deprotection was allowed to proceed for 20 min at 600 rpm. After the deprotection solution was drained, the resin was washed three times with NMP (1 mL), MeOH (1 mL), and NMP (1 mL) at 600 rpm for 3 min each. Then, 0.4 M protected monomer in NMP (0.6 mL), 0.4 M HATU or HCTU in NMP (0.5 mL), and 2 M DIPEA in NMP (0.25 mL) were transferred to the well and mixed for 30 min at 600 rpm. After the solution was drained, the coupling step was repeated, and the resin was washed twice with NMP (1 mL) for 3 min at 600 rpm. The deprotection and coupling steps were repeated until the final amino acid was deprotected, and then the N-terminus of the peptide was acetylated by twice adding 11.63% acetic anhydride in DMF (1.3 mL) and 2 M DIPEA in DMF (0.25 mL) and mixing for 30 min at 600 rpm. Cleavage and precipitation of crude peptide from ether were performed as described above for manual peptide synthesis.
RP-HPLC Purification of Peptides.
Purification was performed by semipreparative (3 mL/min flow rate) or preparative RP-HPLC (20 mL/min flow rate). The general gradient was as follows: 0% solvent B for 2 min, 0−33% solvent B over 18 min, 33−100% solvent B over 2 min, 100% solvent B for 3 min. Pure fractions were combined and concentrated in vacuo, taken up in water, and lyophilized.
CBTag 1.0, Ac-GGHPDPCPKGG-NH2 (4):
Automated synthesis followed by preparative RP-HPLC purification yielded 1.351−4.837 mg (6.2−17%) of a white fluffy solid. Manual synthesis followed by preparative RP-HPLC purification yielded 15.171 mg (68.3%) of a white fluffy solid. Product was 95% pure by LC-MS. 1H NMR (800 MHz, D2O) δ 8.60−8.25 (m, 8H), 7.52 (s, 1H), 7.29 (s, 1H), 7.09 (s, 1H), 4.41 (s, 2H), 4.30−4.25 (m, 1H), 4.01−3.66 (m, 15H), 3.56 (q, J = 6.9 Hz, 2H), 3.25−2.71 (m, 9H), 2.56 (dd, J = 16.1, 7.7 Hz, 1H), 2.26 (tt, J = 12.6, 6.2 Hz, 3H), 2.06−1.72 (m, 17H), 1.67 (s, 3H), 1.53−1.39 (m, 3H). 13C{1H} NMR (126 MHz, D2O) δ 175.2, 174.9, 174.5, 174.3, 174.1, 173.9, 172.5, 172.0, 171.1, 170.5, 170.1, 169.7, 163.3, 163.0, 162.7, 133.7, 128.4, 117.8, 117.5, 115.2, 60.7, 60.6, 54.1, 53.8, 50.7, 48.5, 48.2, 48.0, 42.8, 42.4, 42.3, 39.6, 39.5, 35.4, 30.3, 29.6, 29.5, 26.5, 26.1, 24.9, 24.8, 24.6, 22.2, 21.9. HRMS (ESI) m/z: [M + 2H]2+ calcd for C44H69N15O14S 531.7429; found 531.7436.
PEP-FOLD-4, Ac-GGDPHPCKGG-NH2 (5):
Automated synthesis followed by semipreparative RP-HPLC purification yielded 1.977 mg (7.5%) of a white fluffy solid. Product was 95% pure by LC-MS. HRMS (ESI) m/z: [M + 2H]2+ calcd for C39H62N14O13S2 483.2165; found 483.2170.
PEP-FOLD-1, Ac-GGDPCPHPKGG-NH2 (6):
Automated synthesis (0.06 mmol scale) followed by preparative RP-HPLC purification yielded 0.919 mg (1.4%) of a white fluffy solid. Product was 95% pure by LC-MS. HRMS (ESI) m/z: [M + 2H]2+ calcd for C44H69N15O14S 531.7429; found 531.7428.
