

Abstract
Background
Iron deficiency (ID) compromises the developing nervous system, including the hippocampus, resulting in later-life deficits despite iron repletion. The iron-dependent molecular changes driving these lasting deficits, and the effect of early iron repletion, are incompletely understood. Previous studies have utilized dietary models of maternal-fetal ID anemia (IDA) to address these questions; however, concurrent anemia prevents delineation of the specific role of iron.
Objective
The aim of the study was to isolate the effects of developmental ID on adult hippocampal gene expression and to determine if iron repletion reverses these effects in a mouse model of nonanemic hippocampal neuronal ID.
Methods
Nonanemic, hippocampus-specific neuronal ID was generated by using a Tet-OFF dominant negative transferrin receptor (DN-TFR1) mouse model that impairs cellular iron uptake. Hippocampal ID was reversed with doxycycline at postnatal day 21 (P21) in a subset of mice to create 2 experimental groups, chronically iron-deficient and formerly iron-deficient mice, which were compared with their respective doxycycline-treated and untreated iron-sufficient controls. RNA from adult male hippocampi was sequenced. Paired-end reads were analyzed for differential expression. Differentially expressed genes were analyzed in Ingenuity Pathway Analysis.
Results
A total of 346 genes were differentially expressed in adult, chronically iron-deficient hippocampi compared with controls. ID dysregulated genes in critical neurodevelopmental pathways, including axonal guidance, CDK5, Ephrin receptor, Rac, and Neurotrophin/Trk signaling. Iron repletion at P21 normalized adult hippocampal expression of 198 genes; however, genes involved in cAMP response element-binding protein (CREB) signaling, neurocognition, and neurologic disease remained dysregulated in adulthood.
Conclusions
Chronic ID during development, independent of anemia, alters the adult mouse hippocampal transcriptome. Restoring iron status during a known critical period of hippocampal neurodevelopment incompletely normalized these changes, suggesting a need for additional studies to identify the most effective timeline for iron therapy, and adjunctive treatments that can fully restore ID-induced molecular changes, particularly in human populations in whom chronic ID is endemic.
Keywords: iron deficiency, hippocampus, transcriptome, neurodevelopment, developmental origins of disease
Introduction
Micronutrient deficiencies are estimated to affect 2 billion people worldwide, with pregnant women and children <5 y of age at greatest risk (1). Iron deficiency (ID) is the most common of the micronutrient deficiencies, with an estimated global prevalence of 25% (1, 2). Among those affected, the burden falls disproportionately on pregnant women, infants, and preschool-aged children, with 40–50% of individuals affected (2, 3). A large body of data now supports that ID in these most vulnerable populations is detrimental to neurodevelopment (4, 5). Human cohort studies have found an association between ID in infancy and slower perceptual speed, impaired language abilities, and difficulty with quantitative concepts—all deficits that may contribute to the finding that, in adolescence, formerly iron-deficient individuals have lower scores on reading and arithmetic tests and an increased likelihood to repeat a grade (6). Studies in this same cohort at age 25 y showed an association between early-life ID and decreased likelihood of completing secondary school and pursuing higher education and increased reports of negative emotions (7). Low maternal iron intake during pregnancy is associated with an increased risk of schizophrenia (8) and autism (9). Together, these findings suggest that ID during fetal and early postnatal life has lasting negative effects on neurocognitive function and adult mental health. Furthermore, ID is chronic in many populations around the world, and those children who experience chronic ID suffer more significant neurodevelopmental consequences than those who experience less severe ID (10).
Preclinical rodent models have shown that ID during the fetal and early postnatal periods negatively affects the developing neurocognitive system, particularly the hippocampus. In a rat model of dietary ID anemia (IDA), chronic IDA beginning during gestation results in learning and memory behavioral deficits in adulthood (11). Importantly, adult, formerly IDA rats also show behavioral deficits in hippocampus-dependent learning and memory, despite iron repletion starting at postnatal day (P) 7 (12–15). Adult P65 formerly IDA rats have altered apical dendrite structure in hippocampal area CA1 (16, 17), altered synaptic function (18), and altered expression of genes critical for neuronal morphogenesis, plasticity, and energy metabolism (16, 19, 20). On a genomewide scale, formerly IDA rats continue to have widespread hippocampal gene expression alterations in adulthood despite iron treatment starting at the rodent developmental equivalent of term human birth (21). These altered genes map onto pathways and functions related to schizophrenia, autism, and mood disorders (21).
A major limitation of the rat model of dietary IDA in deciphering the specific role of iron in long-term neurodevelopment is that animals are anemic, with an average hematocrit of 25% during the period of ID (22). Because systemic anemia compromises tissue oxygen delivery, anemia is a confounding factor to understanding the effect of ID itself on the developing central nervous system. We previously developed a conditional dominant negative transferrin receptor (DN-TFR1) genetic mouse model of hippocampal, neuronal-specific ID in order to separate the effects of neuronal ID from those of anemia (23). In this model, a point mutation in transferrin receptor 1 (Tfr1) results in a dominant negative (DN), nonfunctional TFR1 protein, which impairs binding and cellular uptake of transferrin, the major iron carrier. By expressing the DN-TFR1 under a CaMKII-driven Tet transactivator (tTA), ID is restricted to hippocampal neurons and is rapidly reversible with doxycycline (Dox) treatment (23). Left chronically iron deficient (CID), these mice have similar hippocampal structural and functional deficits as the IDA rat model in adulthood, indicating that ID, independent of anemia, does compromise the developing hippocampus (23). When hippocampal ID is reversed at P21, hippocampal dendrite morphology and learning and memory performance are grossly intact in adulthood, suggesting that P21 may be within the critical period for iron in neurodevelopment (23). However, it is unknown whether deficits to neuronal circuitry and gene expression remain.
