

Abstract
Host factors that silence provirus transcription in CD4+ memory T cells help HIV-1 escape eradication by the host immune system and by antiviral drugs1. These same factors, though, must be overcome for HIV-1 to propagate. Here we show that Vpx and Vpr encoded by diverse primate immunodeficiency viruses activate provirus transcription. Vpx and Vpr are adaptor proteins for the DCAF1-CUL4A/B E3 ubiquitin ligase that degrade SAMHD1 and increase reverse transcription2–4. Nonetheless, Vpx and Vpr have effects on reporter gene expression that are not explained by SAMHD1 degradation5–8. A screen for factors that mimic these effects identified the Human Silencing Hub (HUSH) complex, FAM208A (TASOR/RAP140), MPHOSPH8 (MPP8), PPHLN1 (PERIPHILIN), and MORC29–13. Vpx associated with the HUSH complex and decreased steady-state level of these proteins in a DCAF1/CUL4A/B/proteasome-dependent manner14,15. Replication kinetics of HIV-1 and SIVMAC was accelerated to a similar extent by vpx or FAM208A knockdown. Finally, vpx increased steady-state levels of LINE-1 ORF1p, as previously described for FAM208A disruption11. These results demonstrate that the HUSH complex represses primate immunodeficiency virus transcription, and that, to counteract this restriction, viral Vpx or Vpr proteins degrade the HUSH complex.
To examine the effect of vpx on provirus transcription, Jurkat CD4+ T cells were transduced with a lentiviral vector that expresses SIVMAC251 vpx from the spleen focus forming virus (SFFV) promoter, and puromycin acetyltransferase (puroR) from the PPIA (CypA) promoter6,16 (Lenti 1 in Fig. 1a, Supplementary Fig. 1a, and Supplementary Table 1). Puromycin was added on day three to select those cells that had been transduced. On day seven, cells were transduced with a second lentivector bearing a codon-optimized gag-gfp reporter gene expressed from the SFFV promoter, as well as SIVMAC251 vpx expressed from the CypA promoter (Lenti 2 in the Fig. 1a timeline and Supplementary Fig.1a). On day ten, virus-like particles (VLPs) containing Vpx protein were added to the twice-transduced cells (Fig. 1a). On day fourteen, the percent GFP+ cells was assessed by flow cytometry (Supplementary Figure 1b). Vpx increased the percentage of GFP+ cells, whether vpx was transduced before, or concurrent with, reporter gene transduction, or if Vpx protein was delivered in VLPs, after reporter gene transduction (Fig. 1b and Supplementary Figure 1c).
Figure 1. Diverse primate immunodeficiency virus vpx and vpr orthologues activate provirus transcription, whether delivered before, during, or after reporter provirus integration.
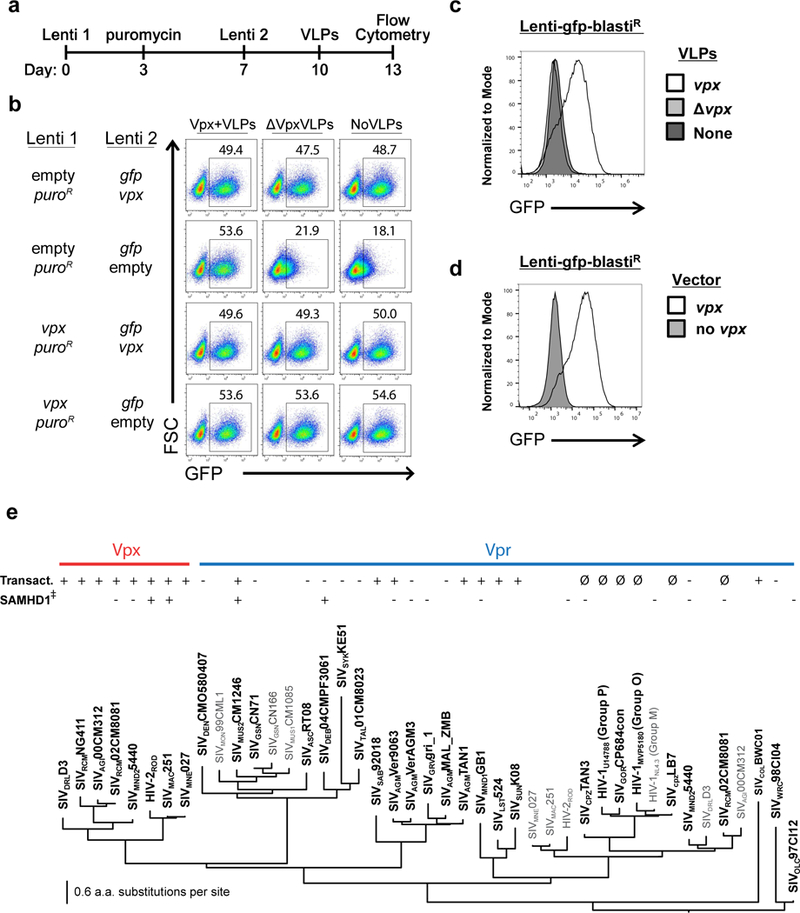
a, Schematic of experimental protocol in (b). b, Flow cytometry plot showing percent GFP+ Jurkat cells after sequential transduction with the indicated lentivectors, followed by exposure to the indicated VLPs. c,d, Histogram of flow cytometry signal in Jurkat cells transduced with gfp-reporter virus, and either exposed to the indicated VLPs (c), or transduced with the indicated vectors (d). e, Maximum likelihood tree showing evolutionary relationship of Vpx and Vpr proteins, generated using RAxML with node support assessed by performing 100 bootstrap replicates. The transactivation activity of Jurkat reporter lines, tested as in (d), and human SAMHD1 degradation activity 4, are indicated. ∅ indicates Vprs that were too toxic (G2 arrest) for assessment. All data shown is representative of at least three biological replicates.
Jurkat T cells were then transduced with a vector in which gag-gfp was expressed from the SFFV promoter, and blasticidin-S deaminase (blastiR) was expressed from the CypA promoter. Four days after blasticidin selection, cells were challenged with Vpx+ VLPs, or transduced and selected with the lentivector encoding vpx and puroR (Lenti 1 in Supplementary Fig. 1a). Four days later the GFP signal was at background levels unless Vpx was provided, either by VLPs (Fig. 1c) or by vpx transduction (Fig. 1d). Using qRT-PCR, reporter gene expression was shown to be increased by vpx (Supplementary Fig. 1d). Gene repression, and reactivation by Vpx, was not specific to the SFFV promoter, since GFP signal was similar when the reporter gene was expressed from the human EEF1A1 (EF1α) promoter or from the Herpes simplex virus type 1 thymidine kinase (TK) promoter (Supplementary Fig. 1e). The presence of the gag open reading frame in the fusion construct did not affect repression or activation since GFP alone was repressed and reactivated by Vpx (Supplementary Fig. 1f). These results demonstrated that Vpx overcomes transcriptional repression of the provirus.
