

Bartonella henselae causes cat scratch disease and several other clinical entities. Infections with B. henselae are frequently occurring; however, the infection is only rarely diagnosed, mainly due to a lack of knowledge in the medical community.
KEYWORDS: Bartonella henselae, serology, IFA, ELISA, development, human, serodiagnostics
ABSTRACT
Bartonella henselae causes cat scratch disease and several other clinical entities. Infections with B. henselae are frequently occurring; however, the infection is only rarely diagnosed, mainly due to a lack of knowledge in the medical community. Microscopic immunofluorescence assays (IFA) are widely used for the serodiagnosis of B. henselae infections but are laborious and time-consuming, and interpretation is subjective. An easy and reliable method for the serological diagnosis of B. henselae infections is needed to overcome the shortcomings of the current IFA. Here, we report the development of an ELISA detecting human anti-B. henselae antibodies from serum samples. By separating the water-insoluble fraction of B. henselae Houston-1 via ion-exchange chromatography, 16 subfractions were generated and tested for immunoreactivity via line blotting. One particular fraction (fraction 24) was selected and spotted on ELISA plates using an industrial production platform. By use of well-characterized human sera from the strictly quality-controlled serum library of the German National Consiliary Laboratory for Bartonella infections, the sensitivity of this ELISA was 100% for PCR-proven infections and 76% for clinically suspected infections at a specificity of 93%. This ELISA is therefore a reliable high-throughput method allowing the serodiagnosis of B. henselae infections.
INTRODUCTION
Infections with Bartonella henselae usually result in cat scratch disease (CSD), a benign and self-limiting but often prolonged lymphadenitis. In immunocompromised patients (e.g., AIDS patients), B. henselae infections can lead to vasculoproliferative diseases such as bacillary angiomatosis and peliosis hepatis (1). Cats are the primary reservoir host of B. henselae, and transmission to humans occurs by cat scratches or cat fleas (2); the role of ticks in the transmission process of B. henselae has been controversially discussed (3, 4). Regional lymphadenopathy is the predominant clinical feature of CSD and develops ∼2 to 3 weeks after inoculation (5). Most often, axillary and epitrochlear (46%), head and neck (26%), and groin (17.5%) lymph nodes are affected (6) and are regularly tender, swollen, and suppurating in ∼13% of the cases (5). Other symptoms are fever (48%), malaise (45%), and skin lesions at the site of the cat scratch (25%) (7). B. henselae infections are the third most common reason for fever of unknown origin (8) and an often ignored pathogen causing “culture-negative” endocarditis (2).
Human infections caused by Bartonella spp. are common. Bartonella-specific antibodies are present in ∼5 to 10% of the population (2, 9, 10). B. henselae infections are responsible for ∼14% of all cervicofacial lymphadenopathies (11, 12). Recently, the prevalence of B. henselae-specific IgG antibodies was determined to be >40% in German forest workers (13). In the United States, ∼6.4 to 9.4/100,000 people suffer from a newly diagnosed B. henselae infection per year (14). From these numbers, a disease load of ∼7,000 cases per year for Germany and of 12,000 for the United States can be calculated. It is suggested that the number of unrecognized cases is probably much higher because of insufficient knowledge about this infection in the medical community. Additionally, many anti-B. henselae seroreactive patients do not report a history of CSD, most likely due to a clinically inapparent course of infection.
Methods for the laboratory identification of B. henselae infections include histological examination (e.g., Warthin-Starry staining), bacterial cultivation (conventional blood agar, cell coculture), molecular (PCR), and serological (indirect immunofluorescence assay [IFA]) approaches (2). The most reliable diagnosis of B. henselae infection is the direct detection of the pathogen via PCR assays and by laborious cultivation techniques. This approach, however, requires invasive procedures (e.g., biopsy, fine needle aspiration) to gain adequate patient material (9, 15) and is, therefore, often avoided.
IFA for specific antibodies is the widely used diagnostic method of choice for the laboratory diagnosis of B. henselae infections. For this purpose, an IFA protocol was established using whole-cell antigen from B. henselae bacteria cocultivated with Vero cells (16) and has been only slightly modified since the description of this technique in the year 1992. Currently, IgG titer levels are internationally accepted to be clearly positive at titers of ≥1:200, ≥1:256, and ≥1:320, depending on the respective serum dilution scheme (10). For IgM, the CDC does not give clear anti-B. henselae IgM cutoff values (10). Previously described anti-B. henselae immunoglobulin ELISAs, however, lack sensitivity and specificity (5, 15, 17, 18). Nevertheless, an ELISA-based B. henselae serodiagnostic test would overcome the shortcomings of the time-consuming IFA, which is, moreover, influenced by interobserver variability. As it allows the automated handling of a high number of serum samples in parallel (18–20), a B. henselae-specific ELISA would also allow seroepidemiological studies, which can currently be performed only on a small scale using classical IFAs (13).
Here, we report the development of a novel, ELISA-based detection method for anti-B. henselae antibodies. This test was systematically established and evaluated by the use of well characterized human serum.
MATERIALS AND METHODS
Collection of human sera.
Sera of patients were collected routinely by physicians or general practitioners for medical reasons to confirm or exclude the clinical diagnosis of cat scratch disease or Bartonella infection and were sent to the German Consiliary Laboratory for Bartonella infections (Frankfurt am Main, appointed by the Robert Koch Institute, Berlin, Germany). Sera were used for routine anti-B. henselae IFA testing and for evaluation of the anti-B. henselae ELISA. This procedure was approved by the ethics committee of the University Hospital Frankfurt (ethics proposal 423/11).
Laboratory characterization of human serum samples.
Laboratory testing was performed under strict quality-controlled criteria (laboratory accreditation according to ISO 15189:2014 standards; certificate D-ML-13102-01-00, valid through 25 January 25 2021) at the Institute for Medical Microbiology and Infection Control, University Hospital, Frankfurt am Main, Germany. Indirect immunofluorescence assays (IFAs) were performed using the Bartonella henselae/Bartonella quintana (IgG) kit (Euroimmun, Lübeck, Germany) with some modifications. Standard serum dilution series from 1:80 to 1:320 and higher were screened for anti-B. henselae IgG antibodies. The results were considered positive when specific fluorescence signals were detected at titers of ≥1:320. Control sera were considered IFA negative at a titer of <1:80. Titers in between these values were considered equivocal.