PEP-FOLD-2, Ac-GGHPDCPKGG-NH2 (7):
Automated synthesis (0.06 mmol scale) combined with manual synthesis (0.02 mmol scale) followed by preparative RP-HPLC purification yielded 1.503 mg (1.9%) of a white fluffy solid. Product was 93.5% pure by LC-MS. As the ratio of labeled to unlabeled peptide after cysteine treatment was not competitive with CBTag 1.0 (Figure S1B), no further purification was performed. HRMS (ESI) m/z: [M + 2H]2+ calcd for C39H62N14O13S 483.2165; found 483.2162.
PEP-FOLD-5, Ac-GGHPEPCPKGG-NH2 (8):
Automated synthesis (0.06 mmol scale) followed by preparative RP-HPLC purification yielded 1.774 mg (2.7%) of a white fluffy solid. Product was 95% pure by LC-MS. HRMS (ESI) m/z: [M + 2H]2+ calcd for C44H69N15O14S 538.7507; found 538.7510.
PEP-FOLD-6, Ac-GGHPECPKGG-NH2 (9):
Automated synthesis (0.06 mmol scale) followed by preparative RP-HPLC purification yielded 33.860 mg (57.6%) of a white fluffy solid. Product was 92% pure by LC-MS. As the ratio of labeled to unlabeled peptide after cysteine treatment was not competitive with CBTag 1.0 (Figure S1F), no further purification was performed. HRMS (ESI) m/z: [M + 2H]2+ calcd for C40H64N14O12S 490.2244; found 490.2246.
PEP-FOLD-7, Ac-GGEPHPCPKGG-NH2 (10):
Automated synthesis (0.06 mmol scale) followed by preparative RP-HPLC purification yielded 2.959 mg (4.6%) of a white fluffy solid. Product was 90.9% pure by LC-MS. As the ratio of labeled to unlabeled peptide after cysteine treatment was not competitive with CBTag 1.0 (Figure S1G), no further purification was performed. HRMS (ESI) m/z: [M + 2H]2+ calcd for C45H71N15O14S 538.7507; found 538.7509.
PEP-FOLD-8, Ac-GGCPKPHPHGG-NH2 (11):
Manual synthesis followed by preparative RP-HPLC purification yielded 0.896 mg (4.0%) of a white fluffy solid. Product was 94.3% pure by LC-MS. As the ratio of labeled to unlabeled peptide after cysteine treatment was not competitive with CBTag 1.0 (Figure S1H), no further purification was performed. HRMS (ESI) m/z: [M + 2H]2+ calcd for C46H71N17O12S 542.7589; found 542.7595.
PEP-FOLD-9, Ac-GGCPKPDPHGG-NH2 (12):
Manual synthesis followed by preparative RP-HPLC purification yielded 1.537 mg (6.9%) of a white fluffy solid. Product was 95% pure by LC-MS. HRMS (ESI) m/z: [M + 2H]2+ calcd for C44H69N15O14S 531.7429; found 531.7434.
PEP-FOLD-11, Ac-GGCPDPHPKGG-NH2 (13):
Manual synthesis followed by preparative RP-HPLC purification yielded 5.329 mg (24.3%) of a white fluffy solid. Product was 83% pure by LC-MS; major impurity was due to deletion of two glycine residues, which was inseparable from the desired product and accounted for 11.2% of the final compound mixture. As missing glycines should not significantly affect reactivity, this compound was used under the assumption that it was effectively 94.2% “pure” though the MALDI-TOF spectra would have to be interpreted carefully. As the ratio of labeled to unlabeled peptide after cysteine treatment was not competitive with CBTag 1.0 (Figure S1J), no further purification was attempted. HRMS (ESI) m/z: [M + 2H]2+ calcd for C44H69N15O14S 531.7429; found 531.7435.
PEP-FOLD-12, Ac-GGKPCPHPDGG-NH2 (14):
Manual synthesis followed by preparative RP-HPLC purification yielded 5.153 mg (23.7%) of a white fluffy solid. Product was 96.9% pure by LC-MS. HRMS (ESI) m/z: [M + 2H]2+ calcd for C44H69N15O14S 531.7429; found 531.7432.
