

Human T-cell leukemia virus type 1 (HTLV-1) is the pathogen that causes adult T-cell leukemia (ATL), which is a unique malignancy of CD4+ T cells. A role for long noncoding RNA (lncRNA) in HTLV-1-mediated cellular transformation has not been described. In this study, we demonstrated that the lncRNA ANRIL was important for maintaining the proliferation of ATL cells in vitro and in vivo. ANRIL was shown to activate NF-κB signaling through forming a ternary complex with EZH2 and p65. Furthermore, epigenetic inactivation of p21/CDKN1A was involved in the oncogenic function of ANRIL. To the best of our knowledge, this is the first study to address the regulatory role of the lncRNA ANRIL in ATL and provides an important clue to prevent or treat HTLV-1-associated human diseases.
KEYWORDS: ATL, HTLV-1, ANRIL, EZH2, NF-κB, adult T-cell leukemia, human T-cell leukemia virus
ABSTRACT
Adult T-cell leukemia (ATL) is a highly aggressive T-cell malignancy induced by human T-cell leukemia virus type 1 (HTLV-1) infection. Long noncoding RNA (lncRNA) plays a critical role in the development and progression of multiple human cancers. However, the function of lncRNA in HTLV-1-induced oncogenesis has not been elucidated. In the present study, we show that the expression level of the lncRNA ANRIL was elevated in HTLV-1-infected cell lines and clinical ATL samples. E2F1 induced ANRIL transcription by enhancing its promoter activity. Knockdown of ANRIL in ATL cells repressed cellular proliferation and increased apoptosis in vitro and in vivo. As a mechanism for these actions, we found that ANRIL targeted EZH2 and activated the NF-κB pathway in ATL cells. This activation was independent of the histone methyltransferase (HMT) activity of EZH2 but required the formation of an ANRIL/EZH2/p65 ternary complex. A chromatin immunoprecipitation assay revealed that ANRIL/EZH2 enhanced p65 DNA binding capability. In addition, we observed that the ANRIL/EZH2 complex repressed p21/CDKN1A transcription through H3K27 trimethylation of the p21/CDKN1A promoter. Taken together, our results implicate that the lncRNA ANRIL, by cooperating with EZH2, supports the proliferation of HTLV-1-infected cells, which is thought to be critical for oncogenesis.
IMPORTANCE Human T-cell leukemia virus type 1 (HTLV-1) is the pathogen that causes adult T-cell leukemia (ATL), which is a unique malignancy of CD4+ T cells. A role for long noncoding RNA (lncRNA) in HTLV-1-mediated cellular transformation has not been described. In this study, we demonstrated that the lncRNA ANRIL was important for maintaining the proliferation of ATL cells in vitro and in vivo. ANRIL was shown to activate NF-κB signaling through forming a ternary complex with EZH2 and p65. Furthermore, epigenetic inactivation of p21/CDKN1A was involved in the oncogenic function of ANRIL. To the best of our knowledge, this is the first study to address the regulatory role of the lncRNA ANRIL in ATL and provides an important clue to prevent or treat HTLV-1-associated human diseases.
INTRODUCTION
Human T-cell leukemia virus type 1 (HTLV-1) is a retrovirus that transforms human T lymphocytes (1). After a long latency period, about 2% to 5% of individuals infected with HTLV-1 develop adult T-cell leukemia (ATL) (1). HTLV-1 carries several regulatory and accessory genes, including tax, rex, HBZ, p12, p13, and p30, in the pX region located between env and the 3′ long terminal repeat (LTR) (1). Among all of the HTLV transcripts, the tax gene is thought to play a critical role in leukemogenesis because of its pleiotropic functions (2). Tax activates transcriptional pathways, including nuclear factor κB (NF-κB), cAMP response element binding protein (CREB), activator protein 1 (AP-1), and serum-responsive factor (SRF) (2). The constitutive activation of the NF-κB pathway in HTLV-1-transformed T cells argues for a critical role of this factor in mediating the development of ATL (3). However, Tax-mediated NF-κB activation may not fully explain ATL biology, because some leukemic cells that no longer express Tax continue to show constitutive NF-κB activation (4–6). Recent reports provide new evidence that elevated expression of NF-κB-inducing kinase (NIK) has a pivotal role in the activation of the alternative NF-κB pathway in ATL independent of Tax expression (7). However, it remains unknown whether other mechanisms underlying the Tax-independent activation of the NF-κB pathway are involved in the development of ATL.
Regulatory noncoding RNAs (ncRNAs), such as microRNAs (miRNAs), small interfering RNAs, and long noncoding RNAs (lncRNAs), play important roles in the development of human diseases (8). lncRNAs, ranging from 200 to 100,000 nucleotides, are involved in a range of biological processes, including modulation of cell growth, apoptosis, stem cell pluripotency, and the immune response, through the modulation of gene expression by epigenetic regulation, chromatin remodeling, transcription, and posttranscriptional processing (9, 10). Additionally, accumulating evidence has shown that lncRNAs play a critical role in tumorigenesis (11). However, the contribution of lncRNAs to the genesis of HTLV-1-induced ATL has not been investigated.
Recently, the lncRNA ANRIL (antisense noncoding RNA in the INK4 locus), which is transcribed from the INK4b-ARF-INK4a gene cluster in the opposite direction, has been identified as a genetic susceptibility locus associated with human disease, in particular cancers (12–14). ANRIL was involved in repression of the p15/CDKN2B-p16/CDKN2A-p14/ARF gene cluster in cis by directly binding to polycomb repressor complex 2 (PRC2), which resulted in increased cell proliferation and suppression of apoptosis (15–17).
PRC2 has been demonstrated to be a functional target of some lncRNAs, e.g., ANRIL, HOTAIR, Fendrr, H19, MALAT1, and COLDAIR (2, 15, 16, 18–21). Moreover, PRC2/lncRNA complex-mediated dynamic control of H3K27 trimethylation (H3K27me3) is central to gene silencing in various cellular processes (9, 22). Enhancer of zeste homolog 2 (EZH2), one of the genes identified to be aberrantly overexpressed in ATL, is a component of PRC2 (23, 24). EZH2 contains a catalytic domain (SET domain) at the COOH terminus that provides the methyltransferase activity, which plays a key role in the epigenetic maintenance of repressive chromatin marks (25, 26). In addition to its known role as a transcriptional suppressor, several studies have also identified a PRC2-independent function of EZH2 in transcriptional activation rather than repression (27–30). In castration-resistant prostate cancer, EZH2 acts as a coactivator for critical transcription factors, including the androgen receptor (AR) (31). This functional switch is dependent on the phosphorylation of EZH2 and requires an intact methyltransferase domain. The activation role of EZH2 was also demonstrated in breast cancer cells, in which EZH2 activates NF-κB targets or NOTCH1 (29, 30). However, the significance and potential role of polycomb group proteins and the associated lncRNA in ATL are still unknown.
In this study, we report that ANRIL interacted with EZH2 to support the proliferation of ATL cells, indicating that dysregulation of ANRIL is associated with the leukemogenesis of ATL.
RESULTS
lncRNA ANRIL is upregulated in ATL.
lncRNAs have been reported to be associated with the development of various cancers (8, 15, 20, 21). To identify lncRNAs that are involved in the development of ATL, we first examined the expression of selected onco-lncRNAs in HTLV-1-infected cell lines. Compared with non-HTLV-1-infected control cells, the levels of ANRIL, H19, and SAF were enhanced in ATL cells, whereas the expression levels of three lncRNAs were found to be either slightly reduced (HOTAIR and TUSC7) or unchanged (MALAT1) (Fig. 1A). Since ANRIL was more highly expressed in ATL cells, our subsequent studies focused on ANRIL. The real-time PCR results verified the enhanced expression of ANRIL in HTLV-1-infected cell lines (Fig. 1B). Next, we examined the expression of ANRIL in primary ATL cells isolated from patients in comparison with peripheral blood mononuclear cells (PBMCs) from healthy subjects under resting conditions. Primary ATL cells expressed ANRIL at levels much higher than those expressed by healthy PBMCs (Fig. 1C).
FIG 1.

ANRIL gene expression in ATL cells. (A) Expression of lncRNAs in ATL cells. Total RNA was extracted from ATL and HTLV-1-immortalized cell lines and T-cell lines not infected with HTLV-1. The levels of HOTAIR, H19, TUSC7, ANRIL, MALAT1, SAF, Tax, and GAPDH mRNAs were analyzed by semiquantitative PCR. (B) lncRNA ANRIL is overexpressed in HTLV-1-associated cell lines. Total RNA was extracted from ATL and HTLV-1-immortalized cell lines (black bars) and T-cell lines not infected with HTLV-1 (open bars). The level of ANRIL mRNA was analyzed by quantitative PCR. (C) High expression level of ANRIL in ATL. CD4-positive cells were isolated from PBMCs of healthy donors and ATL patients, and real-time PCR was performed to analyze the expression of ANRIL mRNA. HD1 and -2 indicate healthy donors, and ATL1 to -6 indicate ATL patients. The Mann-Whitney test using GraphPad Prism software or Student’s t test was used to determine significance where appropriate. *, P < 0.05; **, P < 0.01. Representative results of 3 independent experiments are shown.
