

Influenza virus is a main cause of viral respiratory infection in humans as well as animals, occasionally with high mortality. Circulation of influenza viruses resistant to the matrix protein 2 (M2) inhibitor, amantadine, is highly prevalent. Moreover, the frequency of detection of viruses resistant to the neuraminidase inhibitors, including oseltamivir phosphate (OSV-P) or zanamivir, is also increasing. These issues highlight the need for discovery of new antiviral agents with different mechanisms. Salinomycin as the monovalent cation-proton antiporter exhibited consistent inhibitory effects against influenza A and B viruses. It plays multifunctional roles by blocking endosomal acidification and by inactivating the proton transport function of M2, the key steps for influenza virus uncoating. Notably, salinomycin resulted in marked therapeutic effects in influenza virus-infected mice when combined with OSV-P, suggesting that its chemical derivatives could be developed as an adjuvant antiviral therapy to treat influenza infections resistant or less sensitive to existing drugs.
KEYWORDS: M2 ion channel, antiviral agent, combination treatment, influenza virus, ionophore, salinomycin
ABSTRACT
Screening of chemical libraries with 2,000 synthetic compounds identified salinomycin as a hit against influenza A and B viruses, with 50% effective concentrations ranging from 0.4 to 4.3 μM in cells. This compound is a carboxylic polyether ionophore that exchanges monovalent ions for protons across lipid bilayer membranes. Monitoring the time course of viral infection showed that salinomycin blocked nuclear migration of viral nuclear protein (NP), the most abundant component of the viral ribonucleoprotein (vRNP) complex. It caused cytoplasmic accumulation of NP, particularly within perinuclear endosomes, during virus entry. This was primarily associated with failure to acidify the endosomal-lysosomal compartments. Similar to the case with amantadine (AMT), proton channel activity of viral matrix protein 2 (M2) was blocked by salinomycin. Using purified retroviral Gag-based virus-like particles (VLPs) with M2, it was proved that salinomycin directly affects the kinetics of a proton influx into the particles but in a manner different from that of AMT. Notably, oral administration of salinomycin together with the neuraminidase inhibitor oseltamivir phosphate (OSV-P) led to enhanced antiviral effect over that with either compound used alone in influenza A virus-infected mouse models. These results provide a new paradigm for developing antivirals and their combination therapy that control both host and viral factors.
IMPORTANCE Influenza virus is a main cause of viral respiratory infection in humans as well as animals, occasionally with high mortality. Circulation of influenza viruses resistant to the matrix protein 2 (M2) inhibitor, amantadine, is highly prevalent. Moreover, the frequency of detection of viruses resistant to the neuraminidase inhibitors, including oseltamivir phosphate (OSV-P) or zanamivir, is also increasing. These issues highlight the need for discovery of new antiviral agents with different mechanisms. Salinomycin as the monovalent cation-proton antiporter exhibited consistent inhibitory effects against influenza A and B viruses. It plays multifunctional roles by blocking endosomal acidification and by inactivating the proton transport function of M2, the key steps for influenza virus uncoating. Notably, salinomycin resulted in marked therapeutic effects in influenza virus-infected mice when combined with OSV-P, suggesting that its chemical derivatives could be developed as an adjuvant antiviral therapy to treat influenza infections resistant or less sensitive to existing drugs.
INTRODUCTION
Influenza viruses belonging to the family Orthomyxoviridae harbor a genome comprising eight-segment negative-sense RNAs. These viruses are classified into three types, A, B, and C, based on variations in the nucleoprotein (NP) and matrix protein 1 (M1) (1). Influenza A viruses are further divided into subtypes distinguished by the antigenic properties of two viral surface glycoproteins, hemagglutinin (HA) and neuraminidase (NA). Although inactivated vaccines and antiviral agents are available to prevent or treat influenza A and B, these viruses still cause seasonal epidemics with 300,000 to 500,000 deaths worldwide every year (World Health Organization [http://www.who.int/mediacentre/factsheets/fs211/en/]) (2). Viral infection is initiated by binding of HA to sialic acid receptors on the epithelial cell surface, followed by engulfment of viral particles within endocytic vesicles. For successful viral infection, it is essential for the precursor HA0 to be enzymatically cleaved into two subunits, HA1 and HA2, that are linked by a single disulfide bond (3). The low-pH environment of the endosome activates the proton channel function of matrix protein 2 (M2), which is necessary for release of viral ribonucleoprotein (vRNP) complexes, comprising viral RNA, NP, and viral RNA polymerases PB2, PB1, and PA, from the M1 matrix layer. It also induces irreversible conformational changes in HA which lead to HA2-mediated fusion between the viral envelope and the endosomal membrane (4–6). Each free vRNP is transported to the nucleus, where viral RNA transcription and replication occur. Newly synthesized vRNPs are bound again to M1, which interacts with the nuclear export protein (NEP; formally called nonstructural protein 2 [NS2]) and is exported to the cytoplasm via a chromosomal maintenance 1-dependent pathway (7). Assembly of influenza virus particles requires successive migration of these vRNPs beneath the apical plasma membrane, where all structural protein components of HA, NA, M2, and M1 are arrayed. At the final stage of the viral life cycle, progeny virions bud from the cell surface with the help of NA, which cleaves terminal sialic acid from cell surface glycans.
Two classes of antivirals that target influenza virus proteins NA and M2 have been approved by the U.S. Food and Drug Administration (8, 9). The NA inhibitors, oseltamivir phosphate (OSV-P) and zanamivir, are used globally to treat influenza infection. Although they have potent broad-spectrum efficacy, the emergence of drug-resistant viruses harboring mutations in NA is a major concern. Recently, sporadic human infections caused by oseltamivir-resistant seasonal or influenza A pandemic (H1N1) 2009 viruses were reported (10). Adamantanes (amantadine and rimantadine), which are proton channel M2 inhibitors, can be used to treat influenza A, but not B. However, because almost 100% of currently circulating influenza A viruses are resistant to these drugs, M2 inhibitors are not recommended (11). Adamantane resistance is mainly conferred by Val27-to-Ala (V27A) and/or Ser31-to-Asn (S31N) mutations within M2. To overcome the limitations associated with reduced antiviral efficacy, synthesis of chemical derivatives of adamantanes, which are active against the mutant M2 proteins of influenza A viruses, was investigated (12, 13). Nevertheless, their therapeutic activity was still restricted to influenza A virus. This is not surprising since influenza A virus M2 and influenza B virus M2 have little structural or sequence homology despite their functional similarity as proton channels (14, 15). Thus, antiviral compounds with a high barrier to resistance that inhibit M2 of both types are required.
Ionophores are small molecules that facilitate movement of specific ions across lipid bilayer membranes; they are divided into electrogenic and electroneutral ionophores (16). Electrogenic ionophores, such as valinomycin, carbonyl cyanide m-chlorophenylhydrazone (CCCP), and carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP), transfer a net charge across the membrane. In contrast, electroneutral ionophores, also called carboxyl polyether ionophores, such as monensin, A23187, nigericin, salinomycin, and lasalocid acid, form zwitterionic complexes with cations and facilitate electrically neutral cation exchange diffusion. Polyether ionophores are regarded as promising bioactive molecules due to their broad-spectrum anticancer and antibacterial properties (17, 18). They were additionally reported to possess antiviral activity: zinc ionophores (pyrithione and hinikitiol) are active against picornavirus, herpes simplex virus, and coronavirus (19–21), and the sodium-selective carboxylic ionophore monensin is active against mouse polyomavirus (22). Monensin was suggested to affect the processing and intracellular transport of HA at first, but it was also proposed to stimulate the proton channel of influenza virus M2 (23, 24).
In this study, we found that another monovalent ionophore, salinomycin, which was identified through high-throughput screening (HTS) of chemical libraries, suppressed influenza A and B virus infection in cell culture. Its antiviral activity was mediated via inhibition of endosomal acidification and M2 proton channel activity simultaneously. It is noteworthy that oral administration of salinomycin together with OSV-P to mice that were infected with influenza A virus resistant to amantadine hydrochloride (AMT) or doubly resistant to AMT and OSV-P showed improved antiviral efficacy compared to their separate treatments. If a toxicity-attenuated monovalent ionophore is discovered through chemical modifications, this combination approach might provide an alternative for patients infected with influenza viruses that show reduced or no sensitivity to current existing antivirals.
RESULTS
Activity of salinomycin against influenza viruses.