PEP-FOLD-13, Ac-GGWPHPCPKGG-NH2 (15):
Manual synthesis followed by preparative RP-HPLC purification yielded 5.141 mg (21.9%) of a white fluffy solid. Product was 95% pure by LC-MS. HRMS (ESI) m/z: [M + 2H]2+ calcd for C51H74N16O12S 567.2691; found 567.2696.
PEP-FOLD-14, Ac-GGWPHPGCPKGG-NH2 (16):
Manual synthesis followed by preparative RP-HPLC purification yielded 9.628 mg (39.1%) of a white fluffy solid. Product was 95% pure by LC-MS. HRMS (ESI) m/z: [M + 2H]2+ calcd for C52H77N17O12S 595.7798; found 595.7802.
PEP-FOLD-15, Ac-GGKPCPGWPHGG-NH2 (17):
Automated synthesis followed by preparative RP-HPLC purification yielded 4.893 mg (20.1%) of a white fluffy solid. Product was 95% pure by LC-MS. HRMS (ESI) m/z: [M + 2H]2+ calcd for C52H77N17O12S 595.7798; found 595.7805.
PEP-FOLD-16, Ac-GGHPHPKPCPEGG-NH2 (18):
Automated synthesis followed by preparative RP-HPLC purification yielded 20.41 mg (76.0%) of a white fluffy solid. Product was 91% pure by LC-MS. As the ratio of labeled to unlabeled peptide after cysteine treatment was not competitive with CBTag 1.0 (Figure S1O), no further purification was performed. HRMS (ESI) m/z: [M + 2H]2+ calcd for C56H85N19O16S 655.8066; found 655.8071.
PEP-FOLD-17, Ac-GGHPWPKPCPEGG-NH2 (19):
Automated synthesis followed by preparative RP-HPLC purification yielded 7.203 mg (25.8%) of a white fluffy solid. Product was 95.6% pure by LC-MS. HRMS (ESI) m/z: [M + 2H]2+ calcd for C61H88N18O16S 680.3168; found 680.3176.
PEP-FOLD-18, Ac-GGHPKPGCGFGG-NH2 (20):
Automated synthesis followed by preparative RP-HPLC purification yielded 0.955 mg (4.2%) of a white fluffy solid. Product was 92.5% pure by LC-MS. As the ratio of labeled to unlabeled peptide after cysteine treatment was not competitive with CBTag 1.0 (Figure S1Q), no further purification was performed. HRMS (ESI) m/z: [M + 2H]2+ calcd for C48H72N16O12S 556.2587; found 556.2594.
PEP-FOLD-19, Ac-GGHPKPCGFGG-NH2 (21):
Automated synthesis followed by preparative RP-HPLC purification yielded 1.109 mg (5.1%) of a white fluffy solid. Product was 90.8% pure by LC-MS; major impurity was due to deletion of one glycine residue, which was inseparable from the desired product and accounted for 5.3% of the final compound mixture. As missing glycines should not significantly affect reactivity, this compound was used under the assumption that it was effectively 96.1% “pure” though the MALDI-TOF spectra would have to be interpreted carefully. As the ratio of labeled to unlabeled peptide after cysteine treatment was not competitive with CBTag 1.0 (Figure S1R), no further purification was attempted. HRMS (ESI) m/z: [M + 2H]2+ calcd for C46H69N15O12S 527.7480; found 527.7488.
CBTag 1.0 (K9R), Ac-GGHPDPCPRGG-NH2 (22):
Manual synthesis followed by preparative RP-HPLC purification yielded 13.894 mg of a white fluffy solid containing two peaks by LC-MS (987 and 1090 m/z). These peaks were assumed to correspond to the [M + H]+ for a cysteine deletion and the product, respectively, based on the masses, the successful reactions of chloroacetate and CBTNAc with the product peptide, and the lack of an observed reaction between the impurity and CBTNAc or chloroacetate. By integration of the LC-MS trace, approximately 45.8% (6.365 mg, 27.9% yield) was desired product. A portion of this sample (8.186 mg) was repurified using the following preparative RP-HPLC method: 10% solvent B for 10 min. Combining the >95% pure fractions yielded 1.34 mg (16.4% of 8.186 mg injected mixture; 10% overall yield assuming 16.4%, or 2.274 mg, of the 13.184 mg obtained after the first purification would yield pure product) of a white solid after lyophilization. Product was 97% pure by LC-MS. HRMS (ESI) m/z: [M + 2H]2+ calcd for C44H69N17O14S 545.7460; found 545.7454.