Three categories of human samples were used in this study: (i) samples from patients suffering from a PCR-confirmed Bartonella infection (n = 10), (ii) samples from patients with a clinically suspected B. henselae infection based on the medical history (lymphadenopathy and/or cat scratch) and a positive (≥1:320) IFA result (n = 21), and (iii) not further qualified samples from IFA-positive (≥1:320) blood donors (n = 12). IFA-negative serum samples (<1:80) from healthy blood donors were used as controls (n = 16). For PCR-based detection of Bartonella DNA from human tissue samples, DNA was extracted using the DNeasy blood and tissue kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. A nested PCR for the detection of the Bartonella 16S rRNA genes was performed as previously described (21) using the Taq DNA polymerase kit (Invitrogen, Schwerte, Germany). Further, a PCR detecting the 16S-23S rRNA intergenic transcribed spacer (ITS) region of Bartonella was conducted using the Platinum Taq polymerase kit (Invitrogen). Positive (B. henselae Marseille [22]) and negative (water) controls were always included. DNA was amplified in a Biometra T3000 thermocycler. Products were separated on an agarose gel, stained with ethidium bromide, and visualized under UV light. 16S rRNA gene and ITS-PCR products were sequenced (GATC, Constance, Germany) with both forward and reverse primers to distinguish Bartonella species (23). Sequences were analyzed using the Chromas software (version 2.6; Technelysium, South Brisbane, Australia) and compared to sequences deposited in GenBank (https://www.ncbi.nlm.nih.gov/GenBank/).
Bacterial strains and cultivation.
For the development of the ELISA described herein, B. henselae Houston-1 (ATCC 49882 [16, 24]) and B. henselae Marseille (22, 25) were used. In some experiments, B. henselae San Antonio-3 (designation CHDE161:SA3; kind gift of C. Dehio, Basel, Switzerland [26]) was also used.
B. henselae was grown from deep-frozen stock samples (−80°C) on Columbia blood agar (CBA; Becton Dickinson, Heidelberg, German) in a humidified atmosphere at 37°C and 5% CO2. Bartonella liquid (BaLi) medium (27) was inoculated with ∼1 × 107 CFU of B. henselae and incubated for 72 h at 37°C and 100 rpm in Erlenmeyer flasks.
Bacterial antigen preparation.
B. henselae bacterial cells were harvested by centrifugation for 15 min at 4,990 × g at 4°C and washed once with phosphate-buffered saline (PBS; Thermo Fisher Scientific, Darmstadt, Germany). Pellets were resuspended in PBS containing 1 mM EDTA (Thermo Fisher Scientific) and 4 mM Pefabloc (Roche Diagnostics, Mannheim, Germany). Cell disruption was performed with a homogenizer (Miccra GmbH, Müllheim, Germany) on ice. The resulting suspension was treated with 100 μg/ml gentamicin (Thermo Fisher Scientific) and stored at −20°C. The absence of viable bacteria was determined by cultivating 20 μl of the bacterial suspension on Columbia agar plates for 14 days. The suspension was divided into a PBS-soluble fraction and a PBS-insoluble fraction by centrifugation for 10 min at 11,300 × g at room temperature. The supernatant was collected (soluble part) and the pellet (insoluble part) was washed three times with PBS before it was resuspended in 8 M urea (Sigma-Aldrich, St. Louis) with 20 mM Tris (Sigma-Aldrich) at a pH 8.0.
IEX.
All samples were filtered through a 0.45-μm filter (Sartorius, Göttingen, Germany) prior to use. Ion-exchange chromatography (IEX) was done with an anion-exchange column (HiPrep Q HP 16/10; GE Healthcare, Chicago, IL, USA) on an Äkta Start system (GE Healthcare). Samples were loaded onto the column in 20 mM Tris buffer, pH 8.0. Buffer for the insoluble fraction in PBS contained, in addition, 8 M urea. After the unbound proteins were washed out, elution was performed using a linear sodium chloride (Merck, Darmstadt, Germany) gradient from 0 to 1 M. A new fraction was collected every 5 ml.
Line blotting.
Line blots were used to find IEX fractions containing immunodominant antigens by using reactive human sera. Fractions from the IEX were printed directly from IEX samples without any further processing on nitrocellulose membranes (GE Healthcare) with a dispenser (FrontLine HR microliter contact; BioDot, Irvine, CA, USA) with 0.7 μl/cm and air dried. Membranes were cut into 3-mm strips and stored at 4°C until use. Strips were equilibrated in sample dilution buffer (NovaTec Immundiagnostica, Dietzenbach, Germany) for 5 min prior to use. IFA-positive and IFA-negative human samples were diluted 1:100 (a broadly used standard dilution for Western blotting in serodiagnostics) with sample dilution buffer, and strips were incubated for 1 h, followed by a 30-min incubation with NovaLisa conjugate based on horseradish peroxidase (HRP)-coupled protein A/G (NovaTec Immundiagnostica). After each incubation step, the strips were washed three times with washing buffer (NovaTec Immundiagnostica) and developed with 3,3′,5,5′-tetramethylbenzidine (TMB) solution (NovaTec Immundiagnostica) for 10 min; this was stopped by washing with distilled water. All steps were performed on a rocking platform at room temperature. Quantification of signals from scanned line blots was done using ImageJ software (1.48v; http://rsb.info.nih.gov/ij/index.html).
ELISA procedures.
Medium-binding 96-well ELISA plates (Greiner, Nürtingen, Germany) were coated with all fractions (0.1 μg/well) overnight at 4°C and blocked with bovine serum albumin (BSA) in PBS. Plates were stored at 4°C until use. Samples were diluted 1:100 in sample dilution buffer (NovaTec Immundiagnostica) prior to application onto the ELISA plate and incubated for 1 h at 37°C, followed by 30 min of incubation at room temperature with NovaLisa antibody conjugate (NovaTec Immundiagnostica), which was also used for the line blots. After each incubation step, the wells were washed three times with washing buffer (NovaTec Immundiagnostica). Development was done with 100 μl TMB for 15 min and stopped by adding the same amount of stop solution (NovaTec Immundiagnostica). The results were measured at 450 nm in a plate reader (Anthos Labtec Instruments, Wals-Siezenheim, Austria). Values of strongly positive samples above an optical density of 3.0 (the limit of the device) were set to 3.0. All measurements were performed in duplicate.
Western blot analysis.
Proteins for Western blot analysis were concentrated and desalted by chloroform-methanol precipitation (28) and dissolved directly in SDS sample buffer after drying. SDS-PAGE was performed in a 12% acrylamide gel. To obtain a consistent gel for the Western blots, the sample was loaded in one 7.4-cm-wide pocket next to the prestained protein marker (AppliChem GmbH, Darmstadt, Germany) and separated for 30 min at 60 V. Proteins were transferred via semidry blotting to a nitrocellulose membrane for 60 min with 1 mA/cm2 and cut in 3-mm strips. The outer lines at the side of the blot were not used for Western blots. Further treatment was performed as described for the line blots above.
Statistics.