We screened 2,000 chemicals for activity against influenza viruses A/Puerto Rico/8/1934 (PR8; H1N1), A/Hong Kong/8/1968 (HK; H3N2), and B/Lee/1940 (Lee) by examining cytopathic effects (CPEs) in a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)-based assay with Z′ values of >0.6 (25). Of the 9 primary hits that maintained >80% viability of influenza A or B virus-infected Madin-Darby canine kidney (MDCK) cells at a concentration of 20 μM, three compounds, including atovaquone, Evans blue, and salinomycin, were characterized to suppress viral replication in a dose-dependent manner with selectivity indices of >10 (Table 1). The antiviral tests with amantadine hydrochloride (AMT), ribavirin (RBV), and oseltamivir carboxylate (OSV-C) used as positive controls ensured the reliability of the assay. As shown in our previous report, wild-type PR8 virus was resistant to AMT (26). Sequence analysis of viral cDNA followed by alignment to a reference sequence from GenBank (accession no. EF467824) revealed that PR8 M2 possesses amino acids Ala27 (A27) and Asn31 (N31), which confer AMT resistance. Moreover, among the three hits, salinomycin, which has five ether rings and a terminal carboxylic acid (Fig. 1A), exhibited the most potent activity against the influenza strains PR8, HK, and Lee and showed high 50% effective concentration (EC50) values, ranging from 0.4 to 0.8 μM, and a 50% cytotoxic concentration (CC50) of 35.6 μM (Table 1 and Fig. 1B). To evaluate its broad-spectrum antiviral activity, a CPE assay was performed repeatedly against 17 additional influenza viruses, including 7 A/H1N1 strains (Table 2), 4 A/H3N2, A/H3N8, or A/H9N2 strains (Table 3), and 4 B strains (Table 4). These results showed that salinomycin has inhibitory effects against wild-type influenza A viruses as well as AMT- or OSV-resistant strains or even against avian influenza A/H3N8 or A/H9N2 viruses, with EC50 values between 0.4 to 4.3 μM. Western blot analysis again demonstrated that salinomycin suppressed NP, HA, M1, and M2 expression of PR8 in a dose-dependent manner (Fig. 1C). Consistent with this result, a plaque assay verified salinomycin-mediated reduction in the number of infectious influenza viral particles from the culture supernatant on days 1 and 2 postinfection (p.i.) (Fig. 1D). These data indicate that salinomycin is active against both influenza A and B viruses.
TABLE 1.
Cytopathic-effect-based antiviral assay of selected hit compounds
| Compound | CC50 (μM)a in MDCK cells | EC50 (μM)b
against influenza virus (SIc
) |
||
|---|---|---|---|---|
| PR8d | HKe | Leef | ||
| Atovaquone | >100.0 | 2.4 ± 0.2 (>41.7) | 2.1 ± 0.1 (>47.6) | 2.0 ± 0.1 (>50.0) |
| Evans blue | >100.0 | 10.6 ±1.9 (>9.4) | 5.4 ± 0.5 (>18.5) | 1.5 ± 0.2 (>66.7) |
| Salinomycin | 35.6 | 0.7 ± 0.1 (48.3) | 0.4 ± 0.1 (84.5) | 0.8 ± 0.1 (42.3) |
| AMTg | >100.0 | >100.0 (ND) | 0.9 ± 0.2 (>111.1) | >100.0 (ND) |
| RBVh | >100.0 | 18.8 ± 1.8 (>5.3) | 12.8 ± 3.2 (>7.8) | 13.5 ± 0.5 (>7.4) |
| OSV-Ci | >100.0 | 0.02 ± 0.01 (>5,000) | <0.005 (>20,000) | 0.13 ± 0.03 (>769) |
Concentration at which cell viability was reduced by 50%.
Concentration required to improve viability of influenza virus-infected MDCK cells by 50%.
SI, selectivity index (ratio of CC50 to EC50). ND, not determined.
A/Puerto Rico/8/34 (H1N1).
A/Hong Kong/8/68 (H3N2).
B/Lee/40.
AMT, amantadine hydrochloride.
RBV, ribavirin.
OSV-C, oseltamivir carboxylate.
FIG 1.

Antiviral activity of salinomycin against influenza viruses. (A) Chemical structure of salinomycin with five ether ring systems (rings A to E in red). The representative carbon positions are numbered in blue. (B) CPE-based antiviral assay. MDCK cells were mock infected (no virus) or infected with PR8 (A/H1N1 strain), HK (A/H3N2 strain), or Lee (B strain) at an MOI of 0.001. Cells were then exposed to increasing concentrations of salinomycin (ranging from 0.1 to 33.0 μM) for 3 days at 35°C, prior to analysis using an MTT assay. Cell viability ranged from 0% (cells infected with each virus) to 100% (mock-infected cells). Data are expressed as means ± SD of three replicates. n.d., not detected. (C) Western blot analysis. PR8-infected MDCK cells (MOI, 0.001) were treated with 0.1, 1.0, or 10.0 μM salinomycin. OSV-C (0.1 μM) was used as a control. On day 1 p.i., cell lysates were subjected to immunoblot analysis to detect viral proteins, including NP, HA, M1, and M2, together with a cellular protein (β-actin) used as a loading control. (D) Plaque titration. On days 1 and 2 p.i., the amount of infectious viral particles in the culture supernatant was quantified in a plaque assay from three independent samples. The number of particles from virus-infected cells not treated with salinomycin was set at 100%.
TABLE 2.
Antiviral activity of salinomycin against influenza A/H1N1 strains
| Compound | EC50 (μM) against influenza A/H1N1 virus (SIa
) |
||||||
|---|---|---|---|---|---|---|---|
| A/Brisbane/59/2007 | A/California/7/2009 | A/Korea/01/2009 | rgA/Korea/09/2009Δ53- 60(H275Y)b |
A/Korea/2785/2009c | rgA/Puerto Rico/8/ 1934(H275Y)d |
A/Taiwan/1/1986 | |
| Salinomycin | 1.4 ± 0.3 | 1.9 ± 0.2 | 1.2 ± 0.0 | 1.1 ± 0.2 | 1.7 ± 0.0 | 0.7 ± 0.1 | 3.5 ± 0.1 |
| (41.6) | (30.3) | (48.8) | (53.4) | (34.0) | (86.3) | (16.3) | |
| AMT | 0.1 ± 0.0 | >100.0 | 100.0 | 100.0 | 100.0 | 100.0 | 10.5 ± 2.6 |
| (>90.9) | (ND) | (ND) | (ND) | (ND) | (ND) | (>9.6) | |
| RBV | 28.2 ± 6.9 | 19.3 ± 0.3 | 13.1 ± 2.0 | 6.5 ± 2.5 | 38.7 ± 4.5 | 24.6 ± 0.2 | 50.2 ± 1.9 |
| (>3.6) | (>5.2) | (>7.6) | (>15.5) | (>2.6) | (>4.1) | (>2.0) | |
| OSV-C | 0.14 ± 0.02 | 0.19 ± 0.06 | <0.005 | 4.45 ± 1.97 | 2.95 ± 0.55 | 1.41 ± 0.03 | 0.99 ± 0.21 |
| (>740.7) | (>526.3) | (>20,000) | (>22.5) | (>33.9) | (>70.9) | (>101.5) | |
The selectivity index was calculated from the ratio of CC50, which is recorded in Table 1, to EC50.
Mouse-adapted, OSV-resistant strain generated by reverse genetics to have a truncation between amino acids 53 and 60 and a point mutation, H275Y, in NA of rgA/Korea/09/2009Δ53-60.
OSV-resistant strain isolated from a Korean patient.
OSV-resistant strain generated by reverse genetics to have the H275Y mutation in NA of PR8.
TABLE 3.
Antiviral activity of salinomycin against influenza virus A/H3N2, A/H3N8, and A/H9N2 strains
| Compound | EC50 (μM) against influenza A/H3N2 viruses (SI) |
EC50 (μM) against avian influenza viruses (SI) |
||||
|---|---|---|---|---|---|---|
| A/Brisbane/10/2007 | A/Perth/16/2009 | A/Seoul/11/1988 | A/Victoria/361/ 2011-like |
A/duck/Korea/GJ79/ 2007 (H3N8) |
A/chicken/Korea/MS96/1996 (H9N2) |
|
| Salinomycin | 2.5 ± 0.1 (22.4) | 2.6 ± 0.1 (21.6) | 1.3 ± 0.4 (43.2) | 1.7 ± 0.3 (34.0) | 1.6 ± 0.5 (36.2) | 4.2 ± 0.1 (13.5) |
| AMT | >100.0 (ND) | >100.0 (ND) | 0.1 ± 0.0 (> 1,000) | >100.0 (ND) | 0.5 ± 0.1 (>200.0) | 0.6 ± 0.3 (>181.8) |
| RBV | 6.8 ± 0.8 (>14.7) | 53.5 ± 1.0 (>1.9) | 12.5 ± 3.5 (>8.0) | 17.3 ± 3.0 (>5.8) | 18.5 ± 0.7 (>5.4) | 76.8 ± 4.7 (>1.3) |
| OSV-C | 0.28 ± 0.10 (>357.1) | 0.02 ± 0.01 (>5,714) | 0.01 ± 0.00 (>10,000) | <0.005 (>20,000) | 0.01 ± 0.00 (>13,333) | 0.27 ± 0.01 (>377.4) |
TABLE 4.