Currently, the underlying mechanisms of iron-dependent structural and functional changes to the developing nervous system remain incompletely characterized. Understanding hippocampal gene dysregulation after ID is an important step toward elucidating the mechanisms of adult hippocampal dysfunction. Unlike studies in the IDA rat model (21), whole-transcriptome analysis of the hippocampus has not been performed, to our knowledge, in a genetic mouse model of ID. The objective of the present study was to assess, at the transcriptomic level, 2 translationally relevant questions: whether chronic ID without anemia alters gene expression in the adult hippocampus and whether iron treatment during development completely rescues adult hippocampal gene expression. Although the first question is relevant to human populations globally where chronic ID is endemic, the second question relates to the public health policy of screening for childhood ID by measuring hematocrit.
Methods
Animals
Two transgenic mouse lines were used to generate litters as described in detail previously (23). Briefly, the first transgenic line carried DN-TFR1 in the transgene TRE2-eGFP-DNTFR1 on a B6;CBA background. Mice from this line were mated with a second transgenic line, B6;CBA-Tg(Camk2a-tTA)1Mmay/J (Jackson Laboratory). The resultant pups that were positive for both transgenes expressed DN-TFR1 at levels sufficient to disrupt hippocampal neuronal iron uptake, resulting in iron-deficient CA1 hippocampal neurons with no effect on systemic tissue iron concentrations, as reported previously (23). These mice are referred to as DN. The resultant pups that were positive for only one or neither transgene were considered wild-type (WT) controls, because they do not express DN-TFR1 and have normal hippocampal neuronal iron uptake and storage, as reported previously (23).
Upon weaning at P21, mice of both genotypes (DN and WT) were either maintained on standard nonpurified diet (2018 Teklad Global 18% Protein Rodent Diet; Envigo) or switched to a Dox-containing diet (0.625 g Dox/kg, TD.01306; Envigo). The Dox diet was nutritionally identical to the standard rodent diet, and both diets were nutritionally iron sufficient (200 mg/kg). This resulted in 2 experimental groups: CID (DNnoDox) and formerly iron deficient (FID; DN+P21Dox). The WT Dox-untreated and -treated groups were used as iron-sufficient (IS) controls (WTnoDox and WT+P21Dox, respectively).
Mice were housed in a 12-h light-dark cycle, with ad libitum access to food and water. All of the mice were maintained according to the Animal Use Policies and Guidelines outlined by the University of Minnesota Institutional Animal Care and Use Committee. All protocols were approved by the University of Minnesota Institutional Animal Care and Use Committee and complied with the Guide for the Care and Use of Laboratory Animals (24).
Tissue collection
Adult male mice (2–9 mo old) were killed by intraperitoneal injection of Beuthanasia (10 mg/kg) followed by rapid decapitation. The hippocampus was isolated and immediately flash-frozen in liquid nitrogen. Tissues were stored at −80°C until used.
Preparation of RNA for sequencing
Total RNA was isolated from 1 hippocampal lobe per mouse with the use of the RNAqueous RNA Isolation kit (Ambion). Four mice (n = 4 biological replicates) were used per experimental group (ntotal = 16). The sample size was determined on the basis of previously described power calculations to optimize detection of differentially expressed genes (25). Isolated RNA was sent to the University of Minnesota Genomics Center for library preparation and next-generation sequencing. RNA was first quantified with the use of the RiboGreen RNA Assay kit (Invitrogen) and assessed for quality by using capillary electrophoresis (Agilent BioAnalyzer 2100; Agilent). Barcoded libraries were constructed for each sample with the use of the TruSeq RNA v2 kit (Illumina). Libraries were selected for fragments of ∼200 bp. Sequencing was performed with the use of Illumina HiSeq 2500 to generate 50-bp paired-end reads. Sequencing depth was >10 million reads per sample for all samples.
RNA-sequencing data analysis methods
Sequenced reads were subjected to quality control with the use of FastQC in Galaxy. All of the samples had an average quality score of >36. After quality control, reads were aligned to the mouse reference genome (version mm10) by using the TopHat package (version 2.0.9) in Galaxy (26). The overall read alignment rate was >97% for all samples. Each experimental group (DNnoDox and DN+P21Dox) was compared with its age-matched control group (WTnoDox and WT+P21Dox, respectively) with the use ofCuffDiff (version 2.2.1) in Galaxy (27). Differentially expressed genes were considered those with abs[log2(fold change)] >0.2 and false discovery rate (q value) <0.05 to account for multiple comparisons. Fisher's exact test was performed to determine the significance of the proportion of up- and downregulated genes in experimental groups compared with their controls, with α set at P < 0.05.
Pathway analysis
Genes identified as differentially expressed were analyzed in Ingenuity Pathway Analysis (IPA; Qiagen) to predict canonical pathways, upstream regulators, cellular functions, and diseases altered or affected relative to controls, as described previously (21). Briefly, core analyses were performed in IPA using the following settings: stringent filter, all data sources, direct relations only, experimentally observed confidence, mammalian species, and cells only, with cutoffs of abs [log2(fold change)] >0.2 and a false discovery rate (q value) <0.05 to account for multiple comparisons. In IPA, statistical significance of genes mapping onto canonical pathways, molecular networks, cellular functions, and upstream regulators was determined by Fisher's exact test for each pathway, where a significant P value [P < 0.05; −log(P) > 1.3] indicates that the mapping of the differentially expressed genes onto a given pathway, regulator, function, or disease was not due to chance alone.
qRT-PCR
qRT-PCR was performed on 5–6 mice (biological replicates) per group (ntotal = 23). Sample size was chosen on the basis of our previous reports (21, 28). RNA samples used for qRT-PCR experiments were a mix of samples used in the RNA-Seq experiment and samples from different age-matched mice. RNA was generated as described above. cDNA synthesis was performed with the use of 1.0 μg total RNA and the High Capacity RNA-to-cDNA kit (Applied Biosystems). qPCR was performed in singleplex with the use of TaqMan Universal PCR Master Mix (Applied Biosystems) and TaqMan gene expression assays (ThermoFisher Scientific) on a MX3000P instrument (Stratagene). TaqMan probe IDs are listed in Table 1. TATA-binding protein (Tbp) was used as an endogenous control (Mm01277042_m1), because the expression of this gene remains unaffected by ID. Samples were run in duplicate, normalized to Tbp, and averaged to generate fold change relative to the respective control group. Results were analyzed by t test, and α was set at P < 0.05.