All Vpx proteins tested, including Vpx encoded by immunodeficiency viruses isolated from humans, mandrills, red-capped mangabeys, and macaques, activated repressed proviruses in human cells (Fig. 1e and Supplementary Fig 1g). Conservation of this activity in human cells among such divergent SIV orthologues was surprising given that Vpx encoded by viruses from non-human primates often do not degrade human SAMHD14. Vprs from SIVs isolated from vervet, sabaeus, tantalus, mandrill, mustached, sun-tailed, colobus, and L’Hoest monkeys, also activated repressed proviral reporters in human cells (Fig. 1e and Supplementary Fig. 1g). Results could not be obtained concerning the activity of Vprs encoded by SIVCPZTAN3, HIV-1U14788 (Group P), SIVGORCP684con, HIV-1MVP5180 (Group O), HIV-1NL4–3 (Group M), SIVCPZLB7, and SIVRCM02CM8081, presumably because these orthologues caused cell cycle arrest and toxicity7,17–19 (indicated by ∅ in Fig. 1e). Vpx and Vpr sequence variability is among the highest observed for lentiviral coding sequences20; the sequences shown in Fig. 1e have an average amino acid identity of only 27%. Such diversity likely reflects rapidly evolving, host-pathogen interfaces21, and precluded activity predictions based on amino acid sequence conservation to guide the engineering of loss-of-function mutations.
To gain insight into the mechanism by which Vpx overcomes transcriptional repression of lentiviral transgenes, a loss-of-function screen was performed focusing on genes reported to contribute to silencing of retroviruses and other transcriptional targets9,10,22–26. Jurkat T cells were transduced with lentivectors that confer puromycin resistance and express shRNAs27 targeting either AGO1, AGO2, AGO3, DNMT3A, HDAC1, HP1, SUV39H1, SUV39H2, PIWIL2, TRIM28, SETDB1, FAM208A, MPHOSPH8, PPHLN1, or MORC2. After selection for five days with puromycin, cells were transduced with the Lenti 2 gag-gfp reporter vector without vpx (Supplementary Fig. 1a). Four days later, the change in gfp reporter expression attributable to the knockdown was calculated as a percentage of the activity observed in Jurkat cells expressing vpx (Fig. 2a). A gene was implicated as a transcriptional silencing factor for the provirus reporter gene if the three shRNA targets for that gene differed significantly from that of the luciferase knockdown control. shRNAs targeting each of the three core components of the Human Silencing Hub (HUSH) complex, FAM208A, MPHOSPH8, and PPHLN1, as well as the associated MORC2, increased reporter gene expression (Fig. 2a). The HUSH complex has scored prominently in screens for transcriptional repressors of newly integrated retroviral or HIV-1 vectors, retrotransposons, and LINE-1 elements9,11–13,28. The HUSH complex acts by spreading H3K9me3-modified heterochromatin, though it also is capable of repressing transcription within euchromatin9,11,13,28.
Figure 2. Vpx activates provirus transcription by degrading HUSH complex components.
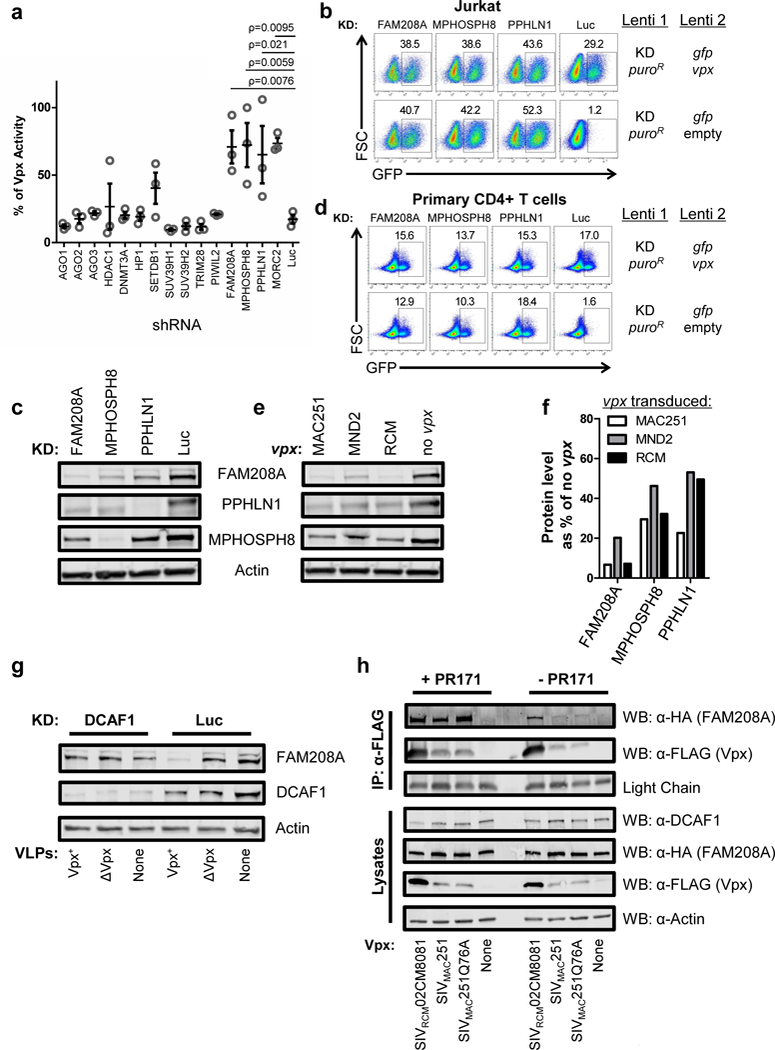
a, Jurkat cells transduced with shRNA-puroR vectors targeting the indicated genes were selected with puromycin, transduced with Lenti 2-Δvpx, and analyzed 5 days later. Plot depicts GFP signal in knockdown lines relative to Jurkats bearing SIVMAC251 vpx (mean ± S.E.M., n=3 shRNA target sites). A gene was implicated as a transcriptional silencing factor for the provirus reporter gene if the three shRNA targets for that gene differed significantly from that of the luciferase knockdown control. P values were determined by 1-way ANOVA with Dunnett’s post-test, relative to luciferase knockdown control. b, Jurkat cells were transduced with the indicated shRNA-puroR vectors and selected with puromycin. Resistant cells were transduced with vpx+ or Δvpx Lenti 2 vector, and analyzed for GFP expression two weeks later (n=4) c, Immunoblot analysis for components of the HUSH complex in Jurkat cells expressing. shRNA constructs used in (b) (n=3). d, CD4+ T cells were activated for 3 days with PHA and then transduced and assayed as in (b). e, Immunoblot analysis of Jurkat lines transduced to express vpx from SIVMAC251, SIVRCMNG411, SIVMND25440, or control (n=3). f, Levels of HUSH components in (e) shown as vpx treated condition relative to control (n=3). g, FAM208A, DCAF1, and Actin immunoblot of Jurkat cells transduced with DCAF1 shRNA-puroR vector or control, that were treated with Vpx+ or ΔVpx VLPs for 18 hrs (n=3). h, HEK293 cells were co-transfected with HA-FAM208A and the indicated FLAG-Vpx constructs. 18 hrs after transfection, cells were either exposed to proteasome inhibitor PR171 or left untreated. 8 hrs after inhibitor treatment cells were lysed, FLAG-Vpx was immunoprecipitated, and immunoblotted for FLAG-Vpx and HA-FAM208A (n=3). Immunoblotting of input lysates are shown below.