All experiments were performed at least two times. As variances within the groups were not normally distributed according to the Shapiro-Wilk normality test, differences between experimental and control groups were analyzed by an unpaired t test with Welch's correlation. A P value of <0.05 was considered statistically significant. Ratios were defined as the quotient of the mean value for patient sera and the mean value for control sera. Differences were defined as the lowest value (e.g., integrated image density) for patient sera minus the highest value for control sera.
RESULTS
Determination of B. henselae protein composition for ELISA-based serodiagnostics.
There is no consensus about which particular B. henselae strain should be used for serodiagnostics of B. henselae infections. Therefore, two different strains (B. henselae Houston-1 and B. henselae Marseille) were evaluated in parallel in the first steps for identifying a functional ELISA antigen composition. An antigen purification strategy allowing for later integration into an industrial manufacturing process of ELISAs was chosen, and both water-soluble and water-insoluble protein compositions were enriched. Crude extracts were separated into water-soluble and water-insoluble fractions and coated on ELISA plates. In a first step, binding of anti-B. henselae-directed antibodies was analyzed by using highly IFA-positive (≥1:1,280) and IFA-negative (<1:80) sera, and the respective ELISA (Fig. 1) demonstrated for both strains that the water-soluble protein composition revealed a better discrimination between IFA-positive and -negative sera than did that of the water-insoluble proteins (B. henselae Houston-1 ratio, 7.88 versus 5.69; B. henselae Marseille ratio, 13.7 versus 4.27, respectively).
FIG 1.
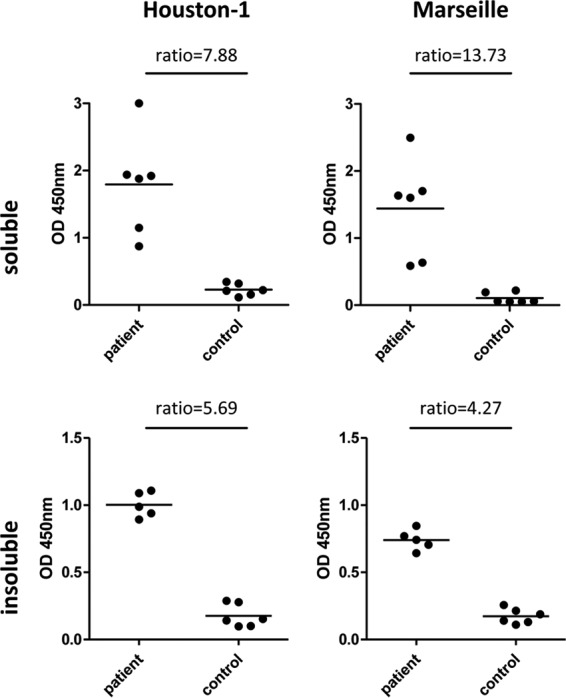
Optical density results of ELISAs (human sera) using crude extracts from different B. henselae strains. Crude protein extracts were prepared from B. henselae Houston-1 and B. henselae Marseille as described in Materials and Methods. ELISA plates were coated with 1.0 μg/ml protein extract. ELISAs were performed with samples of highly positive sera (IFA > 1:1,000) and IFA-negative control sera (each, n = 6). Upper row, PBS-insoluble fraction; lower row, PBS-soluble fraction. Bars represent the mean values.
Selection of a functional protein composition for anti-B. henselae ELISA.
For optimization of the presumptive Bartonella ELISA antigen, the water-soluble proteins of B. henselae Marseille and the water-insoluble proteins of B. henselae Houston-1 were used for further antigen processing by IEX and subsequent line blot analysis. The fractioning and line blot analysis of B. henselae Marseille and, later on, of single fractions by ELISA did not result in an increased performance compared with that of crude protein preparations (data not shown) and was therefore not followed up. In contrast, it was possible to separate the water-insoluble fraction of B. henselae Houston-1 by IEX into 40 subfractions representing the peak areas for analysis of immunoreactivity with IFA-positive human sera. Fractions with no peak (fractions 1 to 5 and 13 to 18) were collected but not printed on line blots. Fractions 6 to 12 and 19 to 26 were printed on line blots and evaluated for seroreactivity. Fractions 27 to 40 showed only minor reactivity in line blots and were not further analyzed (data not shown). Figure 2A shows the chromatogram obtained by IEX from the water-insoluble fraction of B. henselae Houston-1. Only peak-containing fractions were used for subsequent line blot analysis (Fig. 2B). From these fractions, fraction 11 had the highest ratio between IFA-positive and control sera, whereas fraction 24 showed the highest difference, discriminating the most weakly reacting IFA-positive serum from the most strongly reacting IFA-negative serum.
FIG 2.

Ion-exchange chromatography (IEX) of the insoluble fraction of B. henselae Houston-1 and line blot analysis of the respective fractions by using human reactive sera. (A) Chromatogram of the IEX with the PBS-insoluble antigens from B. henselae Houston-1. Shown are the UV signal (solid line) and the gradient for elution (dotted line). Arrows indicate fraction 11 and fraction 24. (B) Line blots with fractions collected after the IEX. Blots were incubated either with IFA-positive patient sera (n = 7, left) or with IFA-negative sera (n = 6, right). The row with the best ratio (fraction 11) and with the best difference (fraction 24) are marked with arrows. (C) Visualization of the reactivity of the line blots with human sera by heat maps. Values are shown ranging from no binding (green) to maximum binding (red). Numbers at the side indicate the number of the particular IEX fraction (ce, crude extract).
Next, ELISA plates were coated with fractions 11 and 24 and tested with IFA-positive and IFA-negative sera. Results were compared with data from ELISA plates coated with the crude insoluble antigens from B. henselae Houston-1, which were tested with the same sera (Fig. 3). Fractions 11 and 24 showed a significantly improved discrimination of IFA-positive and IFA-negative sera in an ELISA format (fraction 11, P = 0.0088; fraction 24, P < 0.0001) compared to that of the crude antigen preparation (P = 0.0320). Based on these findings, it was concluded that IEX fraction 24 from the water-insoluble fraction of B. henselae Houston-1 harbors the most promising protein composition for further ELISA development.
FIG 3.

Immunoreactivity of IEX fractions 11 and 24 compared to that of crude extracts from B. henselae Houston-1. Antigens were prepared as crude extracts (see Materials and Methods) and purified by IEX, resulting in fractions 11 and 24 (see Fig. 2). ELISA results above the range (OD > 3.0) were set to 3.0. ODs obtained with patient sera (IFA positive) were compared to ODs from control samples (IFA negative). P values between IFA-positive and IFA-negative samples are given within the figure.
ELISA performance characteristics of the IEX fractions.