Antiviral activity of salinomycin against influenza B virus strains
| Compound | EC50 (μM) against influenza B viruses (SI) |
|||
|---|---|---|---|---|
| B/Florida/4/2006 | B/Panama/45/1990 | B/Taiwan/2/1962 | B/Wisconsin/1/2010-like | |
| Salinomycin | 0.4 ± 0.1 (160.3) | 0.9 ± 0.1 (29.5) | 2.5 ± 0.1 (22.9) | 3.8 ± 0.4 (15.0) |
| AMT | >100.0 (ND) | >100.0 (ND) | >100.0 (ND) | >100.0 (ND) |
| RBV | 6.7 ± 2.7 (>15.0) | 14.6 ± 2.2 (>6.8) | 16.3 ± 1.1 (>6.2) | 8.3 ± 0.1 (>12.0) |
| OSV-C | 0.11 ± 0.04 (>909.1) | 0.03 ± 0.01 (>4,000) | 0.28 ± 0.04 (>357.1) | 0.19 ± 0.01 (>540.5) |
Changes in nuclear localization of NP.
Next, we evaluated which stage of the virus life cycle is targeted by the hit compound. To investigate the underlying mechanism(s) of action, we added the test compounds to cells at different stages (e.g., during or after adsorption) and monitored changes in antiviral activity by a plaque reduction assay. We found that incubating virus with salinomycin at 4°C for 1 h (viral adsorption) had no effect on viral growth, whereas epigallocatechin gallate (EGCG) (a viral entry blocker) suppressed virus infection markedly by 92.3% (Fig. 2A) (25). It is noteworthy that at 35°C, a temperature at which PR8 follows the typical viral life cycle, salinomycin interfered with viral infectivity reversely proportional to the time of addition. In other words, its treatment for 0 to 5 or 1 to 5 h p.i. resulted in considerable and efficient inhibition, with reductions in plaque numbers of 83.2% and 79.5%, respectively. However, treatment at 2 to 5 or 4 to 5 h p.i. suppressed PR8 replication by only 60.8% and 53.4%, respectively. Taken together, the results of the time-of-addition study indicated that salinomycin could target the early stage of the influenza virus life cycle but not the very first steps, such as adsorption or attachment of viral particles to the cell surface receptors.
FIG 2.

Effects of salinomycin on the early stages of the influenza virus life cycle. (A) Time-of-addition experiments. The experimental process is described on the left. MDCK cells were infected with influenza PR8 virus for 1 h at 4°C. After removal of unadsorbed virus, the cells were incubated for an additional 4 h at 35°C. They were inoculated under different conditions, i.e., in the absence or presence of 10 µM salinomycin or EGCG. In parallel, at 1, 2, and 4 h p.i., the compounds were added to the cell culture medium. At 5 h p.i., the cell monolayers were washed with PBS and overlay medium was added to allow plaque generation. The numbers are expressed as percentages relative to plaque number from the DMSO-treated sample and represent the means ± SD of triplicate samples. (B) Confocal microscopy showing the subcellular distribution of viral NP. MDCK cells were mock infected (no virus) or infected with PR8 virus at an MOI of 2.5 for 4 h at 37°C in the presence of DMSO, salinomycin, EGCG, or RBV. The viral NP protein was detected using an NP-specific monoclonal antibody and an Alexa Fluor 488-conjugated goat anti-mouse secondary antibody (green). Nuclei were counterstained with DAPI (blue). Original magnification, ×400.
To visualize the effect of salinomycin on the early stage, we tested the intracellular distribution of NP as a representative of vRNPs by confocal microscopy at 4 h p.i. (Fig. 2B). At this time point, vRNPs are fully transported to the nucleus through receptor-mediated endocytosis to initiate RNA replication and transcription there (Fig. 2B, DMSO). Interestingly, salinomycin induced cytoplasmic retention of vRNPs, evidently indicating that it affected their nuclear migration. Meanwhile, a control image of EGCG-treated cells revealed no NP signals as a result of suppression of membrane binding or penetration of virions. Relatively weak intensities of nuclear NP in RBV-treated cells mean an inhibitory effect on viral RNA synthesis but not on vRNP shuttling. Hence, these data suggested that salinomycin restricts nuclear transport of vRNPs during the virus entry step.
Endosomal escape ability of influenza virus vRNPs.
Salinomycin is a monovalent cation ionophore isolated from Streptomyces albus and is presumed to prevent formation of proton gradients by vacuolar ATPase (V-ATPase) existing in intracellular organelles and at the plasma membrane (27, 28). Therefore, we asked whether this electroneutral ionophore neutralizes acidic intracellular compartments such as lysosomes, endosomes, or the Golgi apparatus in cells. Live MDCK cells were stimulated with salinomycin for 1 h by using bafilomycin A1 (a V-ATPase inhibitor) or chloroquine (a lysosomal lumen alkalizer) as a positive control. Intracellular vesicles were stained with acridine orange to monitor pH change (Fig. 3). Emission of red fluorescence highlighted low-pH organelles in mock cells (Fig. 3, mock). In contrast, no or weak red fluorescence was observed in the cytoplasm of cells exposed to salinomycin, similar to the case with cells exposed to bafilomycin A1. Moreover, yellow/green fluorescence was detected in chloroquine-treated cells. This result indicated that the antiviral ionophore raised the pH of acidic cytoplasmic compartments. Thus, the data suggested that salinomycin plays a role as a negative competitor of the cellular proton channels and eventually prevents acidification of the endosome-lysosome system, which is prerequisite for membrane fusion by HA2 and for viral uncoating by M2.
FIG 3.

Salinomycin inhibits endosomal acidification. MDCK cells were stimulated with DMSO (mock), salinomycin, bafilomycin A1 (a V-ATPase inhibitor), or chloroquine (an intralysosomal pH neutralizing agent) for 1 h at 35°C. Each sample was then treated with acridine orange (4 μg/ml) for 10 min. After excitation at 488 nm, merged images were captured through 590/720 nm (red) and 493/560 nm (green) band-pass filters.
Next, we investigated whether salinomycin-mediated inhibition of endocytic vesicle acidification affects endosomal trafficking pathways of influenza virus vRNPs. To explore this, A549 cells were infected at the high multiplicity of infection (MOI) of 10 of PR8 in the absence or presence of salinomycin, in which protein synthesis was arrested by addition of cycloheximide (CHX). Cells were incubated for 8 h at 35°C to allow sufficient nuclear import of incoming vRNPs and then costained for viral NP and early endosome antigen 1 (EEA1, an early endosome marker) (Fig. 4A) or lysosomal-associated membrane protein 1 (LAMP1, a late endosome marker) (Fig. 4B). Confocal microscopy visualized that NP (or vRNP) released from both the early and late endosomes migrated to the nucleus (Fig. 4A and B, left columns). However, salinomycin induced aberrant distribution of NP with accumulation in the cytoplasm (Fig. 4A and B, right columns). Addition of CHX supported the notion that the NP localized in the cytoplasm was derived from input influenza virus virions rather than from newly synthesized, nuclear exported viral products. Interestingly, NP complexed with early or late endosomes gathered around the perinuclear region (Fig. 4, merged images). We concluded that salinomycin-mediated defects in endosomal acidification may be linked with the failure of vRNPs to escape from early or late endosomal vesicles, thereby having an adverse effect on endosomal recycling.
FIG 4.

Endosomal escape of vRNP is affected by salinomycin. Influenza virus PR8-infected A549 cells (MOI, 10) were treated for 8 h with DMSO (mock) or 10 μM salinomycin in MEM supplemented with 10 μg/ml of cycloheximide. Cells were then costained for viral NP and the early endosomal marker EEA1 (A) or the late endosomal marker LAMP1 (B). NP protein was visualized with an anti-NP antibody, followed by an Alexa Fluor 633-conjugated secondary antibody (red). EEA1 and LAMP1 were detected using their specific antibodies, followed by an Alexa Fluor 488-conjugated secondary antibody (green). Nuclei were counterstained with DAPI (blue). Original magnification, ×630.
Effects on the viral proton channel M2.