E2F1 induces ANRIL expression in T cells.
To investigate the mechanism of upregulation of ANRIL in ATL cells, we amplified the putative promoter region of ANRIL (encompassing the region spanning positions −851 to +29) [ANRIL(−851/+29)] and cloned it into a pGL3-Basic reporter plasmid. Next, we searched for possible transcription factor binding sites in the promoter region using TESS (Transcription Element Search Software). E2F1, Sp1, and CTCF binding sites were found in this region. A reporter assay was performed to explore whether the putative transcription factors could affect ANRIL promoter activity. As shown in Fig. 2A, overexpression of E2F1 led to an increase of ANRIL(−851/+29)-luciferase (Luc) expression, whereas Sp1 and CTCF had no effect. To investigate whether E2F1 indeed altered ANRIL expression in T cells, we overexpressed E2F1 in Jurkat cells. The results showed that enforced expression of E2F1 markedly upregulated the level of the ANRIL gene transcript (Fig. 2B). Moreover, knockdown of E2F1 diminished ANRIL expression in HPB-ATL-T cells (Fig. 2C). Chromatin immunoprecipitation (ChIP) assays verified the binding of E2F1 to the promoter region of ANRIL in multiple ATL cell lines (Fig. 2D). These results collectively indicate that the enhanced expression of ANRIL in ATL can be attributed to the association of E2F1 with the ANRIL promoter.
FIG 2.

E2F1 induces high expression levels of lncRNA ANRIL in ATL. (A) E2F1 activates transcription of the ANRIL promoter. (Left) Schema of the ANRIL promoter. (Top) Possible transcription factor binding sites. 293FT cells were transfected with the E2F1 reporter plasmid with or without the plasmid expressing E2F1, Sp1, or CTCF. (Bottom) Luciferase activity was measured 48 h after transfection. (Right) Expression levels of E2F1, Sp1, and CTCF were measured 48 h after transfection. (B) E2F1-induced ANRIL expression. Total RNA was extracted from control or E2F1-expressing Jurkat cells. Real-time PCR was performed to analyze the expression of ANRIL mRNA. (C) Quantitative comparison of ANRIL mRNA levels between E2F1 knockdown and control HPB-ATL-T cells by real-time PCR. (D) ChIP analysis of the association of endogenous E2F1 and ANRIL promoters in HPB-ATL-2, HPB-ATL-T, and MT-2 cells. *, P < 0.05; **, P < 0.01.
Knockdown of ANRIL impairs cellular proliferation and induces apoptosis in vitro.
To clarify the potential role of upregulated ANRIL expression in ATL cells, we knocked down ANRIL by short hairpin RNA (shRNA) in HPB-ATL-T and ED cells. Two lentivirally expressed shRNAs directed against ANRIL resulted in a reduction of ANRIL expression compared with the vector control (Fig. 3A and B). MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] assays revealed that reduction of ANRIL expression significantly inhibited cell proliferation in both HPB-ATL-T and ED cell lines (Fig. 3C and D). We performed a similar experiment in an HTLV-1-negative T-cell line, Jurkat, in which ANRIL is undetectable by real-time PCR. As expected, cell proliferation was not affected by inhibition of ANRIL (Fig. 3E). Furthermore, ANRIL knockdown resulted in a 3- to 4-fold decrease in the clonogenic survival of HPB-ATL-T cells (Fig. 3F). shANRIL#2 induced less-efficient knockdown of ANRIL than shANRIL#1, resulting in less inhibition of cell proliferation. To test the specificity of the knockdown, we reintroduced shRNA-resistant ANRIL to cells stably transfected with ANRIL shRNA. As shown in Fig. 3G, shRNA-resistant ANRIL rescued cell proliferation inhibition by ANRIL silencing.
FIG 3.

ANRIL supports proliferation of ATL cells. (A and B) Relative mRNA levels of ANRIL in HPB-ATL-T (A) and ED (B) cells transfected with a recombinant lentivirus expressing the pLKO negative-control or pLKO-shANRIL vector. After puromycin selection, total RNAs were extracted. The level of ANRIL mRNA was analyzed by real-time PCR. (C) The effect of ANRIL shRNAs on proliferation of HPB-ATL-T cells was determined by an MTT assay. OD595, optical density at 595 nm. (D) MTT assay analysis of ANRIL shRNAs or mock-transfected ED cells. (E) ANRIL knockdown does not affect proliferation of an HTLV-1-negative T-cell line, Jurkat. Jurkat cells were transduced by a lentivirus encoding ANRIL shRNA. After puromycin selection, cell proliferation was determined by an MTT assay. (F) The effect of ANRIL shRNAs on proliferation of HPB-ATL-T cells was determined by a colony formation assay. (G) shRNA-resistant ANRIL rescues cell proliferation inhibition by ANRIL silencing. HPB-ATL-T cells were transfected with a recombinant lentivirus expressing the pLKO-shANRIL vector together with the pCSII-ANRIL shRNA-resistant plasmid. After puromycin selection, cell growth was detected by an MTT assay. (H) The effect of ANRIL shRNAs on apoptosis of HPB-ATL-T and ED cells was analyzed by annexin V and 7-AAD staining and a flow cytometry method. The percentages of each fraction are shown. (I) Knockdown of ANRIL induces cell cycle arrest. The cell cycle of ANRIL knockdown ED cells was analyzed by flow cytometry. The bar chart represents the percentage of cells in G1, S, or G2/M phase, as indicated. Statistical analysis was performed by Student’s t test (*, P < 0.05; **, P < 0.01).
In addition, ANRIL knockdown increased the total number of apoptotic cells in both the HPB-ATL-T and ED cell lines (Fig. 3H). Next, we evaluated if the effect of the knockdown of ANRIL on ATL cell proliferation was due to cell cycle arrest. Flow cytometry results indicated that the proportion of cells in G1/S phase increased in ANRIL knockdown cells compared with the corresponding control. This suggested that reduction of ANRIL expression blocked cell cycle progression (Fig. 3I).
Taken together, these observations demonstrate that ANRIL silencing suppressed the proliferation of ATL cells, and this inhibitory effect was partially due to the inhibition of cell cycle progression and the promotion of apoptosis.
ANRIL promotes proliferation of ATL cells in vivo.
To further investigate the ability of ANRIL to stimulate ATL tumorigenesis in vivo, we injected equivalent numbers of ED negative-control and ED ANRIL knockdown cells into NCG mice and analyzed tumor growth. The tumors formed by ED-shANRIL#1 cells were smaller in both size and weight than ED control tumors (Fig. 4A to C). Real-time PCR revealed that tumor nodules originating from ED-shANRIL#1 cells had decreased expression of ANRIL, MYC, VCAM1, CDK6, and CCND1 genes and increased expression of CDKN1A, CTGF, and KLF2 compared to nodules originating from ED control cells (Fig. 4D). Hematoxylin and eosin (H&E) staining of the tumors showed that the degree of cell differentiation was significantly higher in ANRIL knockdown-derived tumors than in control tumors (Fig. 4E).
FIG 4.

ANRIL promotes proliferation of ATL cells in vivo. Negative-control or ANRIL knockdown ED cells were injected into NCG mice (n = 6). Thirty days after injection, all mice were sacrificed. (A) Photographs of dissected tumors from NCG mice. (B) Diagram of average weights of tumors. (C) Diagram of average volumes of tumors. (D) Expression levels of ANRIL, CDKN1A, MYC, CTGF, VCAM1, CDK6, KLF2, and CCND1 were examined by real-time PCR in tumor tissues from NCG mice. (E) Tumor sections under H&E staining. *, P < 0.05; **, P < 0.01.
Overall, these results confirm data from our in vitro transformation assays and suggest that ANRIL has tumor-forming potential.
ANRIL and EZH2 synergistically activate the NF-κB pathway.
Interestingly, as shown in Fig. 4D, the genes that were differentially expressed upon ANRIL depletion are many well-known NF-κB target genes, such as MYC, VCAM1, CDK6, and CCND1. Therefore, we evaluated whether ANRIL could influence the NF-κB pathway in ATL. As shown in Fig. 5A, silencing of ANRIL expression in HPB-ATL-T and ED cell lines resulted in the suppression of NF-κB signaling. Reporter analyses revealed that the expression of ANRIL upregulated Tax-mediated NF-κB activation, while ANRIL had no effect on Tax-mediated transcriptional activation from the HTLV-1 promoter (Fig. 5B and C). Treatment with SN50, an inhibitor of p65 activity, significantly suppressed the ability of Tax and ANRIL to enhance transcriptional activity through κB-responsive elements (Fig. 5B). Moreover, RelA/p65 knockdown impaired ANRIL- and Tax-mediated NF-κB activation, indicating that ANRIL/Tax-mediated NF-κB activation is dependent on p65 (Fig. 5D).
FIG 5.