The effect on reporter gene expression of the most effective shRNA target sequences for the core HUSH complex components FAM208A, MPHOSPH8, and PPHLN1 is shown in Fig. 2b. The effectiveness of the knockdown of each of the HUSH complex components in Jurkat cells was confirmed by immunoblotting lysate with antibodies specific for FAM208A, PPHLN1, or MPHOSPH8 (Fig. 2c). As previously reported9, knockdown of any individual HUSH complex component decreased each of the other components. Similar results on reporter gene expression were obtained in primary human CD4+ T cells (Fig. 2d). Knockdown of each of the HUSH complex components, then, had the same effect as vpx, and, when combined with it, no further in increase reporter gene expression was detected (Fig. 2b and d and Supplementary Fig 2a). These results demonstrate that the HUSH complex is critical for provirus silencing and raise the possibility that Vpx acts as a substrate adaptor targeting HUSH components to the DCAF1/CUL4A/B E3 ubiquitin ligase complex for degradation, in the same way that Vpx targets SAMHD12,3.
To determine if Vpx promotes the degradation of HUSH complex components, lysate from cells transduced to express SIVMAC251, SIVMND25440, or SIVRCMNG411 vpx was immunoblotted with antibodies specific for FAM208A, PPHLN1, or MPHOSPH8. All three Vpx proteins reduced the steady-state level of all three core HUSH complex components (Fig. 2e). In contrast, the HUSH complex associated protein MORC2 increased in response to vpx, or, as previously described10, in response to knockdown of any of the HUSH complex components (Supplementary Fig. 2b-e).
Among the three core HUSH components, FAM208A protein levels were decreased more than the other two components (Fig. 2f), so ongoing experiments focused on the effect of Vpx on FAM208A. Indeed, in addition to the three Vpx proteins assessed in Fig. 2e, the other Vpx and Vpr orthologues shown to have transactivation activity in Fig 1e and Supplementary Fig 1f (HIV-2ROD Vpx, SIVMNE027 Vpx, SIVDRLD3 Vpx, SIVAGMTAN1 Vpr, SIVMND1GB1 Vpr, and SIVLST524 Vpr) all decreased the levels of FAM208A (Supplementary Figs. 2f and g).
To assess whether disruption of FAM208A protein levels by Vpx was dependent upon the DCAF1 adaptor for the CUL4A/B ubiquitin ligase complex, Jurkat T cells were transduced with a lentivector that knocks down DCAF15, or with a control knockdown vector. After selection with puromycin the cells were exposed for 18 hrs to SIV VLPs bearing Vpx, control VLPs that lacked Vpx, or no VLPs. In the DCAF1 knockdown cells, FAM208A protein levels were unchanged by Vpx, indicating that FAM208A disruption by Vpx was dependent upon DCAF1 (Fig. 2g).
Degradation of SAMHD1 requires direct interaction with Vpx or Vpr4. To determine if Vpx similarly associates with proteins of the HUSH complex, HA-tagged FAM208A was co-transfected into HEK293 cells with FLAG-tagged SIVMAC251 Vpx or SIVRCM02CM8081 Vpx. When anti-FLAG antibody was used to immunoprecipitate either of the two Vpx proteins from the soluble cell lysate, HA-FAM208A was detected in the immunoprecipitate (Fig. 2h). FLAG-Vpx mutant (Q76A) is incapable of binding DCAF15,15. Though Vpx-Q76A was unable to transactivate the Lenti-GFP reporter (Supplementary Fig 1g), it was still able to immunoprecipitate HA-FAM208A (Fig. 2h). The strength of the FAM208A signal in the Vpx pull-out increased when the co-transfected HEK293 cells were incubated with the proteasome inhibitor PR171. These results demonstrate that FAM208A associates with Vpx and that the interaction results in proteasome-mediated degradation of FAM208A.
The experiments described above examined the effect of Vpx or Vpr on HIV-1 proviruses in which the reporter gene was transcribed by a heterologous promoter (Figs 1 and 2, and Supplementary Fig. 1). To determine if Vpx is capable of activating a reporter gene driven by the HIV-1 LTR, the TNFα-responsive, J-Lat A1 clonal cell line model of provirus latency was used29, as it was previously shown to be repressed by the HUSH complex9 (Fig. 3a). Transduction with a lentivector expressing SIVmac251 Vpx, or knockdown of FAM208A, caused comparable increase in the percent GFP+ J-Lat A1 cells, and both synergized with TNFα (Fig. 3b and c, Supplementary Fig 3a). Transduction of the J-Lat A1 cell line with lentivectors expressing vpx encoded by SIVRCM02CM8081 or SIVMND25440, as well as with vpr encoded by SIVMND1GB1 or SIVAGMTAN1, caused similar increase in expression of the LTR-driven reporter gene (Supplementary Fig 3b). Delivery of Vpx+ containing VLPs to J-Lat A1 cells also increased expression of GFP in these cells. (Supplementary Fig 3c).
Figure 3. The HIV-1 LTR is activated by Vpx or disruption of FAM208A.