CBTag 1.0 (C7S), Ac-GGHPDPSPKGG-NH2 (23):
Manual synthesis followed by preparative RP-HPLC purification yielded 12.777 mg (58.4%) of a white fluffy solid. Product was 95% pure by LC-MS. HRMS (ESI) m/z: [M + 2H]2+ calcd for C44H69N15O15 523.7543; found 523.7537.
CBTag 1.0−13CBTNAc (amidine) (24):
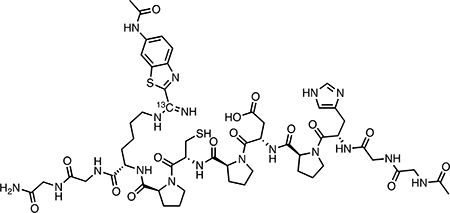
Two methods were used to obtain product.
Method 1.
In a 2 mL Eppendorf tube, 10 mM CBTag 1.0 (10.383 mg) and 5 mM 13CBTNAc (1.067 mg) in approximately 986 μL of 5% DMF in PBS were adjusted to approximately pH 7 and allowed to rotate on a Barnstead/Thermolyne Labquake Rotisserie Shaker (model #C415110) for 40 h at room temperature. At that time, an equal volume of cysteine (10 mM) and DTT (10 mM) adjusted to approximately pH 7 was added to reach final concentrations of 5 mM CBTag 1.0, 2.5 mM 13CBTNAc, 5 mM cysteine, and 5 mM DTT in 1.972 mL of 2.5% DMF in PBS. The tube was allowed to rotate a further 48 h at room temperature. The reaction mixture was then injected directly onto a preparative HPLC column and purified using the following method (20 mL/min flow rate): 0% solvent B for 2 min, 0−33% solvent B over 18 min, 33−100% solvent B over 2 min, 100% solvent B for 3 min. Pure fractions were combined and concentrated in vacuo, taken up in water, and lyophilized to yield 0.993 mg of a white powder. Assuming 13CBTNAc was the limiting reagent and each peptide would only bind 1 equiv of 13CBTNAc, the maximum theoretical yield would be 6.260 mg. Based on this, the percent yield was roughly 15.9%.
Method 2.
In a 2 mL Eppendorf tube, 5 mM CBTag 1.0 (5.220 mg) and 5 mM 13CBTNAc (1.073 mg) in 1 mL of 5% DMF in PBS were adjusted to approximately pH 7 and allowed to rotate on a Barnstead/ Thermolyne Labquake Rotisserie Shaker (model #C415110) for 48 h at room temperature. At that time, an equal volume of cysteine (10 mM) and DTT (200 mM) adjusted to approximately pH 7 was added to reach final concentrations of 2.5 mM CBTag 1.0, 2.5 mM 13CBTNAc, 5 mM cysteine, and 100 mM DTT in 2 mL of 2.5% DMF in PBS. The tube was allowed to rotate a further 48 h at room temperature. The reaction mixture was then injected directly onto a preparative HPLC column and purified using the following method (20 mL/min flow rate): 0% solvent B for 2 min, 0−33% solvent B over 18 min, 33−100% solvent B over 2 min, 100% solvent B for 3 min. Pure fractions were combined and concentrated in vacuo, taken up in water, and lyophilized to yield 1.549 mg of a white powder. Assuming each peptide would only bind 1 equiv of 13CBTNAc, the maximum theoretical yield would be 6.293 mg. Based on this, the percent yield was roughly 35.8%. Final product was >95% pure by LC-MS. 1H NMR (800 MHz, D2O) δ 10.12 (br s, 1H), 8.57−8.16 (m, 7H), 7.61 (d, J = 8.8 Hz, 1H), 7.48 (s, 1H), 7.28 (s, 1H), 7.05 (s, 1H), 4.50−4.26 (m, 8H), 4.00−3.51 (m, 17H), 3.22 (dd, J = 15.5, 5.3 Hz, 1H), 3.06 (ddd, J = 51.4, 14.4, 7.5 Hz, 2H), 2.88−2.64 (m, 8H), 2.51 (dd, J = 16.1, 8.0 Hz, 1H), 2.21 (q, J = 9.3, 8.3 Hz, 4H), 2.03 (s, 2H), 2.01− 1.74 (m, 12H), 1.64−1.46 (m, 3H), 1.24 (d, J = 6.6 Hz, 3H). 13C{1H} NMR (126 MHz, D2O) δ 156.0 [run at low concentration to identify 13C-enriched atoms]. HRMS (ESI) m/z: [2M + 2H]2+calcd for C106 (13C)2 H148N36O30S4 1279.5051; found 1279.5043.