ANRIL/Tax-mediated NF-κB activation is dependent on p65. (A) Inhibition of NF-κB signaling by ANRIL knockdown. ANRIL knockdown HPB-ATL-T (left) and ED (right) cells were cotransfected with κB-Luc and phRL-TK plasmids. Luciferase levels were measured after 48 h. (B) SN50 suppresses Tax- and ANRIL-induced NF-κB activation. Jurkat cells were cotransfected with κB-Luc (0.5 μg), phRL-TK (20 ng), pCG-Tax (0.5 μg), and pCSII-CMV-ANRIL (0.5 μg). Twenty-four hours after transfection, SN50 (20 μM) was added. After 24 h, the cells were harvested and analyzed for luciferase activity. (C) ANRIL does not influence Tax-mediated transcriptional activation from the HTLV-1 promoter. Jurkat cells were cotransfected with WT-Luc, phRL-TK, pCG-Tax, and pCSII-CMV-ANRIL. After 48 h, the cells were harvested and analyzed for luciferase activity. (D) Silencing of RelA/p65 expression impairs ANRIL- and Tax-mediated NF-κB activation. HPB-ATL-T cells were transfected with a recombinant lentivirus expressing pLKO-shRelA/p65. p65 knockdown cells were cotransfected with κB-Luc and phRL-TK plasmids. After 48 h, the expression levels of p65 and β-actin were analyzed by immunoblotting (left), and the cells were harvested and analyzed for luciferase activity (right). All the data shown are relative values of firefly luciferase normalized to the value for Renilla luciferase and expressed as means of results from a triplicate set of experiments (±SD). *, P < 0.05; **, P < 0.01.
It is well established that lncRNA may achieve regulatory goals through interacting with protein partners and modulating the activity of these proteins within the cell. EZH2, which is a functional target of some lncRNAs, was constitutively expressed in all HTLV-1-infected cell lines (Fig. 6A). The association between ANRIL and EZH2 was confirmed to occur endogenously in an ATL cell line, HPB-ATL-T (Fig. 6B). Consistent with data from a previous report (29), EZH2 enhanced p65-induced NF-κB activation (Fig. 6C). Furthermore, in the presence of ANRIL, p65/EZH2-mediated NF-κB activation was dramatically reinforced, whereas it had a less obvious effect without EZH2 (Fig. 6D). To confirm the synergistic effect of ANRIL and EZH2 on NF-κB activation, we measured κB-luciferase activity after suppressing EZH2 expression in ANRIL-silenced cells. Silencing of EZH2 by shRNA further suppressed NF-κB signaling in ANRIL knockdown cells (Fig. 6E). Gene expression analysis revealed suppression of NF-κB target genes, including MYC and VCAM1, upon ANRIL and EZH2 depletion (Fig. 6F). In addition, knockdown of EZH2 synergized with ANRIL knockdown to inhibit cell viability (Fig. 6G). These results collectively indicate that ANRIL and EZH2 could cooperatively activate the NF-κB pathway, resulting in the promotion of cell proliferation in ATL.
FIG 6.

ANRIL/EZH2 activates the NF-κB pathway. (A) Overexpression of EZH2 in ATL. Total proteins were extracted from ATL and HTLV-1-immortalized cell lines. The level of EZH2 protein was analyzed by immunoblotting. (B) ANRIL-associated EZH2 in vivo. Shown are data for radioimmunoprecipitation analysis of the association of endogenous ANRIL RNA and EZH2 protein in HPB-ATL-T cells. (C) EZH2 enhances p65-induced NF-κB activation. Jurkat cells were cotransfected with κB-Luc, phRL-TK, pCMV-p65, and lenti-Myc-EZH2. After 48 h, luciferase activity was measured. (D) ANRIL and EZH2 synergistically reinforce p65-mediated NF-κB activation. Jurkat cells were cotransfected with κB-Luc, phRL-TK, pCSII-CMV-ANRIL, lenti-Myc-EZH2, and pCMV-p65. After 48 h, the cells were harvested, and luciferase activity was determined. (E) Silencing of EZH2 expression suppresses NF-κB signaling in ANRIL knockdown cells. HPB-ATL-T cells were transfected with a recombinant lentivirus expressing the pLKO-shANRIL vector with or without pLKO-shEZH2. After puromycin selection, the expression levels of EZH2 and β-actin were analyzed by immunoblotting (top), and the cells were harvested and analyzed for luciferase activity (bottom). (F) Suppressed expression of selected NF-κB target genes in ANRIL and EZH2 knockdown HPB-ATL-T cells. The levels of MYC and VCAM1 mRNAs were analyzed by real-time PCR. (G) EZH2 silencing synergizes with ANRIL knockdown to inhibit cell viability. HPB-ATL-T cells were transfected with a recombinant lentivirus expressing the pLKO-shANRIL vector together with pLKO-shEZH2. After puromycin selection, cell growth was detected by an MTT assay. (H) Overexpression of p65 rescues cell proliferation inhibition by ANRIL silencing. HPB-ATL-T cells were transfected with a recombinant lentivirus expressing the pLKO-shANRIL vector together with pCSII-CMV-p65. After puromycin selection, cell growth was detected by an MTT assay. *, P < 0.05; **, P < 0.01.
Next, we determined whether p65 was required for the ANRIL-enhanced cell proliferation of ATL cells. An MTT assay showed that the decreased cell proliferation ability mediated by shANRIL was partially rescued by p65 (Fig. 6H). These results together suggest that ANRIL supports the proliferation of ATL cells through activating the NF-κB pathway via EZH2.
ANRIL associates with EZH2 and p65 and enhances NF-κB signaling independent of HMT activity.
To clarify the mechanism by which ANRIL/EZH2 enhances the NF-κB transcriptional response, FLAG-tagged RelA/p65, RelB, or c-Rel and Myc-tagged EZH2 were cotransfected into 293FT cells. Figure 7A demonstrates that EZH2 interacted with p65, c-Rel, and, to a lesser extent, RelB. Because a high degree of homology exists between p65 and c-Rel, our subsequent analyses focused on the p65-EZH2 interaction. The association between p65 and EZH2 was also confirmed in an ATL cell line, HPB-ATL-T (Fig. 7B). Furthermore, ANRIL greatly enhanced the interaction between EZH2 and p65 (Fig. 7C). As ANRIL enhanced the activation of NF-κB signaling by EZH2 and p65, we explored whether ANRIL could form a complex with EZH2 and p65. 293FT cells were transfected with vectors expressing ANRIL, Myc-EZH2, and FLAG-p65, and a serial RNA immunoprecipitation assay was performed. As shown in Fig. 7D, we detected a specific ternary complex only when the three components were coexpressed. To investigate the binding of EZH2/ANRIL/p65 to DNA, we performed a ChIP assay in 293FT cells that were cotransfected with a κB-Luc reporter along with expression vectors for EZH2, ANRIL, and p65. The ChIP assay showed that EZH2 and ANRIL dramatically enhanced the p65 DNA binding capability (Fig. 7E). Moreover, the interaction of p65 with the NF-κB binding site of the MYC gene was diminished in ED cells silencing both ANRIL and EZH2 (Fig. 7F). In addition, we found that silencing of ANRIL could not modulate the phosphorylation of IκBα in ATL cells (Fig. 7G). These results together suggest that ANRIL augments the interaction between EZH2 and p65, thereby potentiating NF-κB signaling.
FIG 7.

ANRIL forms a complex with EZH2/p65 and enhances the NF-κB pathway. (A) EZH2 interacts with p65, RelB, and c-Rel. 293FT cells were cotransfected with lenti-Myc-EZH2 together with FLAG-p65 (left), FLAG-RelB (middle), or FLAG–c-Rel (right). After 48 h, cell lysates were subjected to immunoprecipitation (IP) using anti-c-Myc followed by immunoblotting (IB) using anti-FLAG. (B) The interaction between endogenous EZH2 and p65 in HPB-ATL-T cells was analyzed by an immunoprecipitation assay. (C) ANRIL enhances the interaction between EZH2 and p65. 293FT cells were cotransfected with lenti-Myc-EZH2, pCSII-CMV-ANRIL, and pCMV-p65. Cell lysates were subjected to immunoprecipitation using anti-FLAG followed by immunoblotting with anti-EZH2. (D) ANRIL, EZH2, and p65 can form a ternary complex. pCSII-CMV-ANRIL, lenti-Myc-EZH2, and pCMV-p65 were cotransfected into 293FT cells. Ternary complexes were detected by sequential immunoprecipitation with an anti-FLAG agarose affinity gel and anti-Myc antibody, and coprecipitated RNAs were detected by PCR. (E) ANRIL/EZH2 enhances p65 DNA binding capability. After transfection with lenti-Myc-EZH2, pCSII-CMV-ANRIL, pCMV-p65, and κB-Luc for 48 h, 293FT cells were chromatin immunoprecipitated by anti-FLAG antibody. The precipitated DNAs and 1% of the input cell lysates were amplified by the κB-Luc-specific primers (top, RT-PCR; bottom, real-time PCR). (F) Silencing of ANRIL and EZH2 inhibits p65 DNA binding capability. ANRIL knockdown cells were transfected with a recombinant lentivirus expressing pLKO-shEZH2. After puromycin selection, the binding of p65 to the NF-κB binding site of the MYC gene was analyzed by a ChIP assay. (G) Silencing of ANRIL does not influence phosphorylation levels of IκBα. After knockdown of ANRIL, the phosphorylation level of IκBα was analyzed by immunoblotting. *, P < 0.05; **, P < 0.01.