Next, we asked whether salinomycin influences the function of the viral proteins, such as M2 or HA2, that are involved in endosomal escape of vRNPs. To quantify the proton channel activity of M2, we prepared retroviral Gag-based virus-like particles (VLPs) combined with PR8 M2 by ultracentrifugation of culture supernatants from transfected 293T cells. In this case, AMT-sensitive mutant PR8 M2 (PR8M2-S), which harbors amino acid substitutions V27 and S31, and AMT-resistant wild-type PR8 M2 (PR8M2-R), which harbors amino acids A27 and N31, were independently incorporated into null VLPs to compare their susceptibilities to salinomycin. Dynamic light scattering (DLS) analysis confirmed that their mean diameters were 268.3 ± 8.3 nm for the null, 256.8 ± 2.5 nm for PR8M2-S, and 238.3 ± 5.9 nm for PR8M2-M VLPs with homogeneity in size and shape (data not shown). Western blot analysis showed that the murine leukemia virus (MLV) Gag-derived capsid protein p30 (CA) was included in all VLP preparations; however, M2 was incorporated into PR8M2-S and PR8M2-R chimeric VLPs selectively (Fig. 5A). Individual VLP samples were exposed to acidic conditions (pH 4.5) and real-time activity of pH-gated proton channels was monitored using a fluorescent dye sensitive to membrane potential (Fig. 5B). At the low pH, null VLPs exhibited a basal increase in fluorescence, indicating slow migration of protons across the membrane (Fig. 5B, upper left graph). This is likely caused by cellular proton pumps incorporated within the VLP membrane or by nonselective proton diffusion. The basal proton channel activity of null VLPs did not respond to either AMT or salinomycin. However, the electrogenic ionophore FCCP complexed with VLPs led to a sudden increase in proton transport at 36 s after exposure to low pH buffer. This proton channel activity of FCCP VLPs fell gradually, eventually reaching equilibrium (Fig. 5B, upper right graph). This indicated that FCCP could transport protons bidirectionally. It is noteworthy that salinomycin compensated the effect of FCCP, suggesting that the activity of an electroneutral ionophore is more dominant than that of an electrogenic ionophore when they coexist. In addition, there was no difference between DMSO-treated and AMT-treated FCCP VLPs with respect to proton migration kinetics. Meanwhile, consistent with a previous report (29), VLPs spiked with PR8M2-S and PR8M2-R proteins allowed proton influx under acidic conditions (Fig. 5B, lower left and right graphs, respectively). As expected, PR8M2-S VLPs were sensitive to AMT, whereas PR8M2-R VLPs were not, confirming the feasibility of the M2 activity assay (compare red lines in the lower graphs of Fig. 5B). We found that salinomycin suppressed M2 proton channel activity efficiently, independent of its sensitivity to AMT (compare blue lines in the lower graphs of Fig. 5B). We calculated that it suppressed PR8M2-S and PR8M2-R function by 54% and 72%, respectively, at the endpoint (5 min) of the experiment (Fig. 5C). Similarly, the function of M2 derived from influenza B virus Lee strain (LeeM2) was inhibited by salinomycin but not AMT (Fig. 5A and D). These results suggested that salinomycin expressed antiviral activity against influenza A and B viruses in cells by hindering endocytic pathways in two ways: by preventing acidification of endosomal vesicles (Fig. 3) and by blocking M2-mediated proton migration from virus-carrying endosomes into virions (Fig. 5).
FIG 5.

Salinomycin nullifies the proton channel activity of influenza A virus M2. (A) Western blot analysis of influenza virus M2 and MLV p30. VLPs were purified by ultracentrifugation (final volume, 150 μl; 500-fold concentrated) and loaded onto a 12% SDS-PAGE gel (2 μl per well) for immunoblotting. Gag-derived p30 capsid (CA) protein was probed with an anti-Gag antibody and a secondary HRP-conjugated goat anti-rat IgG. M2 proteins fused with a His tag were visualized by incubation with an anti-6× His tag antibody and an HRP-conjugated goat anti-mouse antibody. (B) Real-time proton channel assay. Channel activity of PR8M2-S- or PR8M2-R-combined VLPs was measured at 6 s intervals for 5 min in the presence of 100 μM salinomycin or AMT after addition of 150 mM MES (pH 4.5). Null and FCCP-treated VLPs were used as controls. Values represent the averages from three independent experiments. (C) Inhibition of M2 channel activity by salinomycin at the experimental endpoint (5 min). The values represent the means ± SD. Statistical analysis was performed using a two-tailed Student’s t test. ***, P < 0.001; ****, P < 0.0001 (compared with the DMSO control). (D) Real-time proton channel assay with LeeM2-combined VLPs. Activity was measured at the same time interval and under the same conditions as for panel B.
Another putative target, HA2, was investigated for whether its membrane fusion activity is regulated by salinomycin. PR8-infected Vero E6 cells were incubated with the antiviral hit by using an HA2-specific antibody as a control to compare changes in HA-mediated cell-cell fusion efficiency in acidic buffer (Fig. 6). Microscopic images revealed large syncytia with multiple nuclei upon influenza virus infection, at pH 5.6. In contrast to the case with anti-HA2 antibody, which is a fusion blocker, efficiency of the formation of large syncytia with multiple nuclei was not affected by salinomycin (Fig. 6A). From 16 randomly selected images, it was determined that the total number of nuclei in syncytia per field was reduced by 61.4% and 79.3% by 0.5 μg/ml and 5.0 μg/ml of the anti-HA2 antibody with statistical significance, but not by salinomycin (Fig. 6B). Thus, HA2, which functions as a fusion protein during endosomal uncoating, did not seem to be a primary target of the electroneutral ionophore salinomycin.
FIG 6.
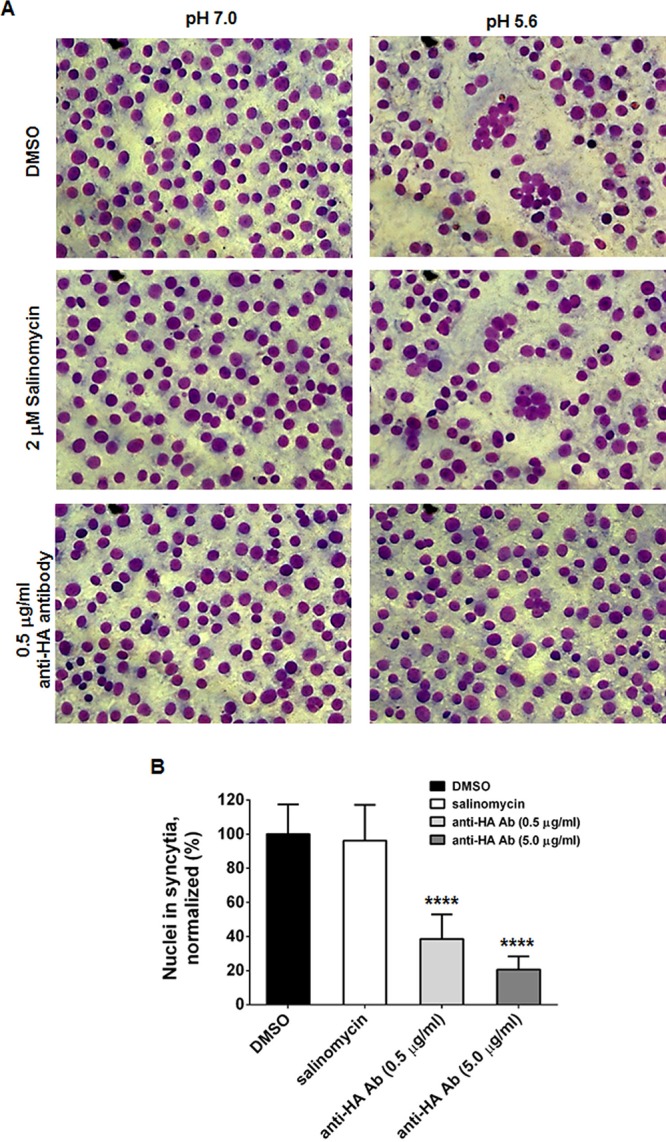
Membrane fusion of cells infected with PR8. (A) Vero E6 cells were infected with PR8 at an MOI of 0.5 at 37°C. At 16 h p.i., cells were preincubated with TPCK-trypsin (5 μg/ml) together with DMSO, 2 μM salinomycin, or 0.5 μg/ml anti-HA2 antibody. Cellular membrane fusion was initiated by exposing samples to the indicated conditions (pH 7.0 or 5.6). After staining with Giemsa, fixed cells were visualized by microscopy. Original magnification, ×200. (B) The relative number of nuclei in syncytia was counted from 16 representative images per sample at pH 5.6. Statistical significance was analyzed by comparing differences between the DMSO-treated group and the compound- or antibody-treated groups. ****, P < 0.0001.
Antiviral activity in vivo.