TABLE 1.
qRT-PCR validation of CREB1-regulated target genes in CID and FID mouse hippocampus1
| Fold-change (relative to IS control) | |||||
|---|---|---|---|---|---|
| CID | FID | ||||
| Gene | Probe ID | RNA-Seq2 | qRT-PCR3 | RNA-Seq2 | qRT-PCR3 |
| Arc | Mm01204954_g1 | 0.51 | 0.65* | 1.49 | 1.48* |
| Btg2 | Mm00476162_m1 | 0.65 | 0.75* | 1.48 | 1.2** |
| Egr1 | Mm00656724_m1 | 0.75 | 0.79 | 1.64 | 1.29* |
| Fos | Mm00487425_m1 | 0.51 | 0.43** | 3.12 | 2.73 |
| Junb | Mm04243546_s1 | 0.65 | 1.02 | 1.58 | 1.23 |
| Nr4a1 | Mm01300401_m1 | 0.59 | 0.67** | 1.42 | 1.05 |
1A list of definitions of gene and protein names used in this table is included in Supplemental Materials. *P < 0.05, **P < 0.01 [compared with the respective IS control group (WTnoDox or WT+P21Dox]. CID, chronically iron deficient; CREB1, cAMP response element-binding protein 1; Dox, doxycycline; FID, formerly iron deficient; ID, identifier; IS, iron-sufficient.
2All target genes had P < 0.05 by RNA-Seq analysis, after correcting for multiple comparisons.
3 n = 5–6/group.
Results
Chronic nonanemic neuronal ID alters expression of genes involved in development and plasticity in the adult hippocampus
To determine the effect of nonanemic hippocampal ID on the neuronal transcriptome, gene expression in the CID hippocampus was compared with age-matched IS controls. A total of 346 genes were differentially expressed in the CID hippocampus, representing ∼1.5% of the sequenced transcriptome (Supplemental Data 1). Of these genes, a significant proportion (58%; P = 0.047) were downregulated (Figure 1). The dysregulated genes mapped onto cellular functions that are critical for neurodevelopment, including shape change in neurites and axons and branching of neurites and axons (Table 2). These neuronal functions were all predicted to have decreased activation (z score <−2.0) in the CID hippocampus based on the observed changes in gene expression. In addition, dysregulated genes mapped onto canonical signaling pathways that are involved in neurodevelopment and synaptic plasticity (Figure 2A), including axonal guidance, CXCR4, CDK5, Ephrin receptor, GABA receptor, Rac, and Neurotrophin/TRK signaling pathways. The majority of these pathways were predicted to have reduced activity (z score <−1.5) in the ID hippocampus.
FIGURE 1.
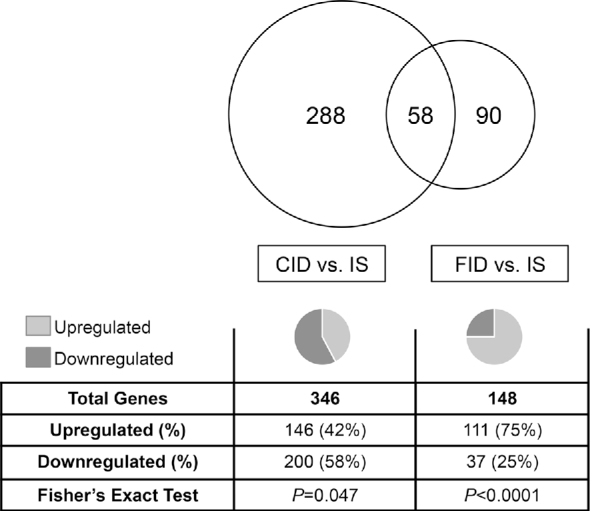
Gene dysregulation in the CID and FID adult hippocampus. Chronic ID starting in late embryonic development results in dysregulation of 346 genes in the adult hippocampus. The majority of these genes are downregulated (P = 0.047). When chronic ID is reversed at P21, 148 genes are dysregulated in the adult hippocampus. The majority are upregulated (P < 0.0001). Of these 148 genes, 58 are genes that are also dysregulated in the CID hippocampus. The remaining 90 dysregulated genes are uniquely dysregulated in the FID hippocampus. CID, chronically iron deficient; FID, formerly iron deficient; ID, iron deficiency; IS, iron sufficient; P, postnatal day.
TABLE 2.