We next evaluated the ANRIL deletion mutants shown in Fig. 8A to determine which region of ANRIL is responsible for activating NF-κB signaling. Luciferase experiments revealed that the only full-length ANRIL could reinforce EZH2/p65-mediated NF-κB activation, whereas the truncated lncRNAs could not, suggesting that the tertiary structure of full-length ANRIL, rather than one or more of the ANRIL domains, was responsible for potentiating NF-κB signaling (Fig. 8A).
FIG 8.

ANRIL/EZH2 enhances the NF-κB pathway independently of HMT activity. (A) Analysis of ANRIL deletion mutants for their effect on p65/EZH2-induced NF-κB activation. (Top) Schematic diagram of ANRIL and its mutants used in this study. (Bottom) Jurkat cells were cotransfected with κB-Luc, phRL-TK, lenti-Myc-EZH2, pCMV-p65, and pCSII-CMV-ANRIL or its mutants. Luciferase activity was measured 48 h after transfection. wt, wild type. (B) ANRIL/EZH2 activates the NF-κB pathway independent of the EZH2 SET domain. (Top) Jurkat cells were cotransfected with κB-Luc, phRL-TK, pCSII-CMV-ANRIL, and pCMV-p65 together with lenti-Myc-EZH2 or lenti-Myc-EZH2ΔSET. Forty-eight hours after transfection, a dual-luciferase reporter assay was performed. (Bottom) The level of H3K27me3 was analyzed by immunoblotting. (C) ANRIL/EZH2 interacts with p65 independent of the EZH2 SET domain. 293FT cells were transfected with expression vectors for p65, pCSII-CMV-ANRIL, and lenti-Myc-EZH2 or lenti-Myc-EZH2ΔSET. After 48 h, cell lysates were subjected to immunoprecipitation using anti-c-Myc followed by immunoblotting using anti-FLAG. (D) Mapping the region of the p65 protein necessary for the interaction with ANRIL/EZH2. (Top) Schema of p65 deletion mutants. NLS, nuclear localization signal. (Bottom) 293FT cells were transfected with lenti-Myc-EZH2 and pCSII-CMV-ANRIL along with full-length or mutant FLAG-p65. Forty-eight hours after transfection, total cell lysates were subjected to immunoprecipitation using anti-FLAG followed by immunoblotting using anti-EZH2. *, P < 0.05; **, P < 0.01.
Accumulating evidence shows that the EZH2 SET domain, which has histone methyltransferase (HMT) activity, is required to induce gene silencing by interacting with ANRIL (2, 16, 25, 26). We asked whether the HMT catalytic function of EZH2 is required for the ANRIL/EZH2 complex to positively regulate NF-κB-dependent transcription in ATL cells. We first compared the abilities of EZH2 and the EZH2 SET domain deletion mutant (EZH2-ΔSET) to activate the NF-κB pathway. As shown in Fig. 8B, lentiviral overexpression of EZH2-ΔSET resulted in a robust induction of NF-κB reporter activity, which was comparable to that of wild-type EZH2. In coimmunoprecipitation (co-IP) experiments in 293FT cells transfected with Myc-tagged EZH2 or EZH2-ΔSET, we detected p65 in both EZH2 and EZH2-ΔSET immunoprecipitates (Fig. 8C). In addition, co-IP experiments revealed that the ANRIL/EZH2 complex interacted with the Rel homology domain of p65 (Fig. 8D).
Taken together, these data indicate that the complex of ANRIL/EZH2/p65 enhances the NF-κB pathway in a histone methyltransferase activity-independent manner.
ANRIL/EZH2 suppresses p21/CDKN1A expression by a histone methylation-dependent mechanism.
We next examined whether ANRIL supports the proliferation of ATL cells through modulating the expression of genes involved in apoptosis and proliferation. Expression levels of cleaved caspase-3, caspase-7, and poly(ADP-ribose) polymerase (PARP) were increased in HPB-ATL-T and ED cells with transfection of shANRIL (Fig. 9A). Consistent with the results of the flow cytometry assay, in which knockdown of ANRIL induced cell cycle arrest, cyclin D1 and cyclin E1 expression levels were decreased in ANRIL knockdown HPB-ATL-T and ED cells (Fig. 9A).
FIG 9.

ANRIL modulates the expression of selected apoptosis- and proliferation-regulating genes. (A) Protein levels of apoptosis- and cell cycle-related genes in ANRIL knockdown HPB-ATL-T (left) and ED (right) cells. The expression levels of cleaved caspase-3, caspase-7, caspase-8, PARP, and cyclins D1 and E1 were analyzed by immunoblotting. (B) Transcriptional changes of apoptosis- and proliferation-related genes in ANRIL knockdown HPB-ATL-T (left) and ED (right) cells. The levels of p15/CDKN2B, p16/CDKN2A, p21/CDKN1A, and KLF2 mRNAs were analyzed by real-time PCR. *, P < 0.05; **, P < 0.01.
Several reports have indicated that ANRIL suppressed the expression of the p15/CDKN2B-p16/CDKN2A-p14/ARF gene cluster and contributed to cancer cell proliferation (15–17). However, in the present study, there was no significant difference in the expression of p15/CDKN2B or p16/CDKN2A in ANRIL knockdown cells compared with that in control cells (Fig. 9B). Consistent with the observations in vivo, we found that knockdown of the ANRIL gene led to increased expression of p21/CDKN1A and KLF2, indicating that p21/CDKN1A or KLF2 could be a novel ANRIL target in ATL (Fig. 9B).
It was previously reported that p21/CDKN1A was downregulated in ATL due to methylation of its promoter region (32). We next investigated if ANRIL could also affect the expression of p21/CDKN1A by histone methylation. After knockdown of EZH2 expression, the mRNA and protein levels of p21/CDKN1A were increased in HPB-ATL-T cells (Fig. 10A and B). The results of a ChIP assay showed that overexpression of EZH2 resulted in the accumulation of H3K27 trimethylation on the promoter of p21/CDKN1A, which was further enhanced by ANRIL (Fig. 10C).
FIG 10.

ANRIL/EZH2 suppresses p21/CDKN1A expression by a histone methylation-dependent mechanism. (A) The efficiency of EZH2 knockdown was analyzed by real-time PCR. HPB-ATL-T cells were transfected with a recombinant lentivirus expressing pLKO-shEZH2. After puromycin selection, the expression of EZH2 was analyzed. (B) EZH2 knockdown induces the expression of p21/CDKN1A mRNA and protein. After knockdown of EZH2, the levels of p21/CDKN1A mRNA (left) and protein (right) were analyzed by real-time PCR and immunoblotting, respectively. (C) ANRIL/EZH2 induces the accumulation of H3K27me3 on the p21/CDKN1A promoter. 293FT cells were transfected with lenti-Myc-EZH2 and pCSII-CMV-ANRIL. Forty-eight hours after transfection, 293FT cells were chromatin immunoprecipitated by H3K27me3 antibody. The precipitated DNAs and 1% of the input cell lysates were amplified by primers specific for the p21/CDKN1A promoter. (D) Downregulation of p21/CDKN1A rescues cell proliferation inhibition by ANRIL silencing. HPB-ATL-2 cells were transfected with a recombinant lentivirus expressing the pLKO-shANRIL vector together with pLKO-shCDKN1A. After puromycin selection, cell growth was detected by an MTT assay. *, P < 0.05; **, P < 0.01.
In order to evaluate the contribution of p21/CDKN1A to ANRIL-induced ATL cell proliferation, we performed a rescue experiment. As shown in Fig. 10D, downregulation of p21/CDKN1A in ANRIL-silenced cells partially rescued the shANRIL-impaired proliferation of HPB-ATL-2 cells. These results suggest that ANRIL/EZH2 supports the proliferation of ATL cells through inhibiting the expression of p21/CDKN1A via a histone methylation-dependent mechanism.
Collectively, these data support a model in which the ANRIL/EZH2 complex not only contributes to the activation of NF-κB signaling but also mediates transcriptional silencing of p21/CDKN1A in ATL. Moreover, we propose that the molecular mechanism through which ANRIL/EZH2 supports the proliferation of ATL cells involves both histone methylation-dependent and -independent mechanisms (Fig. 11).
FIG 11.
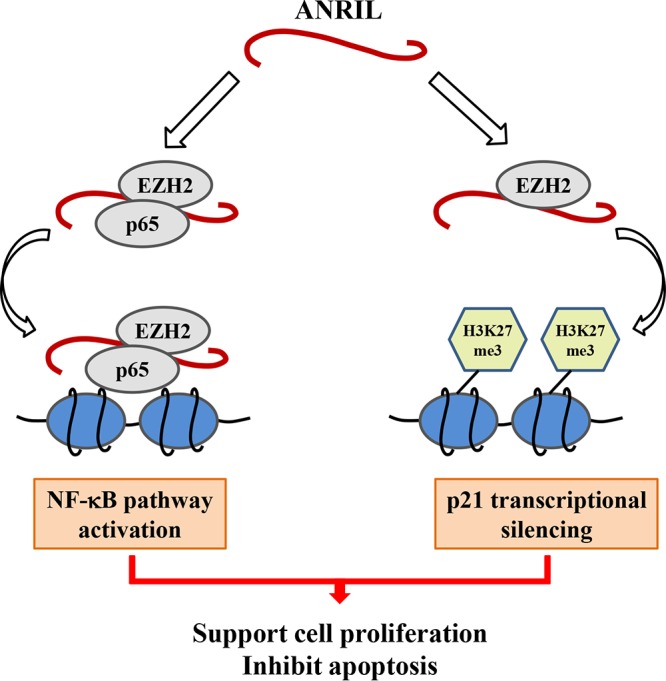
Schematic model that illustrates two functional arms of ANRIL in ATL. ANRIL targeted EZH2 and activated the NF-κB pathway via forming a ternary complex with p65. This activation was independent of EZH2’s histone methyltransferase activity. In addition, the ANRIL/EZH2 complex inhibited the intrinsic apoptosis pathway through suppressing p21/CDKN1A in a methyltransferase activity-dependent manner.