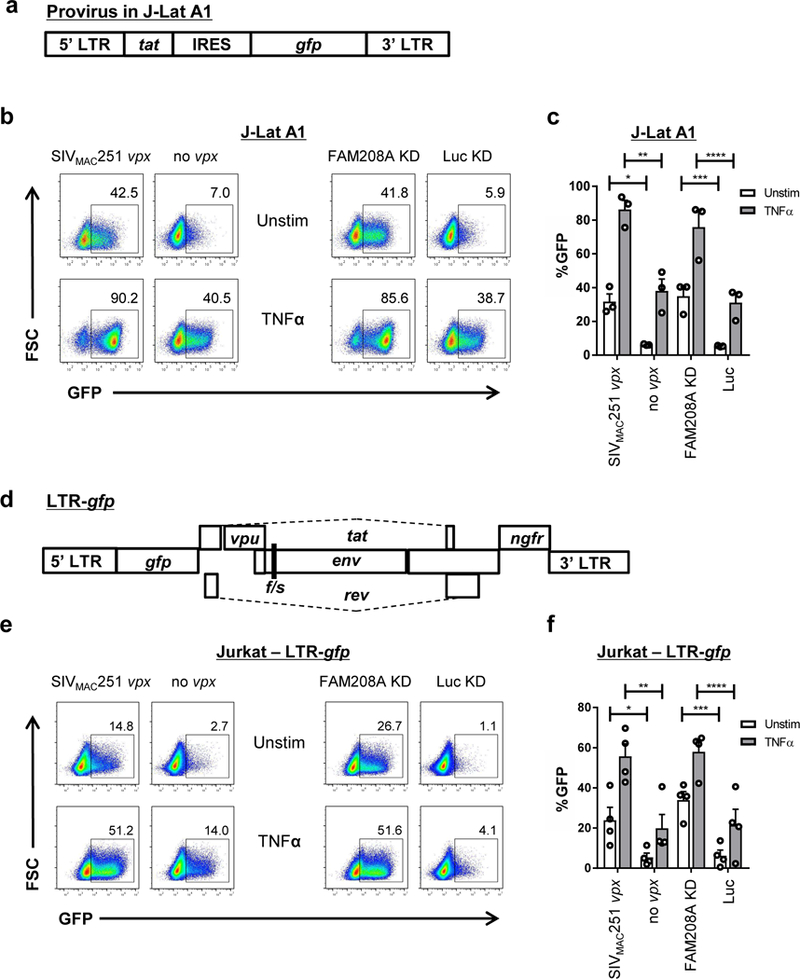
a, Schematic of the HIV-1 minigenome integrated in the J-Lat A1 line. b, J-Lat A1 cells were transduced with Lenti 1 encoding SIVMAC251 vpx or Δvpx control, or with lentivectors expressing shRNA targeting FAM208A or luciferase control. Transduced cells were selected with puromycin, and activated for 24 hrs with 10 ng/ml of TNFα. Representative GFP signal by flow is shown. c, Quantification of results from (b) and additional replicates (mean ± S.E.M., n=3 independent experiments). *, P=0.005, **, P=0.0056, ***, P=0.0055, ****, P=0.016 as determined by two tailed unpaired t test. d, Schematic of the LTR-gfp provirus used to analyze HIV-1 LTR driven gfp expression in pools of cells. e, Jurkat cells transduced with LTR-gfp were kept in culture for at least four weeks and then transduced and assessed by flow cytometry, as in (b). f, Quantification of results from (e) (mean ± S.E.M., n=4 independent experiments) *, P=0.034, **, P=0.0084, ***, P=0.012, ****, P=0.0014 as determined by two tailed unpaired t test.
J-Lat A1 was selected to have a silent HIV-1 LTR-driven provirus with the ability to reactivate in response to TNFα29. The unique provirus within a clone such as J-Lat A1 may be sensitive to position-dependent silencing effects30 and therefore may not accurately reflect the sensitivity of a population of HIV-1 proviruses to transcriptional activation by Vpx or to silencing by FAM208A. To address the effect of Vpx and FAM208A on a population of proviruses with diverse integration sites, Jurkat T cells were transduced with an HIV-1 LTR-driven reporter vector (LTR-gfp) that retains complete LTRs, tat, and rev, but has a frameshift mutation in env, an ngfr reporter gene in place of nef, and gfp in place of gag, pol, vif, and vpr (Fig. 3d). Four weeks after transduction with LTR-GFP, the presence of latent proviruses within the pool of Jurkat cells was confirmed by reactivation with either TNFα or TCR-stimulation (Supplementary Fig. 3d). The Jurkat LTR-gfp cells were then transduced with vectors expressing SIVMAC251 Vpx, or shRNA targeting FAM208A, and selected with puromycin. Compared with control cells, vpx or FAM208A knockdown increased the percentage of GFP+ cells, and exhibited a synergistic effect upon treatment with TNFα (Figs 3e and f, Supplementary Fig 3e). Similar results were obtained in four, independently-generated, biological replicate experiments, in which vpx was delivered, or FAM208A was knocked down, from four to eight weeks after the first LTR-GFP transduction (Fig. 3f). Additionally, expression vectors for SIVMND25440 Vpx, SIVRCM02CM8081 Vpx, SIVMND1GB1 Vpr, or SIVAGMTAN1 Vpr all increased GFP expression in Jurkat LTR-gfp cells (Supplementary Fig. 3f). Knockdown of SETDB1 also increased GFP expression in Jurkat LTR-gfp cells (Supplementary Fig. 3f). In addition, Vpx was able to enhance GFP expression in primary CD4+ T cells that were transduced with the LTR-GFP vector (Supplementary Fig. 3g). Together, these experiments demonstrate that FAM208A contributes to the transcriptional repression of clonal or polyclonal LTR reporter lines, in part due to recruitment of SETDB1, and that primate immunodeficiency viruses counteract this activity via their Vpx and Vpr proteins.
The effect of Vpx or FAM208A knockdown on spreading infection with replication-competent primate immunodeficiency viruses was tested next. Jurkat T cells transduced to express SIVMAC251 vpx, or cells transduced with control vector, were infected with HIV-1-ZsGreen, a replication-competent HIV-1NL4–3 clone, that encodes ZsGreen in place of nef (Supplementary Table 1). Infection was monitored by determining the percent ZsGreen+ cells with flow cytometry, every two days for ten days. Compared with the control, HIV-1 replication kinetics were accelerated by vpx (Fig. 4a, Supplementary Fig. 4a). In similar fashion, HIV-1 infection of Jurkat cells transduced with the FAM208A knockdown vector resulted in faster replication kinetics (Fig. 4b, Supplementary Fig. 4b). The effect of HIV-1 vpr on HIV-1 replication in dividing tissue culture cells8 is perhaps obscured by the toxicity of cell cycle arrest7,17–19. Effects of vpr on HIV-1 are in fact evident when proviral expression is restricted to single-cycle infection7, and vpr offers a selective advantage in vivo since the mutant vpr reading-frame was repaired when virus was injected into chimps or a person7.