Supplementary Material
ACKNOWLEDGMENTS
The authors would especially like to thank Dr. Frances Rodriguez-Rivera, Douglas Fox, Dr. Mason Appel, Dr. Gabrielá de Almeida, Julia McKechnie, Dr. David Spiciarich, Ioannis Mountziaris, and Mireille Kamariza for technical assistance, brainstorming sessions, and, in the case of D.S., I.M., and M.K., helpful comments and suggestions during the writing of this manuscript. We thank Dr. Corey Liu and Dr. Stephen Lynch of the Stanford University Department of Chemistry NMR Facility for useful advice on peptide NMR and for obtaining NMR spectra on the 800 MHz spectrometer. We would also like to thank Dr. Theresa McLaughlin at the Vincent Coates Foundation Mass Spectrometry Laboratory, Stanford University Mass Spectrometry (http://mass-spec.stanford.edu) for acquiring the tandem MS/MS and HRMS data. This work was funded by a grant to C.R.B. from the National Institutes of Health, National Institute of General Medical Sciences (R37 GM058867). S.G.L.K. was supported in part by a National Science Foundation Graduate Research Fellowship (Grant No. DGE 1106400).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.8b00625.
Tables S1−S4 (list of peptide sequences modeled using PEP-FOLD; calculations for LC-MS time-course data), Figures S1−S13 (MALDI-TOF MS spectra for 18 peptides synthesized by SPPS, assay run in the presence of 5 mM glutathione, CBTag 1.0 (K9R), and CBTag 1.0 (C7S), and chloroacetate experiments; analysis of 13C NMR for CBTag 1.0−13CBTNAc product; LC-MS spectra clarifying peak identities prior to time-course assay; MALDITOF MS spectra for 1:1 CBTag 1.0 (K9R)/CBTag 1.0 (C7S) binding assay and cysteine competition; Maestro conformational search results for amidine and condensation reaction products), LC-MS time-course assay spectra and integration results, LC-MS spectra (compound purity), NMR spectra (PDF)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Jing C; Cornish VW Chemical Tags for Labeling Proteins Inside Living Cells. Acc. Chem. Res. 2011, 44, 784–792 and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Spicer CD; Davis BG Selective chemical protein modification. Nat. Commun. 2014, 5, 4740 and references cited therein. [DOI] [PubMed] [Google Scholar]
- (3).Cal PMSD; Bernardes GJL; Gois PMP CysteineSelective Reactions for Antibody Conjugation. Angew. Chem., Int. Ed 2014, 53, 10585–10587 and references cited therein. [DOI] [PubMed] [Google Scholar]
- (4).Stephanopoulos N; Francis MB Choosing an effective protein bioconjugation strategy. Nat. Chem. Biol 2011, 7, 876–884 and references cited therein. [DOI] [PubMed] [Google Scholar]
- (5).Lotze J; Reinhardt U; Seitz O; Beck-Sickinger AG Peptidetags for site-specific protein labelling in vitro and in vivo. Mol. BioSyst. 2016, 12, 1731–1745 and references cited therein. [DOI] [PubMed] [Google Scholar]
- (6).Griffin BA; Adams SR; Tsien RY Specific Covalent Labeling of Recombinant Protein Molecules Inside Live Cells. Science 1998, 281, 269–272. [DOI] [PubMed] [Google Scholar]
- (7).Adams SR; Campbell RE; Gross LA; Martin BR; Walkup GK; Yao Y; Llopis J; Tsien RY New Biarsenical Ligands and Tetracysteine Motifs for Protein Labeling in Vitro and in Vivo: Synthesis and Biological Applications. J. Am. Chem. Soc. 2002, 124, 6063–6076. [DOI] [PubMed] [Google Scholar]