DISCUSSION
The NF-κB signaling pathway, constitutively activated in a variety of cancers, plays a major role in tumor biology as a regulator of key processes of cancer initiation and progression (33). In ATL, Tax-mediated NF-κB activation is reported to be critical for the proliferation of ATL cells and for the transforming activity of HTLV-1 (34). However, ATL cells from most new cases, although infected with HTLV-1, are characterized by a loss of the expression of viral proteins, including Tax, because of host immune surveillance during the long latent period of the virus (1). The fact that NF-κB signaling is strongly and persistently activated in ATL cells implicates other viral and/or cellular factors. It has been reported that NIK may replace Tax in maintaining the constitutive activation of NF-κB, and NIK has been implicated in ATL pathogenesis (7). However, NIK does not seem to be essential for NF-κB activation in all ATL cells, as nuclear NF-κB was not exclusively expressed in NIK-overexpressing cell lines (7). EZH2, a histone methyltransferase subunit of a polycomb repressor complex, is highly expressed in a wide range of neoplasms, including cancers of the breast, lung, and pancreas; sarcomas; and lymphomas (35). The extensive evidence linking EZH2 activity to cancer has prompted interest in the underlying mechanism. We present evidence that EZH2 and its associated lncRNA ANRIL are highly expressed in ATL. EZH2 activated the NF-κB pathway in ATL cells through physical interaction with ANRIL and RelA/p65, and this positive regulation of NF-κB signaling did not require its methyltransferase activity. This constitutive activation by the EZH2/ANRIL/RelA complex may explain how NF-κB is activated in the absence of Tax and advances our understanding of how EZH2 may function as an oncogene. Consistent with our findings, it was recently reported that hepatitis B virus (HBV) infection induces EpCAM expression via RelA in complex with EZH2, and the EZH2/RelA complex acts as an activator of NOTCH1 signaling to accelerate breast cancer growth, supporting that the recruitment of RelA is essential for the oncogenic function of EZH2 (30, 36).
Overexpression of an active form of EZH2 was demonstrated in ATL cells (24). Further study showed that overexpressed EZH2 exhibited histone methyltransferase activity, as ATL cells were strongly positive for H3K27me3 (23). The EZH2-mediated global methylation gain at H3K27 caused silencing of tumor suppressors, transcription factors, and miRNAs, supporting leukemic cell characteristics. In addition to its known roles in histone modification and transcriptional regulation, EZH2 also promotes the tumorigenicity of various cancers independent of its H3K27 methyltransferase activity, and of polycomb repressive complex 2, through transcriptional activation marks (27–30). Our study defines a unique role and mechanism for EZH2 in ATL, whereby EZH2 utilizes polycomb-dependent and -independent mechanisms to protect ATL cells from apoptosis and promote their proliferation.
Recently, Yamagishi et al. reported that overexpression of EZH2 led to polycomb-mediated silencing of miRNA miR-31 and constitutive activation of NF-κB signaling (6). Our results complement this finding and highlight a novel mechanism for NF-κB activation in ATL, where an aberrant overexpression of EZH2 activates NF-κB through both HMT-dependent and -independent pathways. In addition, because EZH2 was undetectable in cells from healthy adults and HTLV-1 carriers, it is likely that deregulation of NF-κB caused by EZH2 is involved in the early steps of ATL oncogenesis. Associations between EZH2 and RelA are essential for NF-κB activation, whereas the epigenetic modification function of EZH2 requires the recruitment of PRC2 subunits, such as SUZ12 and EED. Thus, we speculate that the bimodal function of the EZH2-ANRIL complex depends on the EZH2 binding partner.
Dysregulation of the lncRNA-EZH2 complex has been implicated in viral transformation. For example, lncRNA-HEIH was found to be involved in HBV-related hepatocarcinogenesis through association with the enhancer of EZH2, resulting in the promotion of hepatoma cell proliferation (37). HBx-upregulated lncRNA-UCA1 promotes cell growth and tumorigenesis by recruiting EZH2 and repressing p27Kip1/CDK2 signaling (38). In Kaposi’s sarcoma-associated herpesvirus, lncRNA-PAN RNA binds to protein components of polycomb repression complex 2, EZH2 and SUZ12, and globally influences cellular gene expression, leading to a complete abrogation of the initiation of the viral lytic-phase transcription program (39). Similarly, HIV-expressed lncRNAs recruit and guide a chromatin-remodeling complex (consisting of proteins such as EZH2 and DNMT3a) to the viral promoter, resulting in reduced levels of viral gene expression (40). These findings show that dysregulation of the lncRNA-EZH2 complex is common among different viruses, suggesting that these activities are critical for viral oncogenesis. To our knowledge, all reported instances of virus-induced dysregulation of the lncRNA-EZH2 complex resulted in modulation of global gene expression due to H3K27 methylation. Here we demonstrate that the EZH2-ANRIL complex employs novel HMT-dependent and -independent mechanisms to support the proliferation of ATL cells.
Global changes in the epigenetic landscape are a hallmark of cancer (41). Since genetic changes in specific genes in ATL cells have not been identified, except for p53 and p16, and there is no consistent chromosomal change, it is possible that epigenetic changes, such as DNA methylation, histone modification, and ncRNAs, which dysregulate the transcription of oncogenes, play an important role in oncogenesis (42). In ATL cells, DNA methylation of the promoter regions of tumor suppressor genes (p21 and p16, etc.) and of the 5′ LTR of HTLV-1 is commonly observed and accumulates with disease progression (1, 32, 43). A recent study showed that PRC2-mediated trimethylation at H3K27 (H3K27me3) was significantly and frequently reprogrammed at half of the genes in ATL cells (23). In this study, we identify a novel mechanism of downregulation of p21/CDKN1A due to EZH2/ANRIL-triggered histone H3K27 promoter trimethylation. It has recently become apparent that DNA methylation and histone modification pathways can be dependent on one another (44). Moreover, EZH2 controls CpG methylation through direct physical contact with DNA methyltransferases (45). It is thus likely that, apart from its regulatory function in p16/CDKN1A expression, EZH2 may dysregulate the transcription of other tumor suppresser genes in ATL cells by two dependent epigenetic means to acquire malignant phenotypes. However, it remains unknown how EZH2 selectively regulates its target genes by DNA methylation or histone modification. We speculate that the regulatory effect of EZH2 on the expression of its target gene is dependent on its affinity for DNA and may also depend on its associating transcription factors. Further studies are needed to fully elucidate the role of EZH2/ANRIL in HTLV-1 infection and ATL.
In summary, we show that ANRIL supported the proliferation of ATL cells by interacting with EZH2. HTLV-1 may take advantage of these mechanisms to allow the infected cells to proliferate in vivo. These findings may contribute to the development of novel therapeutic strategies.
MATERIALS AND METHODS
Cell culture and clinical samples.
HTLV-1-infected T-cell lines (MT-2, MT-4, C8166, MT-1, HPB-ATL-2, HPB-ATL-T, ED, and TL-Om1) and HTLV-1-negative T-cell lines (Jurkat, Hut78, and CEM-T4) were grown in RPMI 1640 supplemented with 10% fetal bovine serum (FBS) and antibiotics. 293FT cells were cultured in Dulbecco modified Eagle medium (DMEM) supplemented with 10% FBS and 500 μg/ml G418. Peripheral blood mononuclear cells (PBMCs) were isolated from ATL patients (n = 6) and healthy volunteers (n = 2). Details of clinical samples are shown in Table 1. Clinical samples were obtained and used according to the principles expressed in the Declaration of Helsinki and approved by the Institutional Review Board of Kyoto University. All patients provided written informed consent for the collection and use of samples.
TABLE 1.
List of healthy donors and ATL patients
| Sample | Age of subject (yr) | Gender of subject | ATL type |
|---|---|---|---|
| HD1 | 50 | Female | None |
| HD2 | 38 | Female | None |
| ATL1 | 68 | Female | |
| ATL2 | 52 | Female | Acute |
| ATL3 | 56 | Female | Acute |
| ATL4 | 72 | Female | |
| ATL5 | 45 | Male | |
| ATL6 | 38 | Female | Acute |
Plasmids and reagents.