Prior to the in vivo antiviral study, we investigated whether the combination treatment of salinomycin with an NA inhibitor, OSV, could have the advantage of synergistic interaction. The antiviral effects of salinomycin, OSV-C, and a combination of the two compounds were compared under a multistep condition by CPE assay. EC50 values of salinomycin and OSV-C against wild-type PR8 were 1.81 and 0.07 μM, while those against recombinant PR8 with H275Y mutation in NA [named rgPR8(H275Y)] rescued by reverse genetics were 2.37 and 3.52 μM (data not shown). Isobologram analysis revealed that their combination was synergistic, as the means of the sums of fractional inhibitory concentrations at EC50 levels (FIC50) were 0.79 against wild-type PR8 (Fig. 7A and B) and 0.63 against rgPR8(H275Y) (Fig. 7C and D).
FIG 7.

Isobolograms showing the interaction between salinomycin and OSV-C against wild-type PR8 virus (A and B) and the rgPR8(H275Y) mutant strain (C and D). The numbers on the axes represent normalized FIC50s. The sums of both FIC50 values (ΣFIC50s) of fixed-ratio interaction and their mean values were quantified (B and D). Interactions were classified as synergistic, with ΣFIC50s of <0.8.
Finally, we examined the anti-influenza virus activity of salinomycin or its combination with OSV-P in a mouse model. BALB/c mice were inoculated intranasally with five 50% mouse lethal doses (MLD50) of mouse-adapted PR8 (maPR8) (Fig. 8A). Our preliminary study revealed that oral administration with salinomycin at doses of 1, 5, 10, 20, and 50 mg/kg of body weight per day for 6 days had no therapeutic effect: the body weight of infected mice failed to return to normal and in addition, salinomycin did not increase mean survival time compared with those in the virus-only group (Fig. 8B and C and data not shown). In a positive-control group, daily treatment with OSV-P (10 mg/kg) mitigated virus-induced loss of body weight, resulting in body weight recovery on day 8 and 100% survival at the endpoint. In contrast, a lower dose (0.1 mg/kg) did not show satisfactory therapeutic activity, showing 0% survival. Strikingly, combination of salinomycin (10 mg/kg) and OSV-P (0.1 mg/kg) led to significant attenuation of infection-associated body weight loss (Fig. 8B). Moreover, this combination improved survival rates up to 80% (Fig. 8C). To further investigate whether this enhanced antiviral effect in vivo is reproducible in another mouse model, mice were infected with an OSV-P-resistant influenza virus, rgK/09Δ(H275Y), where the H275Y mutation was reverse genetically introduced into the stalk-truncated NA of the mouse-adapted rgA/Korea/09/2009Δ53-60 strain (30). As expected, salinomycin (10 mg/kg) alone did not show antiviral activity in this model (data not shown). Compared to maPR8, a 10-fold higher dose of OSV-P (100 mg/kg) was consumed to apparently reduce body weight loss or to completely survive rgK/09Δ(H275Y)-infected mice (Fig. 9). There was no statistical significance in body weight changes between the OSV-P (10 mg/kg)-only group and the group treated with OSV-P in combination with salinomycin (Fig. 9A). However, this combination improved mean survival rates from 60% to 100% (Fig. 9B). Notably, therapeutic effect was most potent in the combination of salinomycin with the higher dose of OSV-P (100 mg/kg). Body weight recovery induced by OSV-P (100 mg/kg) was accelerated in the presence of salinomycin with statistical significance between days 4 and 12 p.i., resulting in complete survival in both groups (Fig. 9). It should be also stressed that both strains used for in vivo antiviral studies are resistant to AMT (Tables 1 and 2). Taken together, these studies indicated that salinomycin alone did not induce a marked therapeutic effect, but it was able to boost the antiviral activity of OSV-P when the latter was used at an otherwise ineffective dose or even against an AMT- and OSV-P-resistant strain. This enhanced antiviral effect might be attributed to targeting of various steps of the virus life cycle by two different inhibitors, one that blocks endosomal acidification and M2-mediated vRNP dissociation from the M1 shell and another that suppresses NA-dependent progeny virion release.
FIG 8.
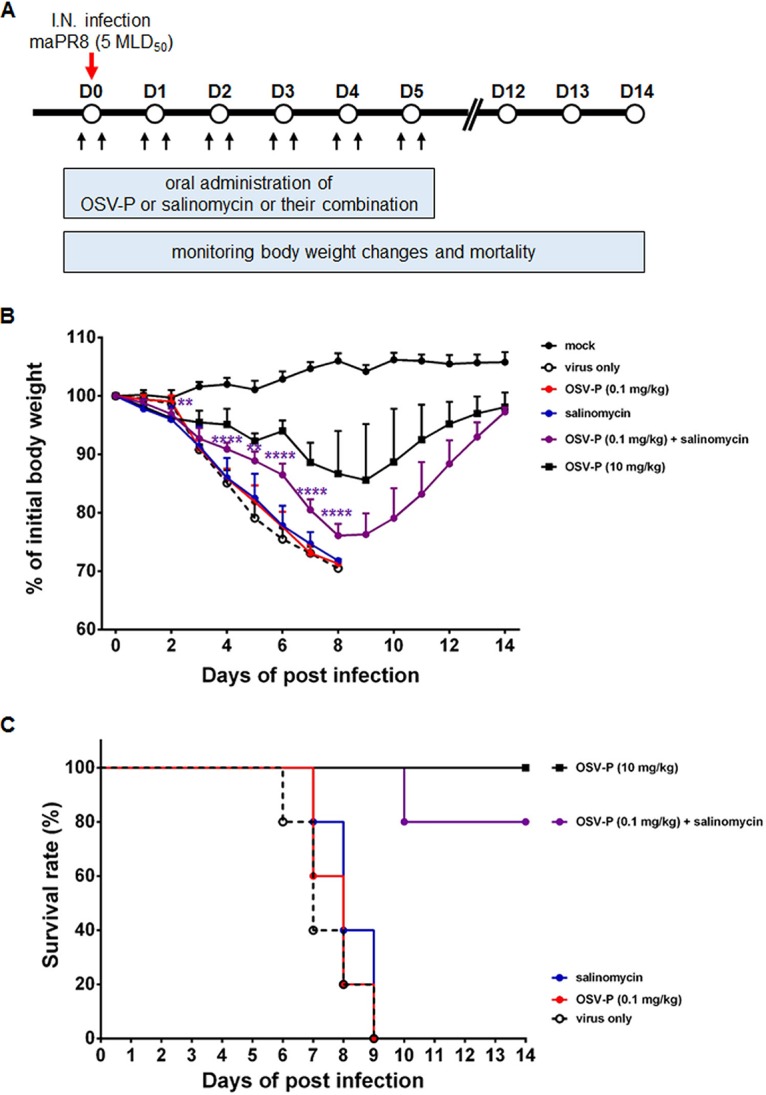
In vivo antiviral activity of salinomycin in combination with OSV-P against maPR8. (A) Schematic presentation of the antiviral study in a mouse model. Mice were inoculated intranasally with 5 MLD50 of mouse-adapted influenza virus PR8 (red arrow). Antivirals were administered orally, once 4 h prior to infection and once 4 h after, and then twice daily for further 5 days (black arrows). Groups of five mice were treated with OSV-P (0.1 mg/kg or 10 mg/kg) or salinomycin (10 mg/kg) alone or with a combination of the two (0.1 mg/kg OSV-P and 10 mg/kg salinomycin). Body weight changes (B) and mortality (C) were monitored from days 0 to 14. Statistical analysis was done using two-tailed Student’s t test relative to the OSV-P (0.1 mg/kg) group. **, P < 0.01; ****, P < 0.0001.
FIG 9.

In vivo antiviral activity of salinomycin in combination with OSV-P against rgK/09Δ(H275Y). The experimental scheme was identical with that of Fig. 8, with minor modifications of OSV-P doses. Groups of five mice were challenged with 5 MLD50 of the mouse-adapted OSV-resistant 2009 pandemic strain and treated with OSV-P (10 mg/kg or 100 mg/kg) alone or in the presence of salinomycin (10 mg/kg). Body weight changes (A) and mortality (B) were monitored from days 0 to 14. Statistical analysis was performed using two-tailed Student’s t test relative to the OSV-P (100 mg/kg) group. *, P < 0.05; **, P < 0.01; ****, P < 0.0001.