To analyze the specificities and sensitivities of fractions 11 and 24, a receiver operating characteristic curve (ROC) analysis was performed. The results for ELISA plates coated with the antigen compositions of the insoluble part of B. henselae Houston-1 fraction 11, fraction 24, and also the crude extract are depicted in Fig. 4. A ROC analysis was used to adjust the cutoff at an optical density (OD) of 0.5 and revealed that fraction 11 shows a sensitivity and specificity of 85% and 93%, respectively, and that fraction 24 shows a sensitivity and specificity of 100% (for PCR-proven infections) and 93%, respectively. In contrast, the crude protein extract of the water-insoluble protein fraction showed only a sensitivity and specificity of 85% and 86%, respectively. Thus, fraction 24 from the water-insoluble protein composition of B. henselae Houston-1 seems to represent the most suitable B. henselae ELISA antigen composition.
FIG 4.
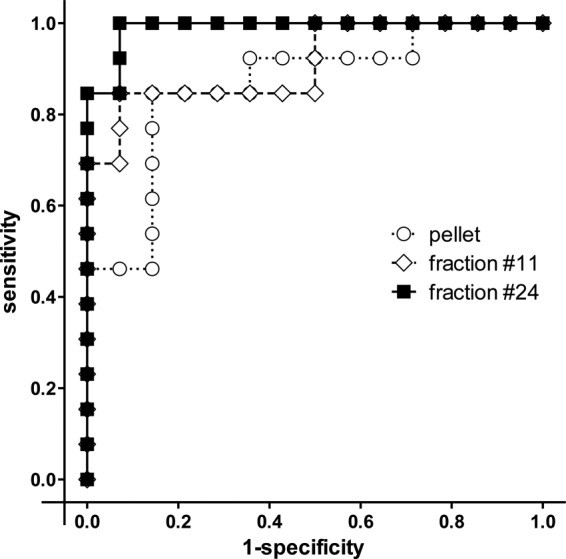
Receiver operating characteristic (ROC) curve analysis of B. henselae Houston-1 pellet and fractions 11 and 24. Results of ELISAs using three different antigen compositions derived from B. henselae Houston-1 (crude pellet, fraction 11, and fraction 24) are shown. IFA-positive (n = 13) and IFA-negative (n = 14) sera were tested on three different ELISA plates. Values are depicted in a ROC diagram according to sensitivity (true-positive rate) and 1 − specificity (false-positive rate). Curves represent the PBS-insoluble crude pellet (dotted line, white circles), fraction 11 (dashed line, white diamonds), and fraction 24 (solid line, black squares).
In the next step, the composition of fraction 24 harboring immunodominant proteins was explored in greater detail. Proteins of fraction 24 were separated by SDS-PAGE, and Western blot analysis was performed with IFA-positive and IFA-negative human sera (Fig. 5), revealing immunoreactive bands at ∼17 kDa (IFA positive, n = 6/8; IFA negative, n = 1/8), 35 kDa (IFA positive, n = 8/8; IFA negative, n = 0/8), and 60 kDa (IFA positive, n = 6/8; IFA negative, n = 0/8). Of the 8 IFA-positive human sera, 4 sera (sera 3, 4, 5, and 6) reacted with all three bands and the remaining 4 human sera (sera 1, 2, 7, and 8) reacted with two of these three bands. Although there are particular recurring bands appearing at these protein sizes, the serum of each patient reacted individually to the antigens of fraction 24. In total, ∼17 protein bands (sizes between 10 and 130 kDa) seem to react in any of the eight tested IFA-positive sera. Generally, in the blots from the IFA-negative control sera, virtually no bands were visible.
FIG 5.

(A) Determination of immunogenic proteins within IEX fraction 24. (A) Proteins of fraction 24 were separated using a 12% denaturing SDS-PAGE gel and blotted onto nitrocellulose membranes. Western blots were incubated either with IFA-positive patient sera (left) or with IFA-negative sera (right) (each, n = 8). (B) Visualization of the reactivity of the Western blots with human sera by heat maps. Values are shown ranging from no binding (green) to maximum binding (red). Categorized areas of protein size are at 17, 19, 25, 33, 45, 59, 74, and 140 kDa.
Performance of the anti-B. henselae fraction 24 ELISA with human sera.
To exclude any influence of the industrially used protein A/G-based conjugate in comparison to anti-human IgG (used in conventional IFA testing), the results of the ELISA were compared using both detection systems. Fraction 24-coated ELISA plates (0,1 μg/well) were incubated with human sera (n = 33) with various IFA titers and developed in parallel using protein A/G conjugate or anti-human IgG. Analysis of the resulting ODs revealed a strong correlation of the two methods (r2 = 0.98) (Fig. 6), demonstrating that the protein A/G conjugate (compared with anti-human IgG) is an appropriate conjugate suitable for industrial use.
FIG 6.
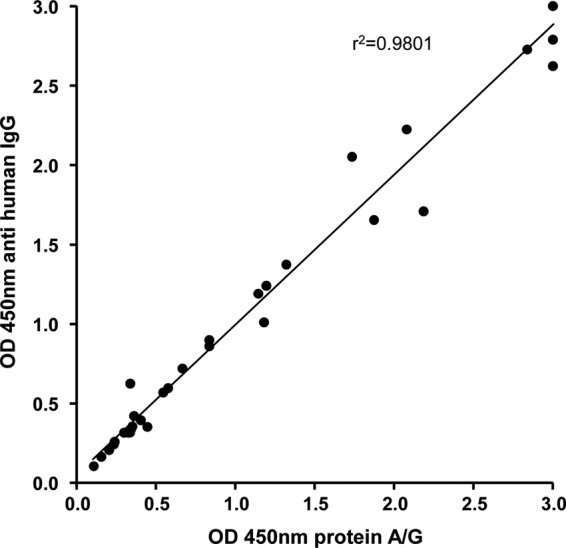
Comparison of the industrially used protein A/G conjugates and anti-human IgG antibodies in the fraction 24 ELISA. A spectrum of IFA-positive and IFA-negative sera (n = 33) were tested in parallel with the B. henselae ELISA plate coated with IEX fraction 24 and the conjugate protein A/G and anti-human IgG antibodies. Optical densities of each sample were plotted in the diagram according to the respective conjugate. A line represents the linear regression of all values.