- (8).Zhang C; Welborn M; Zhu T; Yang NJ; Santos MS; Van Voorhis T; Pentelute BL π-Clamp-mediated cysteine conjugation. Nat. Chem. 2016, 8, 120–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Zhang C; Spokoyny AM; Zou Y; Simon MD; Pentelute BL Enzymatic ‘click’ ligation: selective cysteine modification in polypeptides enabled bypromiscuous glutathione S-transferase. Angew. Chem., Int. Ed. 2013, 52, 14001–14005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Hacker SM; Backus KM; Lazear MR; Forli S; Correia BE; Cravatt BF Global profiling of lysine reactivity and ligandability in the human proteome. Nat. Chem. 2017, 9, 1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Matos MJ; Oliveira BL; Martínez-Sáez N; Guerreiro A; Cal PMSD; Bertoldo J; Maneiro M; Perkins E; Howard J; Deery MJ; Chalker JM; Corzana F; Jimenez-Osé s G; Bernardes GJL Chemo- and Regioselective Lysine Modification on Native Proteins. J. Am. Chem. Soc. 2018, 140, 4004–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Dawson PE; Muir TW; Clark-Lewis I; Kent SB Synthesis of proteins by native chemical ligation. Science 1994, 266, 776–778. [DOI] [PubMed] [Google Scholar]
- (13).Burke HM; McSweeney L; Scanlan EM Exploring chemoselective S-to-N acyl transfer reactions in synthesis and chemical biology. Nat. Commun. 2017, 8, 15655 and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Steffen W; Ko FC; Patel J; Lyamichev V; Albert TJ; Benz J; Rudolph MG; Bergmann F; Streidl T; Kratzsch P; Boenitz-Dulat M; Oelschlaegel T; Schraeml M Discovery of a microbial transglutaminase enabling highly site-specific labeling of proteins. J. Biol. Chem. 2017, 292, 15622–15635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).White EH; McCapra F; Field GF Structure and Synthesis of Firefly Luciferin. J. Am. Chem. Soc. 1963, 85, 337–343. [Google Scholar]
- (16).Gomi K; Kajiyama N Oxyluciferin, a Luminescence Product of Firefly Luciferase, Is Enzymatically Regenerated into Luciferin. J. Biol. Chem. 2001, 276, 36508–36513. [DOI] [PubMed] [Google Scholar]
- (17).Ren H; Xiao F; Zhan K; Kim Y-P; Xie H; Xia Z; Rao J Biocompatible Condensation Reaction for the Labeling of Terminal Cysteine Residues on Proteins. Angew. Chem., Int. Ed. 2009, 48, 9658–9662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Nguyen DP; Elliott T; Holt M; Muir TM; Chin JW Genetically Encoded 1,2-Aminothiols Facilitate Rapid and SiteSelective Protein Labeling via a Bio-orthogonal Cyanobenzothiazole Condensation. J. Am. Chem. Soc. 2011, 133, 11418–11421. [DOI] [PubMed] [Google Scholar]
- (19).Van de Bittner GC; Bertozzi CR; Chang CJ Strategy for Dual-Analyte Luciferin Imaging: In Vivo Bioluminescence Detection of Hydrogen Peroxide and Caspase Activity in a Murine Model of Acute Inflammation. J. Am. Chem. Soc. 2013, 135, 1783–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Jeon J; Shen B; Xiong L; Miao Z; Lee KH; Rao J; Chin F Efficient Method for Site-Specific 18F-Labeling of Biomolecules Using the Rapid Condensation Reaction between 2-Cyanobenzothiazole and Cysteine. Bioconjugate Chem. 2012, 23, 1902–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Ramil CP; An P; Yu Z; Lin Q Sequence-specific 2cyanobenzothiazole ligation. J. Am. Chem. Soc. 2016, 138, 5499–5502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).de Almeida G Improving Tools for Bioorthogonal Chemistry. Ph.D. Dissertation, University of California, Berkeley, Berkeley, CA, 2014. http://digitalassets.lib.berkeley.edu/etd/ucb/text/deAlmeida_berkeley_0028E_14273.pdf (accessed May 29, 2017). [Google Scholar]