DISCUSSION
Several reports suggested that ionophores affect replication of influenza virus. For example, an electrogenic ionophore, CCCP, acts as an artificial M2-like channel to deliver protons to viral particles in influenza virus-infected cells, thereby increasing viral infectivity (31). Similarly, we and others showed that FCCP, a chemical analogue of CCCP, stimulates proton influx into retroviral Gag-based VLPs (29). Among the electroneutral ionophores, monensin is the most actively studied compound regarding its role as a monovalent cation/proton antiporter during influenza virus infection. However, those results were contradictory with respect to viral replication and infectivity. For example, Bron et al. used purified influenza virus and fluorescent pyrene-labeled liposomes to demonstrate that monensin promoted membrane fusion activity. This was an advantage for viral infection; actually a fusion assay showed that monensin was in a manner reverse to that of the M2 blocker, AMT (24). In contrast, Edwardson showed that monensin acts as an inhibitor of terminal glycosylation of HA, eventually interfering with its transport to the plasma membrane via the stacked Golgi cisternae during viral assembly (23). Similarly, Amorim et al. reported that monensin-mediated disruption of vesicular trafficking altered vRNA transport, leading to detection of perinuclear vRNA foci and nuclear retention of NP (32). In this study, we observed that salinomycin inhibited influenza A and B viruses, but via a unique mode of action different from those proposed previously. To the best of our knowledge, this is the first paper to suggest that salinomycin, a representative natural polyether ionophore, targets the uncoating step of an enveloped virus. The findings highlight its multifunctionality: the compound not only neutralizes endosomal and lysosomal vesicles but also nullifies the proton channel activity of viral M2. We studied whether the electroneutral ionophore additionally could inhibit HA2 fusion activity, another key molecule involved in viral uncoating. However, HA2 was not a direct target because HA-mediated cell-cell fusion under acidic conditions was not influenced by salinomycin (Fig. 6). Nevertheless, it is difficult exclude the possibility that it affects the fusion step indirectly by blocking endosomal acidification and by precluding conformational changes in HA.
A report demonstrated the potential benefits of combination therapy of favipiravir (T-705; a viral RNA-dependent RNA polymerase inhibitor) and OSV-P; either compound was ineffective at suboptimal doses, while their combination led to a significant increase in body weight and survival of mice infected with influenza A virus (33). Similarly, triple combination of OSV, AMT and RBV displayed synergistic antiviral activity against multiple influenza viruses (34–36). In agreement with these, we found that oral administration of salinomycin (10 mg/kg per day) and OSV-P (0.1 mg/kg per day) generated therapeutic effects in influenza virus-infected mice; neither drug showed therapeutic efficacy at the same doses when used alone (Fig. 8). The improved antiviral effect might be attributed to inhibition of multiple steps essential for virus replication, such as endosomal acidification, M2 proton channel function, and NA-mediated release of virus from the host cell. Currently, the monovalent ionophore salinomycin is approved for veterinary use as an antiprotozoal agent and is used to treat coccidiosis in poultry in addition to monensin, narasin, and lasalocid (37, 38). Although they were proposed to be effective against human cancers and infectious diseases, their potential toxicity and/or side effects limited their clinical use (39–41). We found that its combination with OSV-P did not cause changes in body weight, behavior, or food uptake of normal mice (data not shown). However, a more systematic toxicology study may be required to examine the presence of delayed or latent toxicity to mammals. We also expect that toxicities could be alleviated by synthesizing less cytotoxic chemical derivatives, as tried in previous studies (42, 43), or by conjugating the active compound to a delivery vehicle specific for virus-infected lung epithelial cells. Particularly, Brogström et al. suggested that modifications to the C20 hydroxy group within the C-ring of salinomycin (Fig. 1A) would be beneficial in the context of selectivity (43). The combined therapeutic approach with salinomycin derivatives and a lower-dose NA inhibitor may also be valuable when the stockpiles of OSV-P are insufficient to respond to an influenza pandemic or when OSV-resistant viruses are circulating globally. Or it might be useful to protect transmission of avian influenza virus from poultry to humans or among poultry by treating chickens or ducks with salinomycin in combination with different antivirals, as it could mitigate the risk of antiviral resistance emergence due to a heightened genetic barrier of multiple mutations (44, 45).
Moreover, the influenza virus strains maPR8 and rgK/09Δ(H275Y) used for the in vivo antiviral studies described here with the naturally occurring carboxylic polyether ionophore are AMT resistant (Table 2). Associated with this, cell culture-based antiviral assays revealed that salinomycin had broad-spectrum antiviral effects against all influenza A and B virus strains independent of their AMT sensitivity (Tables 1 to 4). Functional analysis using purified VLPs spiked with M2 further defined that it inhibited viral M2, irrespective of its responsiveness to AMT (Fig. 5). The results suggest that salinomycin controls fundamentally the proton channel activity of M2 via a mechanism different from that of AMT, an M2 blocker recognizing the ion channel pore and the lipid face of the pore within M2 (46). It is possible that similar to AMT, the ionophore binds directly to a third site within M2. However, based on the biological roles of electroneutral ionophores, it might be more reasonable to surmise that it facilitates countertransport of protons and Na+ ions, which exist abundantly (at a concentration of about 150 mM) under physiological conditions, bidirectionally across lipid bilayers to reach an ionic equilibrium. In the future, we are going to explore antiviral effects of salinomycin against other enveloped viruses that enter cells via endosome-mediated endocytosis and to investigate anti-influenza virus activity of detoxified salinomycin derivatives. The findings described here provide a comprehensive methodology for investigating antiviral effects of ionophores by using salinomycin as a probe and a new paradigm for combination therapy simultaneously targeting viral proteins and a cellular machinery critical for virus uncoating.
MATERIALS AND METHODS
Cells and viruses.
MDCK cells, human lung adenocarcinoma A549 cells, human embryonic kidney 293T cells, and African green monkey kidney epithelial Vero E6 cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA). MDCK and A549 cells were maintained in minimum essential medium (MEM; HyClone, Logan, UT) and RPMI 1640 medium (HyClone), respectively, supplemented with 10% fetal bovine serum (FBS; HyClone) at 37°C. 293T and Vero E6 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; HyClone) supplemented with 10% FBS.
Influenza viruses, including PR8 (A/H1N1), HK (A/H3N2), and Lee (B), were purchased from ATCC. Recombinant rgPR8(H275Y) virus was generated by reverse genetics and site-directed mutagenesis of H275Y in NA of PR8 (25, 47). At M. Park’s laboratory (Korea University, Seoul, Republic of Korea), another recombinant virus, rgK/09Δ(H275Y), was created by genetically replacing His at position 275 with Tyr in NA of the rgA/Korea/09/2009Δ53-60 strain, in which an NA stalk was truncated during mouse adaptation (30). Another mouse-adapted influenza virus, maPR8, was a kind gift from H. J. Kim (Chung-Ang University, Seoul, Republic of Korea) (26). PR8, HK, rgPR8(H275Y), and maPR8 were amplified by infection of 10-day-old embryonated specific-pathogen-fre (SPF) chicken eggs at 37°C for 3 days. Lee was infected into MDCK cells for amplification at 35°C for 3 days in serum-free minimum essential medium (MEM) in the presence of 2 μg/ml of tosyl phenylalanyl chloromethyl ketone (TPCK)-treated trypsin (Sigma-Aldrich, St. Louis, MO). Other influenza viruses used for antiviral assay were obtained from the ATCC, Korea Centers for Disease Control and Prevention (KCDC), and Korea Veterinary Culture Collection (KVCC), and amplified according to the suppliers’ instructions (26). Viruses were harvested by centrifugation of allantoic fluid or culture medium at 1,300 × g for 10 min. They were stocked at −70°C, and viral titers were determined in a plaque assay (48).
Plasmids.
PR8 M2 cDNA (GenBank accession no. EF467824), which encodes two amantadine-resistant amino acid residues, A27 and N31, was synthesized (Bioneer Corp., Daejeon, Republic of Korea) and cloned into the NheI and BamHI sites of pcDNA 3.1/myc-His(−) A (Invitrogen, Carlsbad, CA); the resulting construct was named pcDNA-PR8M2-R. The plasmid pcDNA-PR8M2-S, which expresses amantadine-sensitive PR8M2 harboring amino acids V27 and S31, was prepared using the same method. Plasmid pcDNA-LeeM2 was also cloned by gene synthesis of M2 cDNA derived from the Lee strain (GenBank accession no. DQ792900).
The retroviral packaging plasmid pCgp, which expresses MLV Gag-Pol, was a kind gift from Paula M. Cannon (University of Southern California, CA) (49).
Chemicals used for antiviral assays.
A chemical library comprising 2,000 small molecules, all collected from the Microsource Spectrum Collection (MicroSource Discovery Systems, Gaylordsville, CT), the Prestwick Chemical Library (Prestwick Chemical, Inc., Washington, DC), and Tocriscreen bioactive compounds (Tocris Bioscience, Bristol, UK), was provided by the Korea Chemical Bank (Daejeon, Republic of Korea).