Top dysregulated diseases and functions in the adult chronically iron-deficient mouse hippocampus, annotated by IPA1
| Disease or function annotation | Activation z score | P | Genes | Genes, n |
|---|---|---|---|---|
| Cell survival | −3.47 | 1.16 × 10−3 | Bcl6, Bdnf, Ccnd1, Cebpb, Cebpd, Cxcl12, Egr1, Egr3, Fos, Foxo1, Gfra1, H2afx, Igf2, Il33, Jun, Lcn2, Npnt, Nr4a1, Nrn1, Ntf3, Prkcd, Ptgs2, Smad3, Spp1, Traf3, Vgf | 26 |
| Accumulation of cells | −2.28 | 3.21 × 10−3 | C4a/C4b, Ccnd1, Cdkn1c, Cx3cr1, Dusp1, Egr1, Enpp2, Il33, Krt18, Lcn2, Ltc4s, Ntf3, Ptgds, Tnfrsf25 | 14 |
| Shape change in axons | −2.35 | 8.98 × 10−6 | A2m, Bdnf, Cxcl12, Egr3, Fos, Grasp, Kalrn, Mid1, Tbr1 | 9 |
| Shape change in neurites | −2.28 | 1.31 × 10−8 | A2m, Bdnf, Chn1, Cxcl12, Dock10, Egr3, Fos, Git1, Grasp, Kalrn, Lcn2, Mag, Mid1, Neurod6, Nrn1, Prss12, Ptgs2, Rapgef4, Ryr1, Scn4b, Sulf1, Tbr1, Vgf | 23 |
| Branching of axons | −2.16 | 1.76 × 10−5 | A2m, Bdnf, Cxcl12, Egr3, Fos, Grasp, Kalrn, Mid1 | 8 |
| Branching of neurites | −2.13 | 3.25 × 10−8 | A2m, Bdnf, Chn1, Cxcl12, Dock10, Egr3, Fos, Git1, Grasp, Kalrn, Lcn2, Mag, Mid1, Neurod6, Nrn1, Prss12, Ptgs2, Rapgef4, Ryr1, Scn4b, Sulf1, Vgf | 22 |
1A list of definitions of gene and protein names used in this table is included in Supplemental Materials. IPA, Ingenuity Pathway Analysis.
FIGURE 2.
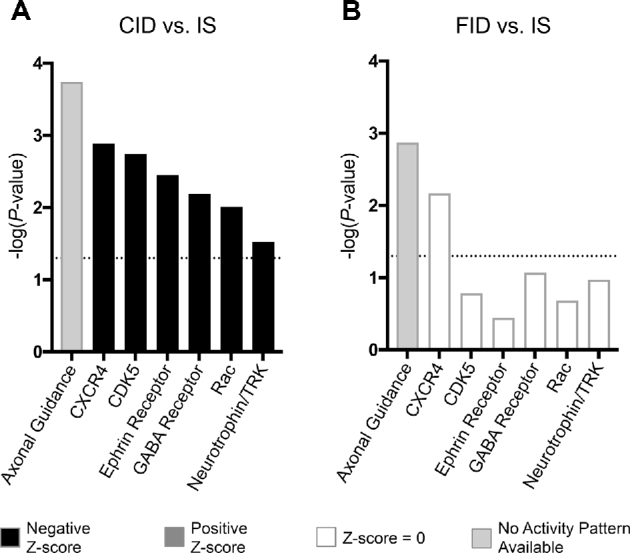
Critical neuronal signaling pathways are altered by ID and rescued by its reversal at P21. In IPA, differentially expressed genes were mapped to canonical signaling pathways. (A) In the adult CID hippocampus, dysregulated genes mapped significantly onto canonical signaling pathways that function in neurodevelopment and neuroplasticity [P < 0.05, −log(P value) > 1.3; dotted horizontal line]. On the basis of the genes that were dysregulated and the directionality of their dysregulation, it was predicted that the majority of these pathways would have a net decrease in activity, indicated by a negative z score (z <−1.5). (B) In the adult FID hippocampus, dysregulated genes no longer mapped significantly to the majority of these same pathways. Dysregulated genes still mapped significantly to axonal guidance signaling and CXCR4 signaling pathways; however, there was no predicted effect on the net function of the pathways. A list of definitions of gene and protein names used in this figure is included in Supplemental Materials. CID, chronically iron deficient; FID, formerly iron deficient; ID, iron deficiency; IPA, Ingenuity Pathway Analysis; IS, iron sufficient.
Adult gene expression is only partially rescued by iron repletion at P21
To determine the effect of ID reversal on hippocampal gene expression in adulthood, iron status was normalized starting at P21, and hippocampal gene expression was assessed in adult FID mice compared with age-matched IS controls. A total of 148 genes were differentially expressed in the adult FID hippocampus despite iron repletion starting at P21, indicating a partial resolution of gene dysregulation compared with the CID hippocampus (Supplemental Data 2). Of these genes, a significant proportion (75%; P < 0.0001) were upregulated (Figure 1). Ninety genes were uniquely dysregulated in the FID hippocampus, and 58 were dysregulated in both the CID and FID hippocampus (Figure 1). With the use of IPA, the same set of neurodevelopmental and plasticity canonical signaling pathways were re-examined in the FID hippocampus. Compared with the CID hippocampus where these pathways were predicted to be significantly downregulated, the majority of these pathways were not affected in the FID hippocampus (Figure 2B). Axonal guidance signaling and CXCR4 signaling pathways had a significant number of genes differentially expressed; however, there was no predicted change in overall pathway activity.
IPA was used to determine the potential functions and effects of the 148 genes that were dysregulated in the adult FID hippocampus. Genes mapped primarily to cellular functions involved in cell death (Table 3). Apoptosis, necrosis, and cell death were all predicted to have reduced activation (z score <−2.0), whereas cellular homeostasis, activation of cells, and cell movement were predicted to have increased activation (z score >2.0).
TABLE 3.