Finally, the performance of the herein described ELISA was evaluated. For this purpose, fraction 24-coated ELISA plates (repeated with three independently produced IEX preparations) were used to analyze human sera for anti-B. henselae antibodies. Human sera were categorized into five different groups: (i) IFA-positive sera from patients suffering from a PCR-proven infection (n = 10), (ii) IFA-positive sera from patients with a CSD-specific medical history (n = 21), (iii) IFA-positive sera without a CSD-specific medical history (n = 12), (iv) IFA-positive sera with a low (1:320) IFA titer (n = 20), and (v) IFA-negative sera (titer, ≤1:80) (n = 16) (Fig. 7). Remarkably, all samples from patients with a PCR-proven Bartonella infection were highly positive in the fraction 24 ELISA (n = 10, 100%), while 16 (76%) of 21 IFA-positive serum samples from patients with a medically validated Bartonella infection were positive. In the group of IFA-positive sera from patients without a documented medical history and no PCR confirmation of the infection, the ELISA detected 10 of the 12 samples (83%) as positive, while 1 was equivocal (8%). When human sera were analyzed from patients without a documented CSD-specific medical history and no PCR confirmation of the infection but with the lowest IFA titer to be categorized as positive (1:320), 30% (n = 6/20) were also rated positive in the ELISA, whereas 25% (n = 5/20) were equivocally close to the cutoff and the remaining 45% (n = 9/20) were negative. From the group of IFA-negative samples, 1 out of 16 samples was positive (6%) in the ELISA and 1 was equivocal. Analysis of IFA titers (over a titer spectrum of 1:80 up to 1:20,480) revealed a strong correlation (r2 = 0.92) with the respective ODs (Fig. 8).
FIG 7.
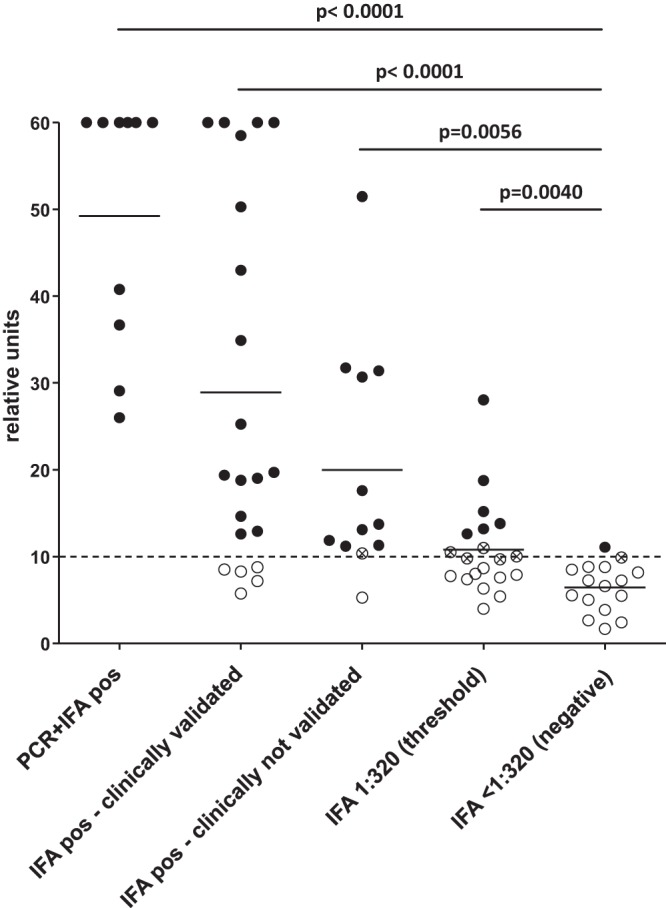
Performance of the B. henselae Houston-1 IEX fraction 24 ELISA with human sera (ntotal = 79). Results of the ELISA are grouped for IFA-positive sera from patients suffering from a PCR-proven infection (n = 10), IFA-positive sera from patients with a CSD-specific medical history (n = 21), IFA-positive sera from patients without a CSD-specific clinical history (n = 12), IFA-positive sera with a low (1:320) IFA titer (n = 20), and IFA-negative (<1:80) sera (n = 16). Values above the range are set to 60 relative units (RU). Bars represent the mean value. Values above 11 RU are considered positive (black circles), and values below 9 RU are considered negative (white circles). Values between 9 and 11 RU are equivocal (white circles with an X). Respective P values between sample categories are given.
FIG 8.
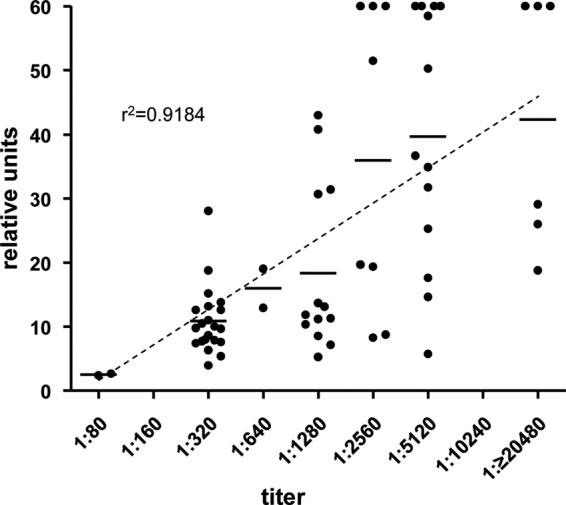
Correlation of human serum IFA titers with ELISA ODs of the B. henselae Houston-1 IEX fraction 24 (ntotal = 65). Results are grouped according to the IFA titers (x axis) and relative OD units [(ODsample/ODcutoff) × 10]. Bars represent mean values. A dashed line represents the linear regression of the mean values.
DISCUSSION
In this study, we established an anti-B. henselae antibody ELISA suitable for large-scale serological laboratory diagnosis of human B. henselae infections. By nature, the widely used IFA is highly laborious, its interpretation is subjective, and the throughput is small. Therefore, an ELISA represents a significant step forward in improving the serological diagnosis of B. henselae infections.
Generally, numerous different IFAs are applied in diagnostic laboratories, providing a simple and reliable method to detect antibodies against a wide variety of pathogens (e.g., viruses, intracellular bacteria, etc.). Since its first description in 1992 (16), the serodiagnosis of B. henselae infections based on IFA has not changed much and the protocol is accepted worldwide. An improved protocol for the generation of B. henselae IFA using cell culture-derived antigen was distributed by the CDC (10), and B. henselae-infected Vero cells turned out to be the most feasible antigen since both the sensitivity (∼90%) and the specificity (∼95%) of IFA testing were higher than those for agar-grown bacteria (9, 16). However, the production of B. henselae IFA antigen is complicated: quality-controlled cell cultures (originally using Vero cells [10]) have to be infected with agar-grown B. henselae (the exact cultivation period on agar is not known). Next, the respective multiplicity of infection (MOI) of the cell culture flask-cultivated Vero cells is not exactly defined, and this MOI is also difficult to be determined in the laboratory. In addition, infected cell cultures need to be cultivated for 24 to 72 h without antibiotics (to allow the growth of the intracellular B. henselae bacteria), resulting in potential contamination problems. Finally, this coculture antigen of bacteria and cells has to be harvested, spotted onto glass slides, and fixed to be stable and to avoid laboratory infections; again, the exact fixation protocol is not clearly defined. So, antigen preparation by cell coculture for B. henselae is naturally a difficult task in an industrial antigen production process.