- (23).Sidhu SS; Weiss GA; Wells JA High Copy Display of Large Proteins on Phage for Functional Selections. J. Mol. Biol. 2000, 296, 487–495. [DOI] [PubMed] [Google Scholar]
- (24).Lamboy JA; Arter JA; Knopp KA; Der D; Overstreet CM; Palermo E; Urakami H; Yu T-B; Tezgel O; Tew G; Guan Z; Kuroda K; Weiss GA Phage Wrapping with Cationic Polymers Eliminates Non-specific Binding between M13 Phage and High pI Target Proteins. J. Am. Chem. Soc. 2009, 131, 16454–16460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Eldridge GM; Weiss GA Hydrazide Reaction Peptide Tags for Site-Specific Protein Labeling. Bioconjugate Chem. 2011, 22, 2143–2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Thevenet P; Shen Y; Maupetit J; Guyon F; Derreumaux P;´ Tuffery, P. PEP-FOLD: an updated de novo structure predictioń server for both linear and disulfide bonded cyclic peptides. Nucleic Acids Res. 2012, 40, W288–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Shen Y; Maupetit J; Derreumaux P; Tuffery P Improved́ PEP-FOLD approach for peptide and miniprotein structure prediction. J. Chem. Theory Comput. 2014, 10, 4745–4758. [DOI] [PubMed] [Google Scholar]
- (28).Sluyterman LAÆ; Wijdenes J Benzoylamidoacetonitrile as an inhibitor of papain. Biochim. Biophys. Acta 1973, 302, 95–101. [DOI] [PubMed] [Google Scholar]
- (29).Brisson J-R; Carey PR; Storer AC Benzoylamidoacetonitrile Is Bound as a Thioimidate in the Active Site of Papain. J. Biol. Chem. 1986, 261, 9087–9089. [PubMed] [Google Scholar]
- (30).Kawakami T; Ogawa K; Goshima N; Natsume T DIVERSE System: De Novo Creation of Peptide Tags for Non-enzymatic Covalent Labeling by In Vitro Evolution for Protein Imaging Inside Living Cells. Chem. Biol. 2015, 22, 1671–1679. [DOI] [PubMed] [Google Scholar]
- (31).Strohalm M; Hassman M; Kosata B; Kodíč ek M mMasš Data Miner: an Open Source Alternative for Mass Spectrometric Data Analysis. Rapid Commun. Mass Spectrom. 2008, 22, 905–908. [DOI] [PubMed] [Google Scholar]
- (32).Strohalm M; Kavan D; Novak P; Volný M; Havlíć ek V̌ mMass 3: A Cross-Platform Software Environment for Precise Analysis of Mass Spectrometric Data. Anal. Chem. 2010, 82, 4648–4651. [DOI] [PubMed] [Google Scholar]
- (33).Niedermeyer THJ; Strohalm M mMass as a Software Tool for the Annotation of Cyclic Peptide Tandem Mass Spectra. PLoS One 2012, 7, e44913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).White EH; Wörther H; Seliger HH; McElroy WD Amino Analogs of Firefly Luciferin and Biological Activity Thereof. J. Am. Chem. Soc. 1966, 88, 2015–2019. [Google Scholar]
- (35).Sharma DK; Adams ST Jr.; Liebmann KL; Miller SC Rapid Access to a Broad Range of 6’-Substituted Firefly Luciferin Analogues Reveals Surprising Emitters and Inhibitors. Org. Lett. 2017, 19, 5836–5839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Kaiser E; Colescott RL; Bossinger CD; Cook PI Color test for detection of free terminal amino groups in the solid-phase synthesis of peptides. Anal. Biochem. 1970, 34, 595–598. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.