AMT and RBV, which are viral M2 and RNA polymerase inhibitors, respectively, were purchased from Sigma-Aldrich. The NA inhibitor OSV-C and its prodrug OSV-P were obtained from U.S. Biological (Swampscott, MA) and Hanmi Pharmaceutical Co. (Gyeonggi-do, Republic of Korea), respectively. The test compound salinomycin and the control compound EGCG were purchased from Sigma-Aldrich. The purity of salinomycin and EGCG was over 95%. Except for screening of the chemical library, high-purity compound was used in all experiments. Other hit compounds, Evans blue and atovaquone, were also purchased from Sigma-Aldrich for reevaluation of their antiviral activity.
Cytopathic effect inhibition assay.
For high-content screening of the chemical library, MDCK cells were seeded in 96-well plates (3 × 104 cells per well) and infected with individual influenza viruses at an MOI of 0.001 in serum-free MEM for 1 h at 35°C or 37°C. After washing with phosphate-buffered saline (PBS), cells were treated with each of the 2,000 compounds diluted in MEM (final concentration, 20 μM) containing 2 μg/ml of TPCK-trypsin. After incubation for 3 days at the same temperature for viral infection, inhibition of influenza virus-induced CPEs was measured by addition of 2.5 mg/ml of 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) (50). Dose responses to the selected hit compounds or the reference antiviral compounds were measured by treating mock- or virus-infected cells with serial dilutions of test compounds. The CC50 and EC50 values were calculated using GraphPad Prism 6 software (GraphPad, La Jolla, CA).
Western blot analysis.
PR8-infected MDCK cells were treated with increasing concentrations of salinomycin (0.1, 1.0, and 10.0 μM) or OSV-C (0.1 μM), and culture lysates were harvested 1 day later. Lysates were loaded onto 12% SDS-PAGE gels (30 μg of total protein per well) and electrotransferred to a polyvinylidene difluoride (PVDF) membrane. Viral NP, HA, M1, and M2 proteins were detected using mouse anti-NP (catalog no. 11675-MM03; Sino Biological, Beijing, China), rabbit anti-HA2 (catalog no. 86001-RM01; Sino Biological), mouse anti-M1 (catalog no. sc-57881; Santa Cruz Biotechnology, Santa Cruz, CA), and mouse anti-M2 (catalog no. sc-32238; Santa Cruz Biotechnology) antibodies, respectively. Cellular β-actin was used as a loading control and detected with a mouse anti-β-actin antibody (catalog no. A1987; Sigma-Aldrich). Horseradish peroxidase (HRP)-conjugated goat anti-mouse or anti-rabbit secondary antibodies were used to probe membrane-bound primary antibodies (Thermo Scientific, Waltham, MA). After addition of a chemiluminescent HRP substrate (SuperSignal West Pico chemiluminescent substrate; Pierce, Rockford, IL), images were obtained using an LAS-4000 luminescent image analyzer (Fujifilm, Tokyo, Japan).
To detect M2-His fusion proteins incorporated into MLV-based VLPs, the purified, 500-fold-concentrated samples (final volume, 150 μl) were loaded onto a 12% SDS-PAGE gel (2 μl per well). Gag-derived p30 capsid (CA) protein was probed with an anti-Gag antibody (catalog no. R187; ATCC) and a secondary HRP-conjugated goat anti-rat IgG. M2 proteins fused with a myc-His tag were visualized by incubation with an anti-6× His tag antibody (catalog no. ab18184; Abcam) and an HRP-conjugated goat anti-mouse antibody.
Plaque reduction assay.
MDCK cells in 6-well plates were inoculated with PR8 (MOI, 0.001) under serum-free culture conditions in the presence of dimethyl sulfoxide (DMSO; 0.2%, vol/vol) or increasing concentrations of salinomycin (0.1, 1.0, and 10.0 μM) and 0.1 μM OSV-C. Cells were cultured at 35°C and supernatants were harvested on days 1 and 2 p.i. To titrate infectious viral particles, fresh MDCK cells were seeded into 48-well plates. On the next day, 10-fold serial dilutions (10−1 to 10−6) of the virus inoculum were treated into MDCK cells for 1 h. After washing with PBS, the cell monolayers were overlaid with overlay medium (MEM containing 0.5% carboxymethylcellulose [CMC; Sigma-Aldrich] and 2 μg/ml of TPCK-trypsin) and incubated for 3 days at 33°C. Viral plaques were visualized by staining with crystal violet (25).
For the time-of-addition experiment, MDCK cells grown to confluence in 48-well plates were infected at 4°C for 1 h with PR8 in the presence of 10 μM salinomycin or EGCG (an entry blocker). After washing with PBS to remove unadsorbed virus and chemicals, the cells were treated at 35°C with medium alone or with the individual compounds for additional 4 h. In parallel, virus-infected cells in the absence of the compounds were treated with the compounds sequentially at 1, 2, and 4 h p.i. At 5 h p.i., all samples were washed with PBS and incubated in overlay medium supplemented with 2 μg/ml of TPCK-trypsin for plaque titration as described above.
Confocal microscopy.
MDCK cells in 4-well chamber slides were infected for 4 h at 37°C with PR8 (MOI, 2.5) in the presence of DMSO (0.2%, vol/vol), salinomycin (10 μM), EGCG (100 μM), or RBV (100 μM, a polymerase inhibitor). The cells were then fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100. After blocking with 1% bovine serum albumin (BSA) and 10% normal goat serum prepared in PBS, the slides were incubated overnight at 4°C with an influenza A virus NP-specific monoclonal antibody (catalog no. sc-80481; Santa Cruz Biotechnology). The anti-NP antibody-bound cells were subsequently labeled for 1 h at room temperature with Alexa Fluor 488-conjugated goat anti-mouse IgG (Invitrogen) and counterstained with Vectashield mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories, Burlingame, CA). Laser scanning confocal microscopy was performed with a Zeiss LSM 700 confocal microscope. Images were analyzed using the ZEN program (Carl Zeiss, Thornwood, NY).
To measure pH changes in cytoplasmic membrane-enclosed vesicles, cells were stained with acridine orange as described previously (51). MDCK cells (8 × 104 per well) were cultured at 37°C in 35-mm glass-bottom dishes (Greiner Bio-One, Frickenhausen, Germany). On the next day, they were treated for 1 h at the same temperature with 100 μM salinomycin, 100 nM bafilomycin A1 (a V-ATPase inhibitor; Sigma-Aldrich), or 100 μM chloroquine (an intralysosomal pH neutralizing agent; Sigma-Aldrich). Acridine orange was added (final concentration, 4 μg/ml) and cells were examined under a confocal microscope. The excitation wavelength was 488 nm, and images were collected in two emission windows: 493 to 560 nm and 590 to 720 nm.
To investigate intracytoplasmic trapping of influenza virus NP, A549 cells seeded in 4-well chamber slides (8 × 104 cells per well) were infected with PR8 at an MOI of 10 at 4°C for 30 min. After washing with PBS, virus-infected cells were incubated at 37°C for 8 h with 0.2% DMSO or 10 μM salinomycin prepared in MEM supplemented with 10 μg/ml of CHX (Sigma-Aldrich), a protein synthesis inhibitor. The cells were then colabeled with antibodies specific for NP and EEA1 (catalog no. sc-33585; Santa Cruz Biotechnology) or for NP and LAMP1 (catalog no. 9691; Cell Signaling Technology). NP was detected with Alexa Fluor 633-conjugated goat anti-mouse IgG (Invitrogen), whereas EEA1 and LAMP1 were detected with Alexa Fluor 488-conjugated goat anti-rabbit IgG (Invitrogen).
Purification of VLPs.
Virus-like particles (VLPs) containing MLV Gag together with either PR8M2-S, PR8M2-R, or LeeM2 were prepared as described previously (29), with some modifications. Briefly, VLPs were generated by cotransfection of 293T cells (3 × 107 cells seeded in 150-mm cell culture dishes) with plasmids pCgp (15 μg) plus pcDNA-PR8M2-S, -PR8M2-R, or -LeeM2 (45 μg) using calcium phosphate. As a control, null VLPs were produced by transfection of cells with pCgp alone. At day 2 posttransfection, cell culture supernatants (total, 80 ml) were centrifuged through a 20% sucrose cushion at 16,500 × g for 2 h at 4°C in an SW-32Ti rotor (Beckman Instruments, Palo Alto, CA). Pellets were resuspended in 1 ml of PBS, followed by ultracentrifugation at 82,000 × g for 1 h at 4°C in an SW-60Ti rotor (Beckman Instruments). After resuspension of the pellet in 150 μl of 10 mM HEPES buffer, pH 7.5, particle homogeneity was measured using DLS (Zetasizer Nano series; Malvern Instruments, Malvern, UK). Two microliters of purified VLPs (about 500-fold concentrated) was separated and analyzed by microchip gel electrophoresis using the Agilent 2,200 TapeStation system with P200 screen tape (Agilent Technologies, Santa Clara, CA).
M2 proton channel assay.