Two different liquid media for cultivation of Bartonella spp. were described some years ago: the so-called BAPGM medium (29), which contains ∼20 different ingredients, and the so-called BaLi medium (27), which has only three ingredients (Schneider′s insect cell culture medium, 10% heat-inactivated fetal calf serum, 5% sucrose). Since BaLi medium is much easier to prepare than BAPGM medium and it has already been shown that cultivation of B. henselae in BaLi medium does not influence the bacterial antigen composition (27), we used this medium for antigen preparation for the ELISA described herein. Compared to cell cultures, liquid B. henselae cultivation allows the production of large amounts of antigen with reproducible quality in a short time (∼72 h). Antigen production with liquid medium is therefore technically easier, avoids background signals in fluorescence microscopy arising from the reaction of autoimmunological antibodies with the cocultivated host cells (e.g., from antinuclear antibodies appearing, e.g., as nuclear dots that look similar to the intracellular perinuclear B. henselae bacteria [30]), and is, moreover, much cheaper than expensive cell cultures.
Previous B. henselae ELISAs suffered from poor sensitivity (36% to 71%) when the specificity was set to sufficient levels (≥93%) and, therefore, failed to achieve a performance that was competitive with that of IFA testing (5, 15, 17). This precluded the launch of commercially available ELISAs for the detection of B. henselae antibodies until the development of the ELISA described in our study. Moreover, the arbitrary selection of single antigens, excluding all other possibly immunogenic proteins from the first setup of such assays (e.g., 17-kDa antigen [17, 31]), and, additionally, the expression of these proteins in Escherichia coli, possibly resulting in unknown posttranslational modifications (affecting the immunogenicity of these proteins), are further potential pitfalls in the selection of an appropriate antigen for anti-B. henselae-specific ELISAs. In our opinion, the restriction to one or two particular B. henselae proteins is a conceptual mistake in the development of an ELISA, as it has been described several times that sera of B. henselae-infected patients react with a broad variety of different antigens with a high interpatient variability (32–34). In the case of IgM, no clear recommendation is given in the original CDC protocols (10), and because of this, a validated cutoff limiting the use of IgM ELISAs does not exist.
We decided to use liquid-grown B. henselae Houston-1 for antigen preparation. This decision was made based on the facts that (i) the water-insoluble fraction of this strain was superior to the Marseille strain in initial experiments (Fig. 1) and (ii) this strain is used for the production of commercially available B. henselae IFA antigens. Therefore, B. henselae Houston-1 antigen in ELISA-based serology should be as similar as possible to current IFA antigens, allowing a direct comparison of the performance of these two technologies. Moreover, since we and others were able to demonstrate that in IFA-positive sera from B. henselae-infected patients, no clear discriminatory bands are present in Western blots (32–34), we did not rely on an arbitrary selection of antigens but identified a complex and reproducible protein composition from the water-insoluble pellet of liquid-grown B. henselae. All of these careful considerations resulted in an industrially applicable antigen preparation able to be used in large-scale ELISA production. Finally, the composition of the ELISA antigen used herein from the water-insoluble compartment of B. henselae should include many different B. henselae proteins (as well as membrane proteins). Currently, we do not know the particular protein composition of fraction 24. As can be seen in Fig. 5, at least 13 different immunoreactive proteins are present (Fig. 5A, positive samples, lane 5). It will be a future task to analyze the composition of fraction 24 in detail, e.g., via mass spectrometry. In the interim, the ELISA described in this study can be used for diagnostic testing and larger epidemiological studies to further clarify the medical importance of B. henselae.
The ELISA described herein has a sensitivity of 100% when using sera of PCR-proven patients and of 76% when using IFA-positive sera from patients with a clinical history suspicious of a B. henselae infection (Fig. 7). It is important to realize that a clinical history of lymphadenitis is per se not specific for a B. henselae infection (e.g., CSD) and neither are positive IFA results specific for B. henselae antibodies, as many IFA cross-reactivities with lymphadenitis-causing pathogens are known (e.g., Bartonella quintana, Coxiella burnetii [35], Chlamydophila pneumoniae [36], Ehrlichia chaffeensis, Mycoplasma pneumoniae, E. coli, Rickettsia spp., and Treponema pallidum [37], and Bordetella pertussis and Borrelia spp. [38]). Therefore, we think that a correct performance evaluation of our ELISA is possible only by restricting the test to PCR-proven serum samples or samples from patients with a conclusive clinical history. This restriction results in 100% sensitivity and 93% specificity, which are the best known performance data of a B. henselae ELISA. When testing 108 human sera for cross-reactivity (data not shown in detail), we found 13 sera to be cross-reactive in our ELISA (B. quintana, n = 1/1; Brucella spp., n = 4/7; Chlamydophila pneumoniae, n = 2/16; Coxiella burnetii, n = 0/10; Epstein-Barr virus, n = 0/13; Mycoplasma pneumoniae, n = 0/26; Treponema pallidum, n = 1/11; Rickettsia spp., n = 5/11; rheumatoid factor, n = 0/8; and antinuclear antibodies, n = 0/5) that were previously tested by certified routine laboratory methods. Since cross-reactivity is a difficult-to-quantify problem in infection serology, the use of certified sera (as done here) results in only a rough estimation of cross-reactivities for our ELISA. It is obvious that the given (and many other potential) cross-reactivities must be evaluated broadly later. It must also be stated that in the group of sera at the anti-B. henselae-positive threshold (IFA titer, 1:320), five sera were evaluated as equivocal, six sera as positive, and nine sera as negative in the ELISA. The reason for this heterogenous reactivity in the ELISA is not clear, but it might be suggested that for sera marginally reaching an IFA threshold titer of 1:320, some of those sera led to a positive IFA result only by cross-reactivity. In particular, these sera were not from PCR-proven infections or even from patients with a clinically suspected B. henselae infection.
The use of the industrially suitable protein A/G as a conjugate did not differ in its performance in the ELISA in comparison to the anti-human IgG. The use of protein A/G conjugate allows the manufacturing of ELISAs in a building-block strategy, which is often used in diagnostic companies to be able to exchange antigens or detection systems. As already mentioned, there are no clear cutoff values for anti-B. henselae IgM (10), and, moreover, detection of IgM is only of limited use in B. henselae infections, which are normally long-lasting. From this, we do not see a medical need to establish IgM-specific ELISAs. However, as the protein A/G conjugate detects IgM simultaneously with IgG when present in the serum sample, it is, however, included in the serological results achieved by the herein-described ELISA.