The sequence of ANRIL was amplified by reverse transcriptase PCR (RT-PCR) and subcloned into pCSII-CMV-GFP. The ANRIL promoter sequence was cloned into the pGL3-Basic vector (pGL3-ANRIL −851/+29). Expression vectors for E2F1, Sp1, Tax, p65, p65 deletion mutants, CTCF, κB-Luc, WT-Luc (reporter vector containing 21 bp of wild-type enhancers in the HTLV-1 5’ LTR), and phRL-TK were prepared as previously described (46, 47). The lenti-Myc-EZH2 (lentivirus vector carrying Myc-tagged EZH2) plasmid was a gift from Xiangping Li of Southern Medical University (48). Expression vectors for FLAG-RelB, FLAG–c-Rel, lenti-Myc–EZH2-ΔSET deletion mutants, pCSII-CMV-ANRIL deletion mutants, and shRNA-resistant pCSII-ANRIL were generated by PCR. SN50 was purchased from Millipore (Billerica, MA, USA).
Quantitative and semiquantitative PCR.
Total RNA was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. cDNA was synthesized using SuperScript III reverse transcriptase (Life Technologies, Grand Island, NY, USA). Quantitative PCR (qPCR) was carried out using Power SYBR green PCR master mix and the StepOnePlus real-time PCR system (Thermo Fisher Scientific, Waltham, MA, USA). Semiquantitative RT-PCR was performed as previously described (46). The specific primers used can be found in Table 2.
TABLE 2.
List of primers for quantitative PCR, semiquantitative PCR, chromatin immunoprecipitation, and RNA immunoprecipitation
| Reaction | Target | Sequencea |
|---|---|---|
| Quantitative PCR | ANRIL | F, CAACATCCACCACTGGATCTTAACA |
| R, AGCTTCGTATCCCCAATGAGATACA | ||
| E2F1 | F, ATGTTTTCCTGTGCCCTGAG | |
| R, AGATGATGGTGGTGGTGAC | ||
| SP1 | F, GCCTCCAGACCATTAAC | |
| R, GCTCCATGATCACCTGGGGCA | ||
| CTCF | F, GATCATGCACAAGCGCAC | |
| R, CCATGGTATTCCGACGTGTA | ||
| p15/CDKN2B | F, GGGAGGGTAATGAAGCTGAG | |
| R, GGCCGTAAACTTAACGACACT | ||
| p16/CDKN2A | F, GGGTCCCAGTCTGCAGTTA | |
| R, GGAGGGTCACCAAGAACCT | ||
| p21/CDKN1A | F, GCAGACCAGCATGACAGATTT | |
| R, GGATTAGGGCTTCCTCTTGGA | ||
| KLF2 | F, CTGCACATGAAACGGCACAT | |
| R, CAGTCACAGTTTGGGAGGGG | ||
| EZH2 | F, CCCTGACCTCTGTCTTACTTGTGGA | |
| R, ACGTCAGATGGTGCCAGCAATA | ||
| MYC | F, GCTGCTTAGACGCTGGATTT | |
| R, CACCGAGTCGTAGTCGAGGT | ||
| CTGF | F, CTTCTGCGATTTCGGCTCC | |
| R, TACACCGACCCACCGAAGA | ||
| VCAM1 | F, TGCCGAGCTAAATTACACATTG | |
| R, CCTTGTGGAGGGATGTACAGA | ||
| CDK6 | F, TGGAGACCTTCGAGCACC | |
| R, CACTCCAGGCTCTGGAACTT | ||
| CCND1 | F, GTTCATGGCCAGCGGGAAGAC | |
| R, CTGCCGTCCATGCGGAAGATC | ||
| 18S | F, AACCCGTTGAACCCCATT | |
| R, CCATCCAATCGGTAGTAGCG | ||
| Semiquantitative PCR | HOTAIR | F, CAGTGGGGAACTCTGACTCG |
| R, GTGCCTGGTGCTCTCTTACC | ||
| H19 | F, TGCTGCACTTTACAACCACTG | |
| R, ATGGTGTCTTTGATGTTGGGC | ||
| TUSC7 | F, GGACCTGCCCTCCATTCTAT | |
| R, GCATGGCATTTTCTCTCCTC | ||
| ANRIL | F, CAGAGCAATTCCAGTGCAAG | |
| R, GATTTGCAAAAACAGCTG | ||
| MALAT1 | F, GAATTGCGTCATTTAAAGCCTAGTT | |
| R, GTTTCATCCTACCACTCCCAATTAAT | ||
| SAF | F, CATCTCAGCCTCTTGGTGTAA | |
| R, ATGGGAGATATGGGATTGAAC | ||
| Tax | F, CCGGCGCTGCTCTCATCCCGGT | |
| R, GGCCGAACATAGTCCCCCAGAG | ||
| E2F1 | F, CACTCGGCTGACGGTGTCGTCGACC | |
| R, GGAGAGCAGGCGCAGCTGCGTAGTA | ||
| EZH2 | F, GTGGAGAGATTATTTCTCAAGATG | |
| R, CCGACATACTTCAGGGCATCAGCC | ||
| GAPDH | F, ATGGGGAAGGTGAAGGTCGGAGTC | |
| R, CCATGCCAGTGAGCTTCCCGTTC | ||
| Chromatin immunoprecipitation | ANRIL-Luc | F, GTAACCCGAATGGGGAAGCC |
| R, GCCGCTTCCTAGAAGACCAG | ||
| κB-Luc | F, TCCAGGGGAATCTCCAGG | |
| R, GTTGCTCTCCAGCGGTTCCA | ||
| CDKN1A promoter | F, CACCAGACTTCTCTGAGCCC | |
| R, AAAGGGCAACCTGATCTCCA | ||
| Myc promoter | F, GTGCATGACCGCATTTCCAA | |
| R, AATCAAAGGTGCTAGACGGG | ||
| RNA immunoprecipitation | ANRIL | F, CAACATCCACCACTGGATCTTAACA |
| R, AGCTTCGTATCCCCAATGAGATACA | ||
F, forward; R, reverse.
Luciferase assay.
Jurkat cells were plated on 12-well plates at 1.2 × 105 cells per well. After 24 h, cells were transfected with the indicated plasmids using Lipofectamine LTX reagent (Invitrogen). Forty-eight hours after transfection, a luciferase reporter assay was performed using a dual-luciferase reporter assay system (Promega, Madison, WI). Each experiment was performed in triplicate, and the data represent the means and standard deviations (SD) of results from 3 independent experiments, each normalized to Renilla activity.
Chromatin immunoprecipitation assay.
293FT cells were transfected with the indicated plasmids. Forty-eight hours after transfection, a ChIP assay was done according to the protocol recommended by Upstate Biotechnology. Precipitated DNA was amplified by PCR using primers specific for the ANRIL-Luc, κB-Luc, or p21/CDKN1A promoter vector. For the endogenous ANRIL and MYC promoters, chromatin samples prepared from HPB-ATL-T cells were subjected to ChIP analysis using anti-E2F1 (Abcam, Cambridge, MA), anti-p65 (Cell Signaling Technology, Danvers, MA, USA), and normal mouse immunoglobulin G (IgG; Santa Cruz Biotechnology). Sequences for the primer set can be found in Table 2.
Knockdown.
Knockdown of ANRIL, E2F1, p65, EZH2, and p21/CDKN1A in ATL cell lines was performed with a lentiviral vector pLKO.1-based shRNA system (Open Biosystems, Lafayette, CO, USA) (47). Stable transfectants were selected in puromycin. The shRNAs specifically targeting ANRIL, E2F1, p65, EZH2, and CDKN1A were shANRIL#1 (5′-GGAATGAGGAGCACAGTGA-3′), shANRIL#2 (5′-AAGCGAGGTCATCTCATTGCTCTAT-3′), shE2F1 (5′-GACGTGTCAGGACCTTCGT-3′), shRelA/p65 (5′-GATTGAGGAGAAACGTAAA-3′), shEZH2 (5′-GACTCTGAATGCAGTTGCTTCAGTACCC-3′), and shCDKN1A (5′-GCGATGGAACTTCGACTTT-3′).
Cell proliferation assay.
Cells were seeded in 96-well plates, and at indicated time points, 10 μl of a 3-(4,5-dimethythiazol-2-yl)-2,5-diphenyl tetrazolium bromide solution was added. Cells were incubated at 37°C for 2 h and then lysed with 100 μl of lysis buffer (4% Triton X-100 and 0.14% HCl in 2-propanol). The absorbance at 595 nm was measured on a microplate reader (Bio-Rad, Hercules, CA).
Colony formation assay.
Stable knockdown cells were plated on 6-well plates at 1,000 cells per well and kept in complete medium for 2 weeks. Colonies were fixed with methanol and stained with methylene blue. Colony formation was determined by counting the number of stained colonies. Triplicate wells were measured for each group.
Measurement of apoptotic cell death.
Apoptotic cells were routinely identified by annexin V (BioLegend, San Diego, CA) and 7-amino-actinomycin D (7-AAD; BD Biosciences, San Jose, CA) staining according to the manufacturers’ instructions and analyzed by flow cytometry (BD FACSVerse; BD Biosciences). Data files were analyzed by using FlowJo software (TreeStar, San Carlos, CA).
In vivo tumorigenicity assay.
Cell suspensions (4 × 106 cells) were injected subcutaneously into the right flank of female NCG mice (NOD-Prkdcem26Cd52 Il2rgem26Cd22/Nju,NOD/ShiLtJNju-based immunodeficient mice) (Model Animal Research Center of Nanjing University, China). Thirty days after injection, all mice were killed. Tumor weights and the expression of ANRIL RNA and its target genes were measured. All animals used in this study were maintained and handled according to protocols approved by Zhejiang Normal University and Huaqiao University.