Proton channel activity of VLPs was measured as described previously (29). MLV Gag-derived VLPs (null) or Gag VLP-containing PR8M2-S, PR8M2-R or LeeM2 were suspended in 10 mM HEPES (pH 7.0) and 150 mM NaCl buffer supplemented with 1% FMP-Blue dye (Molecular Devices, Sunnyvale, CA). As a positive control, null VLPs were mixed with 5 μM FCCP (Sigma-Aldrich), an ionophore that transports protons across the membrane, to generate FCCP VLPs. To measure proton channel activity, each preparation (protein content, 1 μg) was placed in a 96-well black plate and preincubated for 1 h at room temperature with 100 μM AMT or salinomycin. Twenty microliters of 150 mM 2-(N-morpholino)ethanesulfonic acid (MES; Sigma-Aldrich), pH 4.5, was then added, thereby exposing each VLP to acidic conditions. Fluorescence was measured every 6 s for 5 min in a SpectraMax M3 microplate reader (Molecular Devices, Sunnyvale, CA) at an excitation wavelength of 530 nm and an emission wavelength of 565 nm.
Fusion assay.
Formation of syncytia after viral infection was evaluated as described previously (52, 53). Vero E6 cells in a 12-well plate (3 × 105 cells/ml) were infected with PR8 at an MOI of 0.5 at 37°C without TPCK-trypsin. On the next day, virus-infected cells were stimulated with TPCK-trypsin (5 μg/ml) in DMEM at 37°C for 15 min and then incubated with 2 μM salinomycin or an anti-HA2 antibody (0.5 or 5.0 μg/ml; Sino Biological) in DMEM for additional 15 min. After washing with PBS containing 1 mM MgCl2 and 0.1 mM CaCl2 (PBS-CM), the cells were treated again with each compound dissolved in acidic or neutral PBS-CM (pH 5.6 or 7.0, respectively) for 15 min, for which the pH was adjusted with citric acid. Cell-cell fusion was allowed to occur for 3 h at the same temperature in fresh DMEM containing 10% FBS. Cells were then fixed with 96% ethanol, stained with Giemsa (Sigma-Aldrich), and visualized under a microscope at a magnification of ×200.
Determination of the interaction of the two antiviral compounds and the FIC index.
The in vitro interaction of the compounds salinomycin and OSV-C was assessed according to a previous report (54). Prior to the combination treatment, EC50 value of each compound was determined against different strains. Based on their EC50 values, salinomycin and OSV-C were taken alone (5:0 and 0:5) and in fixed ratios of 4:1, 3:2, 2:3, and 1:4. For the combination assay, the top concentrations of the six solutions were prepared to allow the EC50 of the individual compound to position around the middle point in 2-fold serial dilutions, and then six dose-response curves were created.
The FIC50s for each fixed dose ratio were calculated from the individual EC50 values obtained from the dose-response curves (55). The sum of the FIC50s (ΣFIC50s) was represented as isobolograms calculated using the following equation: ΣFIC50 = (EC50 of salinomycin in combination/EC50 of salinomycin alone) + (EC50 of OSV-C in combination/EC50 of OSV-C alone). Interactions were classified as synergistic with ΣFIC50s of <0.8.
In vivo study of antiviral efficacy.
Six- to 7-week-old female BALB/c mice (five per group; Orient Bio Inc., Seongnam, Republic of Korea) were infected intranasally with 5 MLD50 of maPR8 (corresponding to 9.0 × 101 PFU per head) or rgK/09Δ(H275Y) (corresponding to 1.5 × 105 PFU per head). The antiviral compounds, OSV-P and salinomycin, separately and in combination were prepared in 0.5% CMC (Sigma-Aldrich) for oral administration. They were treated once at 4 h before and once 4 h after virus infection, followed by twice-daily treatment 5 more successive days. Changes in body weight were monitored for 15 days from virus infection. Mice that lost more than 30% of their body weight were euthanized in accordance with ethics guidelines approved by the Institutional Animal Care and Use Committee at the Korea Research Institute of Chemical Technology. Kaplan-Meier survival curves were created using GraphPad Prism 6 (GraphPad Software, San Diego, CA).
Statistical analysis.
All the data are presented as means ± the standard deviations (SD). Comparisons between means of different groups were analyzed using an unpaired, two-tailed Student’s t test. P values below 0.05 were considered statistically significant.
ACKNOWLEDGMENTS
This work was supported by a grant from KRICT (KK1803-E00), the Transgovernmental Enterprise for Pandemic Influenza in Korea (TEPIK) (A103001), and the UST ICORE Research Program (2017IC0203). We thank the Korea Chemical Bank for providing chemicals for HTS and for reporting liquid chromatographu-mass spectrometry (LC-MS) and nuclear magnetic resonance (NMR) data.
REFERENCES
- 1.Palese P, Shaw ML. 2007. Orthomyxoviridae: the viruses and their replication, p 1647–1690. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed Lippincott Williams and Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Clem A, Galwankar S. 2009. Seasonal influenza: waiting for the next pandemic. J Glob Infect Dis 1:51–56. doi: 10.4103/0974-777X.52983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen J, Lee KH, Steinhauer DA, Stevens DJ, Skehel JJ, Wiley DC. 1998. Structure of the hemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell 95:409–417. doi: 10.1016/S0092-8674(00)81771-7. [DOI] [PubMed] [Google Scholar]
- 4.Rogers GN, Paulson JC, Daniels RS, Skehel JJ, Wilson IA, Wiley DC. 1983. Single amino acid substitutions in influenza haemagglutinin change receptor binding specificity. Nature 304:76–78. doi: 10.1038/304076a0. [DOI] [PubMed] [Google Scholar]
- 5.Skehel JJ, Wiley DC. 2000. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem 69:531–569. doi: 10.1146/annurev.biochem.69.1.531. [DOI] [PubMed] [Google Scholar]
- 6.Wharton SA, Calder LJ, Ruigrok RW, Skehel JJ, Steinhauer DA, Wiley DC. 1995. Electron microscopy of antibody complexes of influenza virus haemagglutinin in the fusion pH conformation. EMBO J 14:240–246. doi: 10.1002/j.1460-2075.1995.tb06997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neumann G, Hughes MT, Kawaoka Y. 2000. Influenza A virus NS2 protein mediates vRNP nuclear export through NES-independent interaction with hCRM1. EMBO J 19:6751–6758. doi: 10.1093/emboj/19.24.6751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Clercq E. 2006. Antiviral agents active against influenza A viruses. Nat Rev Drug Discov 5:1015–1025. doi: 10.1038/nrd2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hsu J, Santesso N, Mustafa R, Brozek J, Chen YL, Hopkins JP, Cheung A, Hovhannisyan G, Ivanova L, Flottorp SA, Saeterdal I, Wong AD, Tian J, Uyeki TM, Akl EA, Alonso-Coello P, Smaill F, Schunemann HJ. 2012. Antivirals for treatment of influenza: a systematic review and meta-analysis of observational studies. Ann Intern Med 156:512–524. doi: 10.7326/0003-4819-156-7-201204030-00411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen H, Cheung CL, Tai H, Zhao P, Chan JF, Cheng VC, Chan KH, Yuen KY. 2009. Oseltamivir-resistant influenza A pandemic (H1N1) 2009 virus, Hong Kong, China. Emerg Infect Dis 15:1970–1972. doi: 10.3201/eid1512.091057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Y, Liu Q, Wang D, Chen S, Wang S. 2013. Simultaneous detection of oseltamivir- and amantadine-resistant influenza by oligonucleotide microarray visualization. PLoS One 8:e57154. doi: 10.1371/journal.pone.0057154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li F, Ma C, Hu Y, Wang Y, Wang J. 2016. Discovery of potent antivirals against amantadine-resistant influenza A viruses by targeting the M2-S31N proton channel. ACS Infect Dis 2:726–733. doi: 10.1021/acsinfecdis.6b00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rey-Carrizo M, Torres E, Ma C, Barniol-Xicota M, Wang J, Wu Y, Naesens L, DeGrado WF, Lamb RA, Pinto LH, Vazquez S. 2013. 3-Azatetracyclo[5.2.1.1(5,8).0(1,5)]undecane derivatives: from wild-type inhibitors of the M2 ion channel of influenza A virus to derivatives with potent activity against the V27A mutant. J Med Chem 56:9265–9274. doi: 10.1021/jm401340p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Williams JK, Shcherbakov AA, Wang J, Hong M. 2017. Protonation equilibria and pore-opening structure of the dual-histidine influenza B virus M2 transmembrane proton channel from solid-state NMR. J Biol Chem doi: 10.1074/jbc.M117.813998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Williams JK, Tietze D, Lee M, Wang J, Hong M. 2016. Solid-state NMR investigation of the conformation, proton conduction, and hydration of the influenza B virus M2 transmembrane proton