Top dysregulated diseases and functions in the adult formerly iron-deficient mouse hippocampus, annotated by IPA1
| Disease or function annotation | Activation z score | P | Genes | Genes, n |
|---|---|---|---|---|
| Apoptosis | −3.82 | 3.87 × 10−5 | Ace, Ascl1, Btg2, Camk2d, Casp9, Cckbr, Cd200, Cd44, Cnp, Ctgf, Cyr61, Dcn, Dusp1, Egr1, Fmod, Fos, Igf2, Junb, Kitlg, Kl, Klf5, Mef2c, Npas4, Nr4a1, Ntsr1, Pcp4, Prkar2b, Prkca, Ptgds, Sept4, Sfrp1, Slc4a2, Spp1, Trh, Ttr, Vstm2l | 32 |
| Necrosis | −3.23 | 8.18 × 10−5 | Ascl1, Btg2, Camk2d, Casp9, Cd200, Cd44, Cnp, Col6a1, Ctgf, Dcn, Dusp1, Egr1, Fos, Gjb2, Igf2, Junb, Kitlg, Kl, Klf5, Lbp, Mef2c, Npas4, Nr4a1, Pcp4, Prkar2b, Ptgds, Sfrp1, Spp1, Trh, Ttr, Vstm2l | 31 |
| Cellular homeostasis | 2.95 | 4.98 × 10−4 | Atp2b4, Casp9, Cldn1, Cldn2, Dcn, Egr1, F5, Htr2c, Igf2, Junb, Kitlg, Mef2c, Nr4a1, Prkca, Ryr1, Sfrp1, Slc31a1, Slc4a2, Spns2, Spp1 | 20 |
| Cell death | −2.88 | 7.03 × 10−6 | Ace, Antxr1, Ascl1, Atp2b4, Btg2, Camk2d, Casp9, Cckbr, Cd200, Cd44, Cnp, Col6a1, Ctgf, Cyr61, Dcn, Dusp1, Egr1, Fmod, Fos, Gjb2, Igf2, Junb, Kitlg, Kl, Klf5, Klk8, Lbp, Mag, Mef2c, Npas4, Nr4a1, Ntsr1, Pcp4, Prkar2b, Prkca, Ptgds, Ryr1, Sept4, Sfrp1, Slc31a1, Slc4a2, Spp1, Trh, Ttr, Vstm2l | 45 |
| Activation of cells | 2.54 | 2.61 × 10−4 | Ace, Cd200, Cd44, Chrna4, Ctgf, Dusp1, F5, Gjc2, Htr2c, Igf2, Kitlg, Lbp, Nr4a1, Prkca, Rab34, Rab3b, Slc4a2, Spp1 | 18 |
| Cell movement | 2.47 | 9.14 × 10−6 | Ascl1, Atp2b4, Btg2, Cd200, Cd44, Ctgf, Cyr61, Dcn, Efna5, Enpp2, Fnbp1l, Fos, Gjb2, Igf2, Kitlg, Klf5, Lamc2, Lbp, Mef2c, Minos1nbl1/Nbl1, Nr2f2, Nr4a1, Plxnd1, Prkca, Ptgds, Sept4, Sfrp1, Slc4a2, Spns2, Spp1, Sulf1, Wls | 32 |
1A list of definitions of gene and protein names used in this table is included in Supplemental Materials. IPA, Ingenuity Pathway Analysis.
A subset of hippocampal genes are permanently dysregulated by developmental ID
Of the 148 genes that were dysregulated in the FID hippocampus, a subset of 58 genes were also dysregulated in the CID hippocampus, suggesting that expression of these genes is highly sensitive to early-life ID and insensitive to its reversal (Figure 1). Although these genes were dysregulated in both the CID and FID hippocampus, the dysregulation did not always have the same directionality; 67% of genes were dysregulated in the same direction in both conditions, whereas the remaining 33% of genes were dysregulated in opposite directions (Figure 3A). With the use of IPA, these 58 genes mapped onto functions related to neurocognitive function and diseases, including cognition, learning, schizophrenia, and Alzheimer disease (Figure 3B).
FIGURE 3.

Genes permanently dysregulated by ID are involved in neurocognitive function and dysfunction. (A) Of the 58 genes dysregulated in both the CID and FID hippocampus, 9 (15%) are downregulated in both conditions and 30 (52%) are upregulated in both conditions. The remaining 19 genes (33%) are dysregulated with opposite directionality in the 2 conditions. The upward arrows indicate gene upregulation; downward arrows indicate gene downregulation. (B) When mapped to diseases and functions in IPA, these genes map to the functions of cognition and learning as well as schizophrenia and Alzheimer disease. A list of definitions of gene and protein names used in this figure is included in Supplemental Materials. CID, chronically iron deficient; FID, formerly iron deficient; ID, iron deficiency; IPA, Ingenuity Pathway Analysis; IS, iron sufficient.
cAMP response element-binding protein 1 is differentially activated in the chronically and formerly ID hippocampus
IPA was also used to predict which upstream regulators of gene expression might account for the gene dysregulation associated with ID. cAMP response element-binding protein (CREB) signaling was chosen for further analysis because of its critical role in late-stage long-term potentiation, and thus long-term memory. As a transcription factor, CREB serves as the upstream transcriptional regulator of long-term potentiation–associated gene expression changes that underlie memory (29). In addition, CREB is activated by brain-derived neurotrophic factor (BDNF) signaling (30), which is known to be reduced by ID (20). On the basis of the observed gene expression changes, CREB1 activity was predicted to be altered in both the CID and FID hippocampus. In the CID hippocampus (Figure 4A), 13 genes regulated by CREB1 signaling were downregulated, predicting a net decrease in CREB1 activity (z score = −3.4, P < 0.001). In contrast, CREB1 activity was predicted to be increased in the FID hippocampus (z score = 2.6, P < 0.001), where 7 genes known to be regulated by CREB1 signaling were upregulated (Figure 4B). Of these CREB1-regulated genes, 6 were dysregulated in both conditions, but in opposite directions. qRT-PCR was used to validate this subset of 6 CREB1-regulated genes (Table 1).
FIGURE 4.