Figure 4. Vpx counteracts FAM208A restriction of exogenous HIV-1 and SIVMAC239 infection and of endogenous LINE-1.
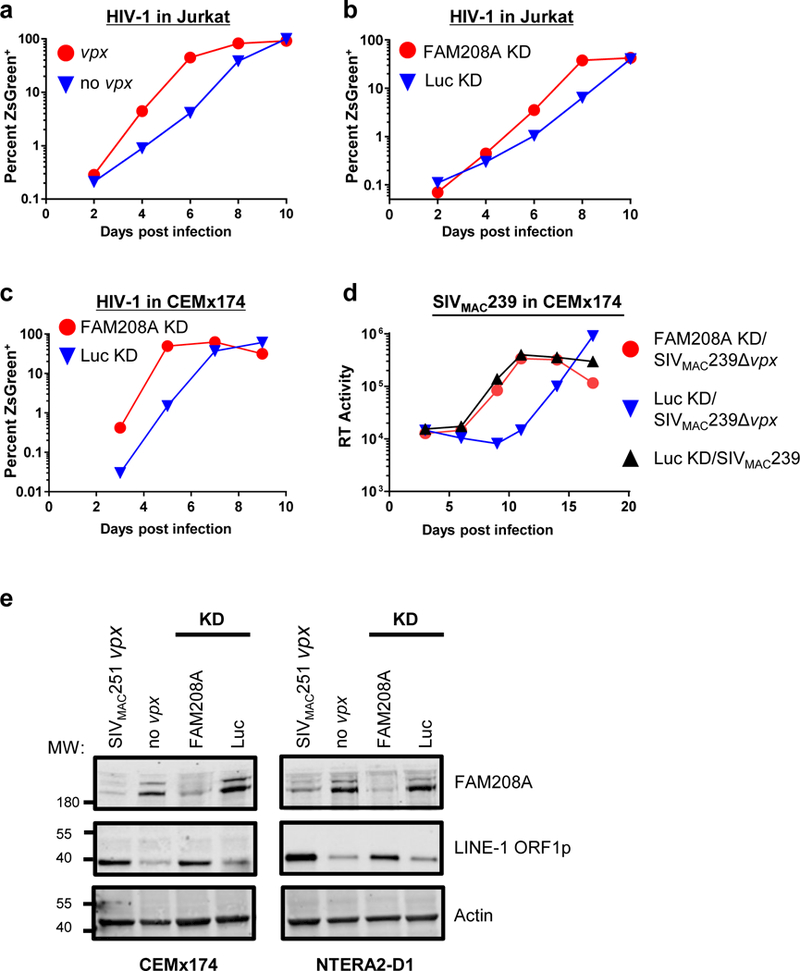
a,b, Replication of HIV-1-ZsGreen in Jurkat cells transduced with SIVMAC251 vpx or control (a), or with lentivectors expressing shRNA targeting FAM208A or Luc control (b). Replication kinetics was measured by flow cytometry for ZsGreen+ cells. c,d, Spreading infection of HIV-1-ZsGreen (c), SIVMAC239 or SIVMAC239Δvpx (d) in CEMx174 cells transduced with FAM208A or Luc control shRNA. Spread of HIV-1-ZsGreen was assessed by flow cytometry as in (a), while spread of SIVmac239 (d) was assessed by measuring the accumulation of reverse transcriptase (RT) activity in the supernatant. All data is representative of at least four repeat experiments. Data from biological replicates was normalized to maximum infection observed in that experiment and the area under the curve was calculated for statistical comparison (see Supplementary Fig. 4). e, FAM208A, LINE-1 ORF1p, and Actin immunoblot, of CEMx174 and NTERA-2D1 cells, transduced with SIVMAC251 vpx or control, or lentivectors expressing shRNA targeting FAM208A or Luc control (n=3).
SIVMAC239 does not replicate in Jurkat cells so CEMx174 cells were used to test the effect of FAM208A and vpx on replication of this virus. As in Jurkat cells, FAM208A knockdown increased HIV-1 replication kinetics in CEMx174 cells (Fig. 4c, Supplementary Fig. 4c). Then, CEMx174 cells transduced with FAM208A or control knockdown vectors were challenged with SIVMAC239 or SIVMAC239-Δvpx and replication was assessed by measuring reverse transcriptase activity in the supernatant. In the absence of vpx, SIVMAC239 replicated slower than the wild-type virus in control knockdown CEMx174 cells (Fig. 4d). This delay in SIVMAC239-Δvpx replication kinetics was not observed when FAM208A was knocked down (Fig. 4d, Supplementary Fig. 4d). These experiments indicate that FAM208A inhibits primate immunodeficiency virus replication and that Vpx antagonizes this restriction, resulting in increased expression from integrated proviruses, permitting virus spread.
Finally, since the HUSH complex limits expression of the autonomous non-LTR retrotransposon LINE-111, the effect of vpx on steady-state levels of LINE-1 ORF1p was examined. CEMx174 and NTERA-2 embryonal carcinoma cells were transduced with FAM208A or control knockdown vectors, or with vpx-expressing or control vectors. For both cell lines, vpx expression resulted in an increase of endogenous LINE-1 ORF1p expression, as did FAM208A knockdown (Fig. 4e).
METHODS
Plasmids.
Sequences encoding 3xFLAG N-terminal-tagged Vpx and Vpr proteins were ordered as codon-optimized, gBlocks Gene Fragments (Integrated DNA Technologies; http://www.idtdna.com/) and cloned into either the pscALPS vector 16 for transduction, or into pcDNA3.1 for transfection. pAPM-D4 is a truncated derivative of the pAPM lentivector 27 that expresses the puromycin acetyltransferase and miR30-based shRNA from the SFFV promoter. Supplementary Table 1 lists all plasmids used here, with corresponding addgene accession numbers, target sites used in particular knockdown vectors, and accession numbers for all the Vpx and Vpr orthologues tested here. All plasmid DNAs and sequences are available at https://www.addgene.org/Jeremy_Luban/.
Cell culture.