Taken together, the ELISA described herein is the first ELISA with a sufficient sensitivity (100% for PCR-proven samples and 76% for clinically suspected samples) and a high specificity (93%). Compared with cell cultures, the use of liquid media and IEX is easy to integrate into an industrial manufacturing process. In diagnostic laboratories, such an ELISA allows the automated, economical, and objective analysis of serum samples for B. henselae antibodies. From a scientific perspective, a B. henselae ELISA will allow high-throughput seroepidemiological analyses, which were highly laborious when using IFA-based technology (13). Finally, our ELISA might also be modified for later use with other Bartonella species or in the analysis of veterinary serum samples.
ACKNOWLEDGMENTS
We thank Corinna Sonntag, Rebecca Kaufmann, Heike Podlich, Agnes Hillebrecht, and Carmen Jung (University Hospital Frankfurt, Germany) and Elif Schluefter, Jennifer Voelger, Elke Heck, and Tina Latz (NovaTec Immundiagnostica, Dietzenbach, Germany) for expert technical assistance; we also thank Nadine Osorio-Villazan and Dirk Saeuberlich (Hessenagentur, Wiesbaden, Germany) for excellent management support.
This project (HA project no. 490/16-02) was funded in the framework of Hessen ModelProjekte, financed with funds from LOEWE—Landes-Offensive zur Entwicklung Wissenschaftlich-ökonomischer Exzellenz, Förderlinie 3: KMU-Verbundvorhaben (State Offensive for the Development of Scientific and Economic Excellence) and by the Robert Koch Institute, Berlin, Germany (Bartonella Consiliary Laboratory; grant 1369-354).
REFERENCES
- 1.Anderson BE, Neuman MA. 1997. Bartonella spp. as emerging human pathogens. Clin Microbiol Rev 10:203–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaiser PO, Riess T, O'Rourke F, Linke D, Kempf VA. 2011. Bartonella spp.: throwing light on uncommon human infections. Int J Med Microbiol 301:7–15. doi: 10.1016/j.ijmm.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 3.Telford SR III, Wormser GP. 2010. Bartonella spp. transmission by ticks not established. Emerg Infect Dis 16:379–384. doi: 10.3201/eid1603.090443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Regier Y, Ballhorn W, Kempf VA. 2017. Molecular detection of Bartonella henselae in 11 Ixodes ricinus ticks extracted from a single cat. Parasit Vectors 10:105. doi: 10.1186/s13071-017-2042-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Herremans M, Bakker J, Vermeulen MJ, Schellekens JF, Koopmans MP. 2009. Evaluation of an in-house cat scratch disease IgM ELISA to detect Bartonella henselae in a routine laboratory setting. Eur J Clin Microbiol Infect Dis 28:147–152. doi: 10.1007/s10096-008-0601-8. [DOI] [PubMed] [Google Scholar]
- 6.Carithers HA. 1985. Cat-scratch disease. An overview based on a study of 1,200 patients. Am J Dis Child 139:1124–1133. [DOI] [PubMed] [Google Scholar]
- 7.Zangwill KM, Hamilton DH, Perkins BA, Regnery RL, Plikaytis BD, Hadler JL, Cartter ML, Wenger JD. 1993. Cat scratch disease in Connecticut. Epidemiology, risk factors, and evaluation of a new diagnostic test. N Engl J Med 329:8–13. [DOI] [PubMed] [Google Scholar]
- 8.Jacobs RF, Schutze GE. 1998. Bartonella henselae as a cause of prolonged fever and fever of unknown origin in children. Clin Infect Dis 26:80–84. doi: 10.1086/516256. [DOI] [PubMed] [Google Scholar]
- 9.Fischer SF, Al Dahouk S, Brandt C, Donoso-Mantke O, Frangoulidis D, Hofmann J, Jansen A, Krüger DH, Kimmig P, Kurth A, Niedrig M, Nitsche A, Nöckler K, Roggendorf M, Ross RS, Schacht E, Mayer-Scholl A, Scholz HC, Splettstoesser WD, Stark K, Wagner-Wiening C, Zöller L, Kempf VAJ. 2012. MiQ 33: Zoonosen, 1st ed Urban & Fischer Verlag, Munich, Germany. [Google Scholar]
- 10.Centers for Disease Control and Prevention. 1999. Serodiagnosis of emerging infectious diseases: Bartonella and Ehrlichia infections (course manual). Centers for Disease Control and Prevention, Atlanta, GA, USA. [Google Scholar]
- 11.Ridder GJ, Boedeker CC, Technau-Ihling K, Grunow R, Sander A. 2002. Role of cat-scratch disease in lymphadenopathy in the head and neck. Clin Infect Dis 35:643–649. doi: 10.1086/342058. [DOI] [PubMed] [Google Scholar]
- 12.Ridder GJ, Boedeker CC, Technau-Ihling K, Sander A. 2005. Cat-scratch disease: otolaryngologic manifestations and management. Otolaryngol Head Neck Surg 132:353–358. doi: 10.1016/j.otohns.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 13.Jurke A, Bannert N, Brehm K, Fingerle V, Kempf VA, Kompf D, Lunemann M, Mayer-Scholl A, Niedrig M, Nockler K, Scholz H, Splettstoesser W, Tappe D, Fischer SF. 2015. Serological survey of Bartonella spp., Borrelia burgdorferi, Brucella spp., Coxiella burnetii, Francisella tularensis, Leptospira spp., Echinococcus, Hanta-, TBE- and XMR-virus infection in employees of two forestry enterprises in North Rhine-Westphalia, Germany, 2011-2013. Int J Med Microbiol 305:652–662. doi: 10.1016/j.ijmm.2015.08.015. [DOI] [PubMed] [Google Scholar]
- 14.Nelson CA, Saha S, Mead PS. 2016. Cat-scratch disease in the United States, 2005-2013. Emerg Infect Dis 22:1741–1746. doi: 10.3201/eid2210.160115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bergmans AM, Peeters MF, Schellekens JF, Vos MC, Sabbe LJ, Ossewaarde JM, Verbakel H, Hooft HJ, Schouls LM. 1997. Pitfalls and fallacies of cat scratch disease serology: evaluation of Bartonella henselae-based indirect fluorescence assay and enzyme-linked immunoassay. J Clin Microbiol 35:1931–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Regnery RL, Olson JG, Perkins BA, Bibb W. 1992. Serological response to “Rochalimaea henselae” antigen in suspected cat-scratch disease. Lancet 339:1443–1445. doi: 10.1016/0140-6736(92)92032-B. [DOI] [PubMed] [Google Scholar]
- 17.Loa CC, Mordechai E, Tilton RC, Adelson ME. 2006. Production of recombinant Bartonella henselae 17-kDa protein for antibody-capture enzyme-linked immunosorbent assay. Diagn Microbiol Infect Dis 55:1–7. doi: 10.1016/j.diagmicrobio.2005.10.020. [DOI] [PubMed] [Google Scholar]