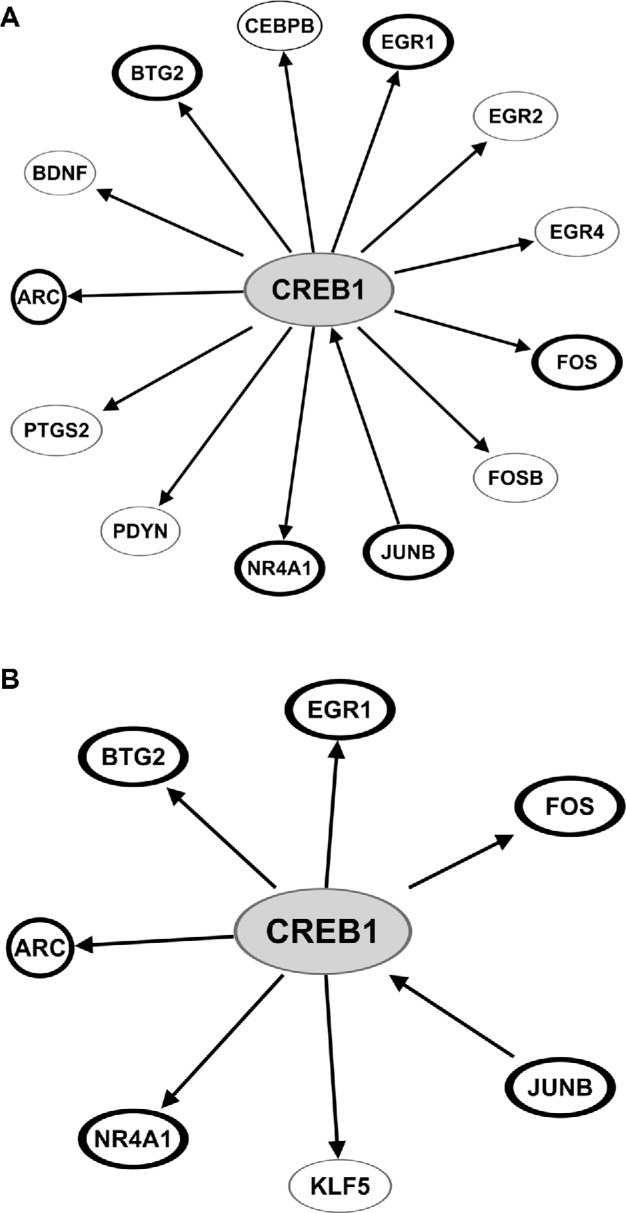
CREB1 signaling is predicted to be altered by ID. (A) Thirteen genes involved in CREB1 signaling were found to be significantly downregulated in the CID hippocampus. On the basis of these gene expression changes, CREB1 activity is predicted by IPA to be significantly decreased (z score = −3.42, P < 0.001). (B) In the FID hippocampus, 7 CREB1-regulated genes were found to be significantly upregulated, which predicts a significant increase in CREB1 activity (z score = 2.61, P < 0.001). Genes outlined in bold are dysregulated in both CID and FID hippocampus. A list of definitions of gene and protein names used in this figure is included in Supplemental Materials. CID, chronically iron deficient; CREB1, cAMP response element-binding protein 1; FID, formerly iron deficient; ID, iron deficiency; IPA, Ingenuity Pathway Analysis.
Discussion
With the use of a unique nonanemic mouse model of hippocampal neuronal-specific ID, we showed that chronic hippocampal ID, without anemia, results in altered expression of ∼1.5% of the adult hippocampal transcriptome. These altered genes map to canonical signaling pathways and functions that are critical for normal neurodevelopment and plasticity, predicting a net decrease in these functions in the iron-deficient hippocampus. These observed gene expression changes are consistent with findings from our primary hippocampal neuronal culture model of pure neuronal ID, which shows decreased branching and complexity of developing dendritic arbors (31) as well as our previous findings with the use of this mouse model, which show compromised apical dendrite structure in hippocampal area CA1 and poorer spatial learning and memory in permanently iron-deficient mice (23).
The vast majority of previous studies of ID have utilized a rat model of dietary ID in which animals become anemic. Although the formerly IDA rat hippocampus exhibits significant, transcriptome-wide changes in gene expression in adulthood (21), it has been unclear how many of these gene expression changes are due to ID itself rather than anemia. The present results indicate that although a smaller percentage of the hippocampal transcriptome is dysregulated by nonanemic ID than by IDA (1.5% and 5.7%, respectively) (21), ID alone is sufficient to disrupt hippocampal gene expression. Translationally, this is important for 2 reasons. First, nonanemic ID is most prevalent in populations that are at risk of suffering its neurodevelopmental effects, including preschool-aged children (32), pregnant women (33), and certain infant populations such as premature infants, infants of diabetic mothers, and intrauterine growth–restricted infants who are at risk of nonanemic ID from birth due to low iron endowment (34). Second, iron is prioritized to RBCs over other tissues, including the central nervous system (CNS) (35). Thus, chronic ID may be present in the CNS long before it affects prioritized tissues such as RBCs and is detected peripherally as anemia (36). Our results indicate that individuals experiencing this type of nonanemic ID, particularly during neurodevelopment, are potentially at risk of transcriptome-level changes to the CNS that may affect CNS function in adulthood.
One question that has remained unanswered is the exact timing of the critical period for iron in hippocampal development. Defining this period is critical for understanding when treatment with iron will completely restore hippocampal function and thus result in no long-term deficits. In previous studies, normalizing iron status within the putative critical period for iron in neurodevelopment (P10–P30) was sufficient to prevent gross hippocampal morphologic and functional abnormalities in adulthood (23, 37). However, normalization of this set of variables does not preclude the possibility that more subtle deficits remain in the FID hippocampus. The present transcriptomic analysis indicates that reversal of chronic hippocampal ID at P21 does not fully normalize the hippocampal transcriptome in adulthood. Although many critical neuronal signaling pathways are no longer predicted to be altered in the FID hippocampus, those genes that do remain dysregulated map to functions such as learning, cognition, and schizophrenia, and predict altered function of key neuronal actors such as CREB. Importantly, these predicted diseases and functions are consistent with the human cohort literature, which shows that low maternal iron intake during pregnancy is associated with increased risk of schizophrenia (8) and that early-life ID is associated with increased risk of learning and cognition deficits (6).