Cells were cultured at 37°C in 5% CO2 humidified incubators and monitored for mycoplasma contamination using the Mycoplasma Detection kit (Lonza LT07–318). HEK293 cells (ATCC) were used for viral production and were maintained in DMEM supplemented with 10% FBS, 20 mM L-glutamine (ThermoFisher), 25 mM HEPES pH 7.2 (SigmaAldrich), 1 mM sodium pyruvate (ThermoFisher), and 1x MEM non-essential amino acids (ThermoFisher). Jurkat cells and CEMx174 cells (ATCC) were cultured in RPMI-1640 supplemented with 10% heat inactivated FBS, 20 mM L-glutamine, 25 mM HEPES pH 7.2, 1 mM sodium pyruvate, 1x MEM non-essential amino acids and Pen/Strep (ThermoFischer) (RPMI-FBS complete). J-Lat A1 cells 29 (NIH AIDS Reagent Program, catalogue #9852, donated by Eric Verdin) were cultured in RPMI-FBS complete media. NTERA-2 D1 (ATCC), a pluripotent human embryonal carcinoma cell line, was cultured in DMEM supplemented with 10% FBS.
Leukopaks were obtained from anonymous, healthy, blood bank donors (New York Biologics, Southhampton, NY). As per NIH guidelines (http://grants.nih.gov/grants/policy/hs/faqs_aps_definitions.htm), experiments with these cells were declared non-human subjects research by the University of Massachusetts Medical School Institutional Review Board. PBMCs were isolated from leukopaks by gradient centrifugation on Histopaque-1077 (Sigma-Aldrich). CD4+ T cells were enriched from PBMCs using anti-CD4 microbeads (Miltenyi) and were >95% CD4+. CD4+ T cells were cultured in RPMI-FBS complete media in the presence of 50 U/mL hIL-2 (NIH AIDS Reagent Program, catalogue #136).
Vector production.
HEK293 cells were seeded at 75% confluency in 6-well plates and transfected with 6.25 μL Transit LT1 lipid reagent (Mirus) in 250 μL Opti-MEM (Gibco) with 2.25 μg total plasmid DNA. Full replicating virus was produced by transfection of 2.25μg of the indicated plasmid. Lenti-GFP reporters, LTR-GFP reporter, and shRNA lentivectors were produced by transfection of the lentivector, psPAX2 gagpol expression plasmid, and the pMD2.G VSV G expression plasmid, at a DNA ratio of 4:3:1. Vpx containing SIV-VLPs were produced by transfection at a 7:1 plasmid ratio of SIV3+ to pMD2.G, and ΔVpx SIV VLPs were produced the same way using SIV3+ ΔVpx plasmid. 12 hrs after transfection, media was changed to the specific media for the cells that were to be transduced. Viral supernatant was harvested 2 days later, filtered through a 0.45 μm filter, and stored at 4oC.
Reverse Transcriptase assay.
Virions in the transfection supernatant were quantified by a PCR-based assay for reverse transcriptase activity 27. 5 μl transfection supernatant were lysed in 5 μL 0.25% Triton X-100, 50 mM KCl, 100 mM Tris-HCl pH 7.4, and 0.4 U/μl RNase inhibitor (RiboLock, ThermoFisher). Viral lysate was then diluted 1:100 in a buffer of 5 mM (NH4)2SO4, 20 mM KCl, and 20 mM Tris–HCl pH 8.3. 10 μL was then added to a single-step, RT PCR assay with 35 nM MS2 RNA (IDT) as template, 500 nM of each primer (5’-TCCTGCTCAACTTCCTGTCGAG-3’ and 5’-CACAGGTCAAACCTCCTAGGAATG-3’), and hot-start Taq (Promega) in a buffer of 20 mM Tris-Cl pH 8.3, 5 mM (NH4)2SO4, 20 mM KCl, 5 mM MgCl2, 0.1 mg/ml BSA, 1/20,000 SYBR Green I (Invitrogen), and 200 μM dNTPs. The RT-PCR reaction was carried out in a Biorad CFX96 cycler with the following parameters: 42°C 20 min, 95°C 2 min, and 40 cycles [95°C for 5 s, 60°C 5 s, 72°C for 15 s and acquisition at 80°C for 5 s]. 3 part vector transfections typically yielded 106 RT units/μL.
Transductions.
For generating pools of shRNA knockdown Jurkat and CEMx174 lines, cells were plated at 106 cells/mL in RPMI-FBS complete and transduced with 107 RT units of viral vector per 106 cells, followed by selection with 1 μg/ml puromycin (InvivoGen, cat# ant-pr-1). To generate stable gag-gfp or gfp expressing Jurkat cells, cells were transduced as for shRNA KD above, followed by selection with 5 μg/mL blasticidin (InvivoGen, cat# ant-bl-1) at day 3 after transduction.
CD4+ T cells were stimulated in RPMI-FBS complete, with 50 U/ml IL-2 and 5 μg/mL PHA-P (Sigma, cat# L-1668). After 3 days, T cells were washed and replated at 3 × 106 cells/mL in RPMI-FBS complete, with 50 U/ml IL-2. Cells were transduced with 108 RT units of viral vector per 106 cells followed by selection in 2 μg/mL puromycin.. After selection, cells were re-plated in RPMI-FBS complete with 50 U/ml IL-2 at 3 × 106 cells/mL in RPMI-FBS complete and transduced again with the indicated GFP vectors, 108 RT units of viral vector per 106 cells. Transduced T cells were analyzed two weeks after the 2nd transduction.
Lentiviral Infections.
5 × 105 Jurkat or CEMx174 cells were incubated with 5 × 107 RT units of HIV-1NL4.3, SIVMAC239, or SIVMAC239Δvpx virus stocks produced in HEK-293 cells for 12 hrs in RPMI-FBS complete media, followed by a wash in media and replated in 1 mL of media. Cells were split every 2–3 days and analyzed. For monitoring of HIV-1 ZsGreen infection, when cells were split, aliquots were fixed in BD Cytofix followed by analysis of GFP+ cells by flow cytometry to determine infection levels. For monitoring of SIV infections, 50 μL aliquots of supernatant were analyzed for RT activity using the above described RT assay.
Re-activation assays.
LTR-driven GFP re-activation assays were performed with 10 ng/ml hTNFα (Invivogen, cat# rcyc-htnf), or with 1 μg/ml soluble α-CD3 and α-CD28 antibody. α-CD3 antibody (clone OKT3) and α-CD28 antibody (clone CD28.2) were provided by Lisa Cavacini (MassBiologics, Mattapan, Massachusetts).
qRT-PCR.