Histological analyses.
Tissue samples were fixed in 10% formalin in phosphate buffer and then embedded in paraffin. Hematoxylin and eosin (H&E) staining was performed according to standard procedures. Images were captured using a confocal laser scanning microscope (TCS SP5 AOBS; Leica).
Immunoprecipitation and immunoblotting.
293FT cells were transfected with the indicated plasmids by using Lipofectamine 3000 reagent (Invitrogen). Tagged proteins were isolated from transfected 293FT cells by immunoprecipitation with anti-c-Myc (clone 9E10; Sigma-Aldrich, St. Louis, MO) or anti-FLAG M2 (Sigma-Aldrich) antibody and analyzed by immunoblotting as described above. Other antibodies used were as follows: anti-mouse IgG and anti-rabbit IgG (GE Healthcare Life Sciences); anti-EZH2, anti-p65, anti-p-IκBα, anti-IκBα, anti-caspase-3, anti-caspase-7, anti-caspase-8, anti-PARP, anti-p21, anti-cyclin D1, and anti-cyclin E1 (Cell Signaling Technology, Danvers, MA, USA); anti-trimethyl-histone H3 (Lys27) (Millipore); anti-hemagglutinin (anti-HA; Sigma-Aldrich); and anti-β-actin (Bioworld, St. Louis Park, MN, USA).
RNA immunoprecipitation.
RNA immunoprecipitation was performed using a MagnaRIP RNA binding protein immunoprecipitation kit (Millipore) according to the manufacturer’s instructions. For sequential RNA immunoprecipitation, transfected 293FT cells were lysed in radioimmunoprecipitation assay buffer and immunoprecipitated with anti-FLAG M2 agarose affinity gel. The precipitates were eluted with FLAG peptide, the eluate was diluted with radioimmunoprecipitation assay buffer and immunoprecipitated with anti-Myc antibody, and coprecipitated RNAs were detected by PCR. The EZH2 antibody used for RNA immunoprecipitation was purchased from Cell Signaling Technology. The primers used to detect ANRIL expression can be found in Table 2.
Statistical analyses.
The Mann-Whitney test using GraphPad Prism software or unpaired Student’s t test was used to determine significance where appropriate. Significance was set at an alpha value of 0.05.
ACKNOWLEDGMENTS
Zaowen Song, Wencai Wu, Mengyun Chen, Jun-ichirou Yasunaga, Masao Matsuoka, and Tiejun Zhao conceived and designed the experiments; Zaowen Song, Wencai Wu, Mengyun Chen, Wenzhao Cheng, Juntao Yu, Jinyong Fang, and Tiejun Zhao performed the experiments; Zaowen Song, Wencai Wu, Mengyun Chen, Wenzhao Cheng, Juntao Yu, Lingling Xu, and Tiejun Zhao analyzed the data; and Zaowen Song, Wencai Wu, Mengyun Chen, and Tiejun Zhao wrote the paper.
This work was supported by grants from the National Natural Science Foundation of China (no. 31470262 and 31200128 to Tiejun Zhao), a grant from the Zhejiang Province Public Welfare Technology Application Research Project (China) (no. 2015C33149 to Tiejun Zhao), a grant from the Qianjiang Talent Program of Zhejiang Province (China) (Tiejun Zhao), and a grant from the Research Program on Emerging and Reemerging Infectious Diseases from the Japan Agency for Medical Research and Development (AMED) (Masao Matsuoka).
We thank Caryl Antalis for proofreading of the manuscript.
We declare no competing interests.
REFERENCES
- 1.Matsuoka M, Jeang KT. 2007. Human T-cell leukaemia virus type 1 (HTLV-1) infectivity and cellular transformation. Nat Rev Cancer 7:270–280. doi: 10.1038/nrc2111. [DOI] [PubMed] [Google Scholar]
- 2.Journo C, Douceron E, Mahieux R. 2009. HTLV gene regulation: because size matters, transcription is not enough. Future Microbiol 4:425–440. doi: 10.2217/fmb.09.13. [DOI] [PubMed] [Google Scholar]
- 3.Lavorgna A, Harhaj EW. 2014. Regulation of HTLV-1 tax stability, cellular trafficking and NF-kappaB activation by the ubiquitin-proteasome pathway. Viruses 6:3925–3943. doi: 10.3390/v6103925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Higuchi M, Matsuda T, Mori N, Yamada Y, Horie R, Watanabe T, Takahashi M, Oie M, Fujii M. 2005. Elevated expression of CD30 in adult T-cell leukemia cell lines: possible role in constitutive NF-kappaB activation. Retrovirology 2:29. doi: 10.1186/1742-4690-2-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hironaka N, Mochida K, Mori N, Maeda M, Yamamoto N, Yamaoka S. 2004. Tax-independent constitutive IkappaB kinase activation in adult T-cell leukemia cells. Neoplasia 6:266–278. doi: 10.1593/neo.03388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamagishi M, Nakano K, Miyake A, Yamochi T, Kagami Y, Tsutsumi A, Matsuda Y, Sato-Otsubo A, Muto S, Utsunomiya A, Yamaguchi K, Uchimaru K, Ogawa S, Watanabe T. 2012. Polycomb-mediated loss of miR-31 activates NIK-dependent NF-kappaB pathway in adult T cell leukemia and other cancers. Cancer Cell 21:121–135. doi: 10.1016/j.ccr.2011.12.015. [DOI] [PubMed] [Google Scholar]
- 7.Saitoh Y, Yamamoto N, Dewan MZ, Sugimoto H, Martinez Bruyn VJ, Iwasaki Y, Matsubara K, Qi X, Saitoh T, Imoto I, Inazawa J, Utsunomiya A, Watanabe T, Masuda T, Yamamoto N, Yamaoka S. 2008. Overexpressed NF-kappaB-inducing kinase contributes to the tumorigenesis of adult T-cell leukemia and Hodgkin Reed-Sternberg cells. Blood 111:5118–5129. doi: 10.1182/blood-2007-09-110635. [DOI] [PubMed] [Google Scholar]
- 8.Esteller M. 2011. Non-coding RNAs in human disease. Nat Rev Genet 12:861–874. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- 9.Mercer TR, Dinger ME, Mattick JS. 2009. Long non-coding RNAs: insights into functions. Nat Rev Genet 10:155–159. doi: 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- 10.Batista PJ, Chang HY. 2013. Long noncoding RNAs: cellular address codes in development and disease. Cell 152:1298–1307. doi: 10.1016/j.cell.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huarte M. 2015. The emerging role of lncRNAs in cancer. Nat Med 21:1253–1261. doi: 10.1038/nm.3981. [DOI] [PubMed] [Google Scholar]
- 12.Congrains A, Kamide K, Ohishi M, Rakugi H. 2013. ANRIL: molecular mechanisms and implications in human health. Int J Mol Sci 14:1278–1292. doi: 10.3390/ijms14011278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sherborne AL, Hosking FJ, Prasad RB, Kumar R, Koehler R, Vijayakrishnan J, Papaemmanuil E, Bartram CR, Stanulla M, Schrappe M, Gast A, Dobbins SE, Ma Y, Sheridan E, Taylor M, Kinsey SE, Lightfoot T, Roman E, Irving JA, Allan JM, Moorman AV, Harrison CJ, Tomlinson IP, Richards S, Zimmermann M, Szalai C, Semsei AF, Erdelyi DJ, Krajinovic M, Sinnett D, Healy J, Gonzalez Neira A, Kawamata N, Ogawa S, Koeffler HP, Hemminki K, Greaves M, Houlston RS. 2010. Variation in CDKN2A at 9p21.3 influences childhood acute lymphoblastic leukemia risk. Nat Genet 42:492–494. doi: 10.1038/ng.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Turnbull C, Ahmed S, Morrison J, Pernet D, Renwick A, Maranian M, Seal S, Ghoussaini M, Hines S, Healey CS, Hughes D, Warren-Perry M, Tapper W, Eccles D, Evans DG, Breast Cancer Susceptibility Collaboration (UK), Hooning M, Schutte M, van den Ouweland A, Houlston R, Ross G, Langford C, Pharoah PD, Stratton MR, Dunning AM, Rahman N, Easton DF. 2010. Genome-wide association study identifies five new breast cancer susceptibility loci. Nat Genet 42:504–507. doi: 10.1038/ng.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kotake Y, Nakagawa T, Kitagawa K, Suzuki S, Liu N, Kitagawa M, Xiong Y. 2011. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15(INK4B) tumor suppressor gene. Oncogene 30:1956–1962. doi: 10.1038/onc.2010.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yap KL, Li S, Munoz-Cabello AM, Raguz S, Zeng L, Mujtaba S, Gil J, Walsh MJ, Zhou MM. 2010. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol Cell 38:662–674. doi: 10.1016/j.molcel.2010.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aguilo F, Zhou MM, Walsh MJ. 2011. Long noncoding RNA, polycomb, and the ghosts haunting INK4b-ARF-INK4a expression. Cancer Res 71:5365–5369. doi: 10.1158/0008-5472.CAN-10-4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, Chang HY. 2010. Long noncoding RNA as modular scaffold of histone modification complexes. Science 329:689–693. doi: 10.1126/science.1192002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grote P, Wittler L, Hendrix D, Koch F, Wahrisch S, Beisaw A, Macura K, Blass G, Kellis M, Werber M, Herrmann BG. 2013. The tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev Cell 24:206–214. doi: 10.1016/j.devcel.2012.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhuo C, Jiang R, Lin X, Shao M. 2017. LncRNA H19 inhibits autophagy by epigenetically silencing of DIRAS3 in diabetic cardiomyopathy. Oncotarget 8:1429–1437. doi: 10.18632/oncotarget.13637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hirata H, Hinoda Y, Shahryari V, Deng G, Nakajima K, Tabatabai ZL, Ishii N, Dahiya R. 2015. Long noncoding RNA MALAT1 promotes aggressive renal cell carcinoma through Ezh2 and interacts with miR-205. Cancer Res 75:1322–1331. doi: 10.1158/0008-5472.CAN-14-2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee JT. 2012. Epigenetic regulation by long noncoding RNAs. Science 338:1435–1439. doi: 10.1126/science.1231776. [DOI] [PubMed] [Google Scholar]