Interestingly, we found that 33% of the genes dysregulated in both the CID and FID hippocampus were dysregulated in opposite directions. A possible explanation for this phenomenon is that CID tissue reaches a new homeostatic set point by P21, and that re-introduction of iron disrupts this homeostasis by iron-overloading a tissue that has adapted, via compensatory changes in gene expression, to low iron concentrations. In this case, re-introduction of iron could result in a rebound dysregulation of genes in the opposite direction of that in the iron-deficient condition. In addition, we found that 90 genes were dysregulated uniquely in the FID hippocampus. Together, these data raise the possibility that iron repletion of previously iron-deficient tissue may have effects of its own. Further research is needed to understand the functional implications of these gene expression changes and the ideal timing for iron therapy in order to avoid any potential lasting or deleterious effects of iron repletion.
Finally, the mechanisms by which developmental iron status drives the reported long-term gene expression changes are incompletely understood. Epigenetic modification of DNA and chromatin is one mechanism by which environmental factors in early life, such as nutrition, can program gene expression into adulthood (38). Several proteins involved in the establishment of epigenetic modifications require iron for their enzymatic activity, including the JmjC ARID-domain containing histone demethylase (JARID) proteins, which require iron for their enzymatic removal of methyl groups from lysine residues of histone tails (39, 40). We previously reported that the formerly IDA rat hippocampus undergoes differential histone methylation at the BDNF-IV promoter compared with IS controls, concomitant with decreased BDNF expression, likely due to decreased JARID enzymatic activity during ID (28). Because epigenetic modifications regulate gene expression throughout the genome, alteration of iron-dependent epigenetic modifications presents a plausible mechanism by which broad, genomewide changes in gene expression could be indirectly driven by ID. Further investigation of these iron-dependent epigenetic modifications will advance our understanding of the underlying mechanisms by which nonanemic ID enacts its lasting effects.
Supplementary Material
Acknowledgments
The authors’ responsibilities were as follows—AB, MKG, and PVT: designed the research and wrote the manuscript; AB, PVT, and SJBF: conducted the research; SJBF: provided essential materials; AB: analyzed data and performed statistical analysis; MKG: had primary responsibility for final content; and all authors: have read and approved the final manuscript.
Notes
Supported by Eunice Kennedy Shriver National Institute of Child Health and Human Development grants R21-HD054490 and R01-HD029421 and the University of Minnesota Masonic Children's Fund (to MKG).
Author disclosures: AB, SJBF, MKG, and PVT, no conflicts of interest.
Supplemental Data 1 and 2 are available from the “Supplementary data” link in the online posting of the article and from the same link in the online table of contents at https://academic.oup.com/jn/.
Abbreviations used:
- BDNF
brain-derived neurotrophic factor
- CID
chronically iron deficient
- CNS
central nervous system
- CREB
cAMP response element-binding protein
- DN
dominant negative
- DN-TFR1
dominant negative transferrin receptor
- Dox
doxycycline
- FID
formerly iron deficient
- ID
iron deficiency
- IDA
iron deficiency anemia
- IPA
Ingenuity Pathway Analysis
- IS
iron-sufficient
- P
postnatal day
- Tfr1
transferrin receptor 1
- WT
wild-type
References
- 1. Bailey RL, West KP, Black RE. The epidemiology of global micronutrient deficiencies. Ann Nutr Metab 2015;66(Suppl 2):22–33. [DOI] [PubMed] [Google Scholar]
- 2. McLean E, Cogswell M, Egli I, Wojdyla D, de Benoist B. Worldwide prevalence of anaemia: WHO Vitamin and Mineral Nutrition Information System, 1993–2005. Public Health Nutr 2009;12:444–54. [DOI] [PubMed] [Google Scholar]
- 3. Stevens GA, Finucane MM, De-Regil M, Paciorek CJ, Flaxman SR, Branca F, Peña-Rosas JP, Qar Z, Bhutta A, Ezzati M. Global, regional, and national trends in haemoglobin concentration and prevalence of total and severe anaemia in children and pregnant and non-pregnant women for 1995–2011: a systematic analysis of population-representative data. Lancet Glob Health 2013;1:e16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Doom JR, Georgieff MK. Striking while the iron is hot: understanding the biological and neurodevelopmental effects of iron deficiency to optimize intervention in early childhood. Curr Pediatr Rep 2014;2:291–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lozoff B. Iron deficiency and child development. Food Nutr Bull 2007;28:S560–71. [DOI] [PubMed] [Google Scholar]
- 6. Lozoff B, Jimenez E, Hagen J, Mollen E, Wolf AW. Poorer behavioral and developmental outcome more than 10 years after treatment for iron deficiency in infancy. Pediatrics 2000;105:E51. [DOI] [PubMed] [Google Scholar]
- 7. Lozoff B, Smith JB, Kaciroti N, Clark KM, Guevara S, Jimenez E. Functional significance of early-life iron deficiency: outcomes at 25 years. J Pediatr 2013;163:1260–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Insel BJ, Schaefer CA, McKeague IW, Susser ES, Brown AS. Maternal iron deficiency and the risk of schizophrenia in offspring. Arch Gen Psychiatry 2008;65:1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schmidt RJ, Tancredi DJ, Krakowiak P, Hansen RL, Ozonoff S. Maternal intake of supplemental iron and risk of autism spectrum disorder. Am J Epidemiol 2014;180:890–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lozoff B, Clark KM, Jing Y, Armony-Sivan R, Angelilli ML, Jacobson SW. Dose-response relationships between iron deficiency with or without anemia and infant social-emotional behavior. J Pediatr 2008;152:696–702, e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Unger EL, Hurst AR, Georgieff MK, Schallert T, Rao R, Connor JR, Kaciroti N, Lozoff B, Felt B. Behavior and monoamine deficits in prenatal and perinatal iron deficiency are not corrected by early postnatal moderate-iron or high-iron diets in rats. J Nutr 2012;142:2040–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schmidt AT, Waldow KJ, Grove WM, Salinas JA, Georgieff MK. Dissociating the long-term effects of fetal/neonatal iron deficiency on three types of learning in the rat. Behav Neurosci 2007;121:475–82. [DOI] [PubMed] [Google Scholar]