Total RNA was isolated from Jurkat cells using Trizol reagent followed by purification of RNA with RNeasy Plus Mini (Qiagen) with Turbo DNase (ThermoFisher) in order to limit DNA contamination. First-strand synthesis used Superscript III Vilo Master mix (Invitrogen) with random hexamers. qPCR was performed in 20 μL using SYBR green reagent (Applied Biosystems) with primers designed against gag, gfp, and gapdh for normalization.. Amplification was on a CFX96 Real Time Thermal Cycler (Bio-Rad) using the following program: 95°C for 10 min, then 45 cycles of 95°C for 15 s and 60°C for 60 s. Cells not transduced with Lenti-GFP vector were used as negative control and the housekeeping gene GAPDH was used to normalize expression levels. The primer sequences used were: gag primers (Forward: 5’-GCTGGAAATGTGGAAAGGAA-3’; Reverse: 5’-AGTCTCTTCGCCAAACCTGA-3’), gfp primers (Forward: 5’-GCAGAGGTGAAGTTCGAAGG-3’; Reverse: 5’-CCAATTGGTGTGTTCTGCTG-3’), gapdh primers (Forward: 5’-AGGGCTGCTTTTAACTCTGGT-3’; Reverse: 5’-CCCCACTTGATTTTGGAGGGA-3’).
Flow cytometry.
Cells were fixed in BD Cytofix Buffer prior to data acquisition on a BD C6 Accuri. Data was analyzed in FlowJo 10.
Western Blot.
Cells were washed in PBS, counted, normalized for cell number, and lysed directly in 1x SDS-PAGE sample buffer. Samples were run on NuPage 4–12% Bis-Tris gels followed by blotting onto nitrocellulose membranes. Primary antibodies used: FAM208A (Atlas, HPA00875), MPHOSPH8 (Proteintech, 16796–1-AP), PPHLN1 (Sigma, HPA038902), SETDB1 (Proteintech 11231–1-AP), DCAF1 (Proteintech, 11612–1-AP), FLAG (Novus, NB600–345), FLAG (Sigma, F1804, used for IP), MORC2 (Bethyl Labs, A300–149A), LINE-1 ORF1p (Millipore, MABC1152),and HA (Biolegend, 901501).
Vpr and Vpx phylogeny.
The following Vpr and Vpx amino acid sequence alignments were obtained from the Los Alamos National Laboratories (LANL) HIV sequence database: 2016 HIV-1/SIVcpz Vpr, 2016 HIV-2/SIVsmm Vpr, 2016 HIV-2/SIVsmm Vpx, 2016 other SIV Vpr, and 2016 other Vpx. Consensus sequences were generated for HIV-1 group M subtypes A, B, C, D, F, G, H, I, J, and those designated U in the LANL database, as well as group N. A master alignment was scaffolded from the above alignments and re-aligned by hand. Redundant SIV and HIV-2 Vpr and Vpx sequences were removed, and the sequences of individual HIV-1 isolates were replaced with the consensus sequences. This was used to generate a master phylogeny using RAxML 8.2.11, as implemented in Geneious (https://www.geneious.com) with gamma LG substitution model and Rapid Bootstrapping with search for best scoring tree algorithm. This master tree was utilized to identify major relationships and identify a reduced number of sequences to retain while maintaining the overall phylogenic structure. Vpx and Vpr sequences from the following viral isolates were retained: HQ179987, L20571, M15390, AF208027, AB731738, KP890355, M15390, AF208027, AB731738, KP890355, U58991, M30931, L40990, KJ461715, KF214240, U42720, AY169968, DQ373065, DQ373064, DQ374658, FJ919724, AJ580407, KM378563, KM378563, FJ424871, M66437, AF468659, AF468658, AF188116, M76764, LC114462, M27470, AY159322, AY159322, U79412, U79412, AY340701, AY340700, EF070329, KF304707, FM165200, HM803690, HM803689, AF382829, AF349680, HM803690, HM803689, AF349680, HQ378594, JX860432, JX860430, JX860426, JX860432, M83293, M83293, FR751162, AY523867, AM182197, AM713177, U26942, and the HIV-1 group M clade B consensus. These sequences were used to generate a phylogeny using the same method as above. Superfluous taxa were pruned from this phylogeny using Mesquite 3.4 and the resulting tree was visualized in FigTree v1.4.3.
Sampling.
At least three biological replicates were performed for all experiments. The screen for factors mediating silencing of the Lenti-GFP vector utilized 3 target sequences for each candidate gene. Flow cytometry plots in the figures show representative data taken from experiments performed at the same time. HIV-1and SIV spreading experiments were repeated 4–5 times each as indicated and representative data of one such experiment is shown.
Statistics.
Information regarding the statistical tests utilized, and the n values, are found in the figure legends. Statistical analysis of the knockdown screen of factors involved in silencing of Lenti-GFP was analyzed by one-way ANOVA with Dunnett post test comparing 3 shRNA target sites to control knockdown conditions. All statistics presented were performed using PRISM 5.0 (GraphPAD Software, La Jolla, CA).
Supplementary Material
Acknowledgements
The authors wish to dedicate these experiments to the memory of Jan Svoboda (1934–2017), whose demonstration that cells may carry Rous sarcoma virus genetic information in the absence of any infectious virus production provided support to the proviral hypothesis. We thank Lisa Cavacini for antibodies, and Nathaniel Landau for SIV plasmids. The following reagents were obtained through the AIDS Reagent Program, Division of AIDS, NIAID, NIH: J-Lat Tat-GFP Cells (A1) from Dr. Eric Verdin, and SIVmac239 SpX and SIVmac239 SpX ΔVpx from Dr. Ronald C. Desrosiers.
Funding: This research was supported by National Institutes of Health (USA) grants R01AI111809, RO1AI117839, and DP1DA034990 to J.L.
Footnotes
Data Availability: all data needed to evaluate the conclusions in the paper are present in the paper or in the supplementary table. The plasmids described in Supplementary Table 1, along with their complete nucleotide sequences, are available at https://www.addgene.org/Jeremy_Luban/.
Competing interests: None declared.