- 23.Fujikawa D, Nakagawa S, Hori M, Kurokawa N, Soejima A, Nakano K, Yamochi T, Nakashima M, Kobayashi S, Tanaka Y, Iwanaga M, Utsunomiya A, Uchimaru K, Yamagishi M, Watanabe T. 2016. Polycomb-dependent epigenetic landscape in adult T-cell leukemia. Blood 127:1790–1802. doi: 10.1182/blood-2015-08-662593. [DOI] [PubMed] [Google Scholar]
- 24.Sasaki D, Imaizumi Y, Hasegawa H, Osaka A, Tsukasaki K, Choi YL, Mano H, Marquez VE, Hayashi T, Yanagihara K, Moriwaki Y, Miyazaki Y, Kamihira S, Yamada Y. 2011. Overexpression of Enhancer of zeste homolog 2 with trimethylation of lysine 27 on histone H3 in adult T-cell leukemia/lymphoma as a target for epigenetic therapy. Haematologica 96:712–719. doi: 10.3324/haematol.2010.028605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Volkel P, Dupret B, Le Bourhis X, Angrand PO. 2015. Diverse involvement of EZH2 in cancer epigenetics. Am J Transl Res 7:175–193. [PMC free article] [PubMed] [Google Scholar]
- 26.Margueron R, Reinberg D. 2011. The Polycomb complex PRC2 and its mark in life. Nature 469:343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi B, Liang J, Yang X, Wang Y, Zhao Y, Wu H, Sun L, Zhang Y, Chen Y, Li R, Zhang Y, Hong M, Shang Y. 2007. Integration of estrogen and Wnt signaling circuits by the polycomb group protein EZH2 in breast cancer cells. Mol Cell Biol 27:5105–5119. doi: 10.1128/MCB.00162-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jung HY, Jun S, Lee M, Kim HC, Wang X, Ji H, McCrea PD, Park JI. 2013. PAF and EZH2 induce Wnt/beta-catenin signaling hyperactivation. Mol Cell 52:193–205. doi: 10.1016/j.molcel.2013.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee ST, Li Z, Wu Z, Aau M, Guan P, Karuturi RK, Liou YC, Yu Q. 2011. Context-specific regulation of NF-kappaB target gene expression by EZH2 in breast cancers. Mol Cell 43:798–810. doi: 10.1016/j.molcel.2011.08.011. [DOI] [PubMed] [Google Scholar]
- 30.Gonzalez ME, Moore HM, Li X, Toy KA, Huang W, Sabel MS, Kidwell KM, Kleer CG. 2014. EZH2 expands breast stem cells through activation of NOTCH1 signaling. Proc Natl Acad Sci U S A 111:3098–3103. doi: 10.1073/pnas.1308953111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT, Wu X, Stack EC, Loda M, Liu T, Xu H, Cato L, Thornton JE, Gregory RI, Morrissey C, Vessella RL, Montironi R, Magi-Galluzzi C, Kantoff PW, Balk SP, Liu XS, Brown M. 2012. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science 338:1465–1469. doi: 10.1126/science.1227604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Watanabe M, Nakahata S, Hamasaki M, Saito Y, Kawano Y, Hidaka T, Yamashita K, Umeki K, Taki T, Taniwaki M, Okayama A, Morishita K. 2010. Downregulation of CDKN1A in adult T-cell leukemia/lymphoma despite overexpression of CDKN1A in human T-lymphotropic virus 1-infected cell lines. J Virol 84:6966–6977. doi: 10.1128/JVI.00073-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karin M. 2006. Nuclear factor-kappaB in cancer development and progression. Nature 441:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 34.Yamaoka S, Inoue H, Sakurai M, Sugiyama T, Hazama M, Yamada T, Hatanaka M. 1996. Constitutive activation of NF-kappa B is essential for transformation of rat fibroblasts by the human T-cell leukemia virus type I Tax protein. EMBO J 15:873–887. doi: 10.1002/j.1460-2075.1996.tb00422.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamaguchi H, Hung MC. 2014. Regulation and role of EZH2 in cancer. Cancer Res Treat 46:209–222. doi: 10.4143/crt.2014.46.3.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fan H, Zhang H, Pascuzzi PE, Andrisani O. 2016. Hepatitis B virus X protein induces EpCAM expression via active DNA demethylation directed by RelA in complex with EZH2 and TET2. Oncogene 35:715–726. doi: 10.1038/onc.2015.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang F, Zhang L, Huo XS, Yuan JH, Xu D, Yuan SX, Zhu N, Zhou WP, Yang GS, Wang YZ, Shang JL, Gao CF, Zhang FR, Wang F, Sun SH. 2011. Long noncoding RNA high expression in hepatocellular carcinoma facilitates tumor growth through enhancer of zeste homolog 2 in humans. Hepatology 54:1679–1689. doi: 10.1002/hep.24563. [DOI] [PubMed] [Google Scholar]
- 38.Hu JJ, Song W, Zhang SD, Shen XH, Qiu XM, Wu HZ, Gong PH, Lu S, Zhao ZJ, He ML, Fan H. 2016. HBx-upregulated lncRNA UCA1 promotes cell growth and tumorigenesis by recruiting EZH2 and repressing p27Kip1/CDK2 signaling. Sci Rep 6:23521. doi: 10.1038/srep23521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rossetto CC, Tarrant-Elorza M, Verma S, Purushothaman P, Pari GS. 2013. Regulation of viral and cellular gene expression by Kaposi’s sarcoma-associated herpesvirus polyadenylated nuclear RNA. J Virol 87:5540–5553. doi: 10.1128/JVI.03111-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saayman S, Ackley A, Turner AW, Famiglietti M, Bosque A, Clemson M, Planelles V, Morris KV. 2014. An HIV-encoded antisense long noncoding RNA epigenetically regulates viral transcription. Mol Ther 22:1164–1175. doi: 10.1038/mt.2014.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sharma S, Kelly TK, Jones PA. 2010. Epigenetics in cancer. Carcinogenesis 31:27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Taylor GP, Matsuoka M. 2005. Natural history of adult T-cell leukemia/lymphoma and approaches to therapy. Oncogene 24:6047–6057. doi: 10.1038/sj.onc.1208979. [DOI] [PubMed] [Google Scholar]
- 43.Nosaka K, Maeda M, Tamiya S, Sakai T, Mitsuya H, Matsuoka M. 2000. Increasing methylation of the CDKN2A gene is associated with the progression of adult T-cell leukemia. Cancer Res 60:1043–1048. [PubMed] [Google Scholar]
- 44.Cedar H, Bergman Y. 2009. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet 10:295–304. doi: 10.1038/nrg2540. [DOI] [PubMed] [Google Scholar]
- 45.Vire E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, Morey L, Van Eynde A, Bernard D, Vanderwinden JM, Bollen M, Esteller M, Di Croce L, de Launoit Y, Fuks F. 2006. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 439:871–874. doi: 10.1038/nature04431. [DOI] [PubMed] [Google Scholar]
- 46.Zhao T, Yasunaga J, Satou Y, Nakao M, Takahashi M, Fujii M, Matsuoka M. 2009. Human T-cell leukemia virus type 1 bZIP factor selectively suppresses the classical pathway of NF-kappaB. Blood 113:2755–2764. doi: 10.1182/blood-2008-06-161729. [DOI] [PubMed] [Google Scholar]
- 47.Zhao T, Satou Y, Sugata K, Miyazato P, Green PL, Imamura T, Matsuoka M. 2011. HTLV-1 bZIP factor enhances TGF-beta signaling through p300 coactivator. Blood 118:1865–1876. doi: 10.1182/blood-2010-12-326199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lu J, He ML, Wang L, Chen Y, Liu X, Dong Q, Chen YC, Peng Y, Yao KT, Kung HF, Li XP. 2011. MiR-26a inhibits cell growth and tumorigenesis of nasopharyngeal carcinoma through repression of EZH2. Cancer Res 71:225–233. doi: 10.1158/0008-5472.CAN-10-1850. [DOI] [PubMed] [Google Scholar]