- 13. Schmidt AT, Alvarez GC, Grove WM, Rao R, Georgieff MK. Early iron deficiency enhances stimulus-response learning of adult rats in the context of competing spatial information. Dev Cogn Neurosci 2012;2:174–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pisansky MT, Wickham RJ, Su J, Fretham S, Yuan LL, Sun M, Gewirtz JC, Georgieff MK. Iron deficiency with or without anemia impairs prepulse inhibition of the startle reflex. Hippocampus 2013;23:952–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Felt B, Beard J, Schallert T, Shao J, Aldridge J, Connor J, Georgieff M, Lozoff B. Persistent neurochemical and behavioral abnormalities in adulthood despite early iron supplementation for perinatal iron deficiency anemia in rats. Behav Brain Res 2006;171:261–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brunette KE, Tran PV, Wobken JD, Carlson ES, Georgieff MK. Gestational and neonatal iron deficiency alters apical dendrite structure of CA1 pyramidal neurons in adult rat hippocampus. Dev Neurosci 2010;32:238–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jorgenson LA, Wobken JD, Georgieff MK. Perinatal iron deficiency alters apical dendritic growth in hippocampal CA1 pyramidal neurons. Dev Neurosci 2003;25:412–20. [DOI] [PubMed] [Google Scholar]
- 18. Jorgenson LA, Sun M, O'Connor M, Georgieff MK. Fetal iron deficiency disrupts the maturation of synaptic function and efficacy in area CA1 of the developing rat hippocampus. Hippocampus 2005;15:1094–102. [DOI] [PubMed] [Google Scholar]
- 19. Carlson ES, Stead JDH, Neal CR, Petryk A, Georgieff MK. Perinatal iron deficiency results in altered developmental expression of genes mediating energy metabolism and neuronal morphogenesis in hippocampus. Hippocampus 2007;17:679–91. [DOI] [PubMed] [Google Scholar]
- 20. Tran PV, Fretham SJB, Carlson ES, Georgieff MK. Long-term reduction of hippocampal brain-derived neurotrophic factor activity after fetal-neonatal iron deficiency in adult rats. Pediatr Res 2009;65:493–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tran PV, Kennedy BC, Pisansky MT, Won KJ, Gewirtz JC, Simmons RA, Georgieff MK. Prenatal choline supplementation diminishes early-life iron deficiency-induced reprogramming of molecular networks associated with behavioral abnormalities in the adult rat hippocampus. J Nutr 2016;146:484–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Carlson ES, Magid R, Petryk A, Georgieff MK. Iron deficiency alters expression of genes implicated in Alzheimer disease pathogenesis. Brain Res 2008;1237:75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fretham SJB, Carlson ES, Wobken J, Tran PV, Petryk A, Georgieff MK. Temporal manipulation of transferrin-receptor-1-dependent iron uptake identifies a sensitive period in mouse hippocampal neurodevelopment. Hippocampus 2012;22:1691–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. National Research Council ; Committee for the Update of the Guide for the Care and Use of Laboratory Animals, Institute for Laboratory Animal Research. Guide for the care and use of laboratory animals. Washington (DC): National Academies Press; 2011. [Google Scholar]
- 25. Ching T, Huang S, Garmire LX. Power analysis and sample size estimation for RNA-Seq differential expression. RNA 2014;20:1684–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 2013;14:R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 2010;28:511–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tran PV, Kennedy BC, Lien YC, Simmons RA, Georgieff MK. Fetal iron deficiency induces chromatin remodeling at the Bdnf locus in adult rat hippocampus. Am J Physiol Regul Integr Comp Physiol 2015;308:R276–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron 2002;35:605–23. [DOI] [PubMed] [Google Scholar]
- 30. Cunha C, Brambilla R, Thomas KL. A simple role for BDNF in learning and memory? Front Mol Neurosci 2010;3:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bastian TW, von Hohenberg WC, Mickelson DJ, Lanier LM, Georgieff MK. Iron deficiency impairs developing hippocampal neuron gene expression, energy metabolism, and dendrite complexity. Dev Neurosci 2016;38:264–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Asobayire FS, Adou P, Davidsson L, Cook JD, Hurrell RF. Prevalence of iron deficiency with and without concurrent anemia in population groups with high prevalences of malaria and other infections: a study in Côte d'Ivoire. Am J Clin Nutr 2001;74:776–82. [DOI] [PubMed] [Google Scholar]
- 33. Engmann C, Adanu R, Lu TS, Bose C, Lozoff B. Anemia and iron deficiency in pregnant Ghanaian women from urban areas. Int J Gynecol Obstet 2008;101:62–6. [DOI] [PubMed] [Google Scholar]
- 34. Siddappa AM, Rao R, Long JD, Widness JA, Georgieff MK. The assessment of newborn iron stores at birth: a review of the literature and standards for ferritin concentrations. Neonatology 2007;92:73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wallace DF. The regulation of iron absorption and homeostasis. Clin Biochem Rev 2016;37:51–62. [PMC free article] [PubMed] [Google Scholar]
- 36. Rao R, Ennis K, Lubach GR, Lock EF, Georgieff MK, Coe CL. Metabolomic analysis of CSF indicates brain metabolic impairment precedes hematological indices of anemia in the iron-deficient infant monkey. Nutr Neurosci 2018;21:40–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fretham SJB, Carlson ES, Georgieff MK. The role of iron in learning and memory. Adv Nutr 2011;2:112–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Barrett JR. Programming the future: epigenetics in the context of DOHaD. Environ Health Perspect 2017;125:A72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen Z, Zang J, Whetstine J, Hong X, Davrazou F, Kutateladze TG, Simpson M, Mao Q, Pan CH, Dai S et al. Structural insights into histone demethylation by JMJD2 family members. Cell 2006;125:691–702. [DOI] [PubMed] [Google Scholar]
- 40. Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature 2006;439:811–6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.