REFERENCES
- 1.Siliciano JD, Kajdas J, Finzi D, Quinn TC, Chadwick K, Margolick JB, Kovacs C, Gange SJ & Siliciano RF Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 9, 727–728 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Hrecka K, Hao C, Gierszewska M, Swanson SK, Kesik-Brodacka M, Srivastava S, Florens L, Washburn MP & Skowronski J Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 474, 658–661 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Laguette N, Sobhian B, Casartelli N, Ringeard M, Chable-Bessia C, Ségéral E, Yatim A, Emiliani S, Schwartz O & Benkirane M SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 474, 654–657 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lim ES, Fregoso OI, McCoy CO, Matsen FA, Malik HS & Emerman M The ability of primate lentiviruses to degrade the monocyte restriction factor SAMHD1 preceded the birth of the viral accessory protein Vpx. Cell Host Microbe 11, 194–204 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pertel T, Reinhard C & Luban J Vpx rescues HIV-1 transduction of dendritic cells from the antiviral state established by type 1 interferon. Retrovirology 8, 49 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reinhard C, Bottinelli D, Kim B & Luban J Vpx rescue of HIV-1 from the antiviral state in mature dendritic cells is independent of the intracellular deoxynucleotide concentration. Retrovirology 11, 12 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goh WC, Rogel ME, Kinsey CM, Michael SF, Fultz PN, Nowak MA, Hahn BH & Emerman M HIV-1 Vpr increases viral expression by manipulation of the cell cycle: a mechanism for selection of Vpr in vivo. Nat. Med. 4, 65–71 (1998). [DOI] [PubMed] [Google Scholar]
- 8.Miller CM, Akiyama H, Agosto LM, Emery A, Ettinger CR, Swanstrom RI, Henderson AJ & Gummuluru S Virion-Associated Vpr Alleviates a Postintegration Block to HIV-1 Infection of Dendritic Cells. J. Virol. 91, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tchasovnikarova IA, Timms RT, Matheson NJ, Wals K, Antrobus R, Göttgens B, Dougan G, Dawson MA & Lehner PJ GENE SILENCING. Epigenetic silencing by the HUSH complex mediates position-effect variegation in human cells. Science 348, 1481–1485 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tchasovnikarova IA, Timms RT, Douse CH, Roberts RC, Dougan G, Kingston RE, Modis Y & Lehner PJ Hyperactivation of HUSH complex function by Charcot-Marie-Tooth disease mutation in MORC2. Nat. Genet. (2017). doi:10.1038/ng.3878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu N, Lee CH, Swigut T, Grow E, Gu B, Bassik MC & Wysocka J Selective silencing of euchromatic L1s revealed by genome-wide screens for L1 regulators. Nature 553, 228–232 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu L, Oliveira NMM, Cheney KM, Pade C, Dreja H, Bergin A-MH, Borgdorff V, Beach DH, Bishop CL, Dittmar MT & McKnight A A whole genome screen for HIV restriction factors. Retrovirology 8, 94 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blewitt ME, Vickaryous NK, Hemley SJ, Ashe A, Bruxner TJ, Preis JI, Arkell R & Whitelaw E An N-ethyl-N-nitrosourea screen for genes involved in variegation in the mouse. Proc. Natl. Acad. Sci. U. S. A. 102, 7629–7634 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Le Rouzic E, Belaïdouni N, Estrabaud E, Morel M, Rain J-C, Transy C & Margottin-Goguet F HIV1 Vpr arrests the cell cycle by recruiting DCAF1/VprBP, a receptor of the Cul4-DDB1 ubiquitin ligase. Cell Cycle 6, 182–188 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Srivastava S, Swanson SK, Manel N, Florens L, Washburn MP & Skowronski J Lentiviral Vpx accessory factor targets VprBP/DCAF1 substrate adaptor for cullin 4 E3 ubiquitin ligase to enable macrophage infection. PLoS Pathog. 4, e1000059 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neagu MR, Ziegler P, Pertel T, Strambio-De-Castillia C, Grütter C, Martinetti G, Mazzucchelli L, Grütter M, Manz MG & Luban J Potent inhibition of HIV-1 by TRIM5-cyclophilin fusion proteins engineered from human components. J. Clin. Invest. 119, 3035–3047 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Re F, Braaten D, Franke EK & Luban J Human immunodeficiency virus type 1 Vpr arrests the cell cycle in G2 by inhibiting the activation of p34cdc2-cyclin B. J. Virol. 69, 6859–6864 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang F, Re F, Sebastian S, Sazer S & Luban J HIV-1 Vpr induces defects in mitosis, cytokinesis, nuclear structure, and centrosomes. Mol. Biol. Cell 15, 1793–1801 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He J, Choe S, Walker R, Di Marzio P, Morgan DO & Landau NR Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J. Virol. 69, 6705–6711 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCarthy KR & Johnson WE Plastic proteins and monkey blocks: how lentiviruses evolved to replicate in the presence of primate restriction factors. PLoS Pathog. 10, e1004017 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fregoso OI, Ahn J, Wang C, Mehrens J, Skowronski J & Emerman M Evolutionary toggling of Vpx/Vpr specificity results in divergent recognition of the restriction factor SAMHD1. PLoS Pathog. 9, e1003496 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weinberg MS & Morris KV Transcriptional gene silencing in humans. Nucleic Acids Res. 44, 6505–6517 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chéné I. du, Basyuk E, Lin Y-L, Triboulet R Knezevich A, Chable-Bessia C, Mettling C, Baillat V, Reynes J, Corbeau P Bertrand E, Marcello A, Emiliani S, Kiernan R& Benkirane M Suv39H1 and HP1γ are responsible for chromatin-mediated HIV-1 transcriptional silencing and post-integration latency. EMBO J. 26, 424–435 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wolf D & Goff SP TRIM28 mediates primer binding site-targeted silencing of murine leukemia virus in embryonic cells. Cell 131, 46–57 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Peterlin BM, Liu P, Wang X, Cary D, Shao W, Leoz M, Hong T, Pan T & Fujinaga K Hili Inhibits HIV Replication in Activated T Cells. J. Virol. 91, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang GZ & Goff SP Transcriptional Silencing of Moloney Murine Leukemia Virus in Human Embryonic Carcinoma Cells. J. Virol. 91, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pertel T, Hausmann S, Morger D, Züger S, Guerra J, Lascano J, Reinhard C, Santoni FA, Uchil PD, Chatel L, Bisiaux A, Albert ML, Strambio-De-Castillia C, Mothes W, Pizzato M, Grütter MG & Luban J TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 472, 361–365 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robbez-Masson L, Tie CHC, Conde L, Tunbak H, Husovsky C, Tchasovnikarova IA, Timms RT, Herrero J, Lehner PJ & Rowe HM The HUSH complex cooperates with TRIM28 to repress young retrotransposons and new genes. Genome Res. (2018). doi:10.1101/gr.228171.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jordan A, Bisgrove D & Verdin E HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J. 22, 1868–1877 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen H-C, Martinez JP, Zorita E, Meyerhans A & Filion GJ Position effects influence HIV latency reversal. Nat. Struct. Mol. Biol. 24, nsmb.3328 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.