- 18.Tsuruoka K, Tsuneoka H, Kawano M, Yanagihara M, Nojima J, Tanaka T, Yamamoto M, Ichihara K. 2012. Evaluation of IgG ELISA using N-lauroyl-sarcosine-soluble proteins of Bartonella henselae for highly specific serodiagnosis of cat scratch disease. Diagn Microbiol Infect Dis 74:230–235. doi: 10.1016/j.diagmicrobio.2012.06.028. [DOI] [PubMed] [Google Scholar]
- 19.Litwin CM, Martins TB, Hill HR. 1997. Immunologic response to Bartonella henselae as determined by enzyme immunoassay and Western blot analysis. Am J Clin Pathol 108:202–209. doi: 10.1093/ajcp/108.2.202. [DOI] [PubMed] [Google Scholar]
- 20.Giladi M, Kletter Y, Avidor B, Metzkor-Cotter E, Varon M, Golan Y, Weinberg M, Riklis I, Ephros M, Slater L. 2001. Enzyme immunoassay for the diagnosis of cat-scratch disease defined by polymerase chain reaction. Clin Infect Dis 33:1852–1858. doi: 10.1086/324162. [DOI] [PubMed] [Google Scholar]
- 21.Dauga C, Miras I, Grimont PA. 1996. Identification of Bartonella henselae and B. quintana 16s rDNA sequences by branch-, genus- and species-specific amplification. J Med Microbiol 45:192–199. doi: 10.1099/00222615-45-3-192. [DOI] [PubMed] [Google Scholar]
- 22.Riess T, Andersson SG, Lupas A, Schaller M, Schäfer A, Kyme P, Martin J, Wälzlein JH, Ehehalt U, Lindroos H, Schirle M, Nordheim A, Autenrieth IB, Kempf VA. 2004. Bartonella adhesin a mediates a proangiogenic host cell response. J Exp Med 200:1267–1278. doi: 10.1084/jem.20040500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maggi RG, Breitschwerdt EB. 2005. Potential limitations of the 16S-23S rRNA intergenic region for molecular detection of Bartonella species. J Clin Microbiol 43:1171–1176. doi: 10.1128/JCM.43.3.1171-1176.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alsmark CM, Frank AC, Karlberg EO, Legault BA, Ardell DH, Canback B, Eriksson AS, Naslund AK, Handley SA, Huvet M, La Scola B, Holmberg M, Andersson SG. 2004. The louse-borne human pathogen Bartonella quintana is a genomic derivative of the zoonotic agent Bartonella henselae. Proc Natl Acad Sci U S A 101:9716–9721. doi: 10.1073/pnas.0305659101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Drancourt M, Birtles R, Chaumentin G, Vandenesch F, Etienne J, Raoult D. 1996. New serotype of Bartonella henselae in endocarditis and cat-scratch disease. Lancet 347:441–443. doi: 10.1016/S0140-6736(96)90012-4. [DOI] [PubMed] [Google Scholar]
- 26.Lu YY, Franz B, Truttmann MC, Riess T, Gay-Fraret J, Faustmann M, Kempf VA, Dehio C. 2013. Bartonella henselae trimeric autotransporter adhesin BadA expression interferes with effector translocation by the VirB/D4 type IV secretion system. Cell Microbiol 15:759–778. doi: 10.1111/cmi.12070. [DOI] [PubMed] [Google Scholar]
- 27.Riess T, Dietrich F, Schmidt KV, Kaiser PO, Schwarz H, Schäfer A, Kempf VA. 2008. Analysis of a novel insect cell culture medium-based growth medium for Bartonella species. Appl Environ Microbiol 74:5224–5227. doi: 10.1128/AEM.00621-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wessel D, Flugge UI. 1984. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal Biochem 138:141–143. doi: 10.1016/0003-2697(84)90782-6. [DOI] [PubMed] [Google Scholar]
- 29.Maggi RG, Duncan AW, Breitschwerdt EB. 2005. Novel chemically modified liquid medium that will support the growth of seven Bartonella species. J Clin Microbiol 43:2651–2655. doi: 10.1128/JCM.43.6.2651-2655.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kempf VA, Schaller M, Behrendt S, Volkmann B, Aepfelbacher M, Cakman I, Autenrieth IB. 2000. Interaction of Bartonella henselae with endothelial cells results in rapid bacterial rRNA synthesis and replication. Cell Microbiol 2:431–441. doi: 10.1046/j.1462-5822.2000.00072.x. [DOI] [PubMed] [Google Scholar]
- 31.Ferrara F, Di Niro R, D'Angelo S, Busetti M, Marzari R, Not T, Sblattero D. 2014. Development of an enzyme-linked immunosorbent assay for Bartonella henselae infection detection. Lett Appl Microbiol 59:253–262. doi: 10.1111/lam.12286. [DOI] [PubMed] [Google Scholar]
- 32.Eberhardt C, Engelmann S, Kusch H, Albrecht D, Hecker M, Autenrieth IB, Kempf VA. 2009. Proteomic analysis of the bacterial pathogen Bartonella henselae and identification of immunogenic proteins for serodiagnosis. Proteomics 9:1967–1981. doi: 10.1002/pmic.200700670. [DOI] [PubMed] [Google Scholar]
- 33.McCool TL, Hoey JG, Montileone F, Goldenberg HB, Mordechai E, Adelson ME. 2008. Discovery and analysis of Bartonella henselae antigens for use in clinical serologic assays. Diagn Microbiol Infect Dis 60:17–23. doi: 10.1016/j.diagmicrobio.2007.07.017. [DOI] [PubMed] [Google Scholar]
- 34.Wagner CL, Riess T, Linke D, Eberhardt C, Schäfer A, Reutter S, Maggi RG, Kempf VA. 2008. Use of Bartonella adhesin A (BadA) immunoblotting in the serodiagnosis of Bartonella henselae infections. Int J Med Microbiol 298:579–590. doi: 10.1016/j.ijmm.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 35.La Scola B, Raoult D. 1996. Serological cross-reactions between Bartonella quintana, Bartonella henselae, and Coxiella burnetii. J Clin Microbiol 34:2270–2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maurin M, Eb F, Etienne J, Raoult D. 1997. Serological cross-reactions between Bartonella and Chlamydia species: implications for diagnosis. J Clin Microbiol 35:2283–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McGill SL, Regnery RL, Karem KL. 1998. Characterization of human immunoglobulin (Ig) isotype and IgG subclass response to Bartonella henselae infection. Infect Immun 66:5915–5920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McGill SL, Friman G, Wesslen L, Hjelm E. 2005. Broad spectrum analysis of Bartonella intra- and intergenus cross reactivity by immunoblot and immunofluorescence assay, p 46. Fourth Int Congr Bartonella Emerging Pathog, Uppsala, Sweden, 26–28 August 2005. [Google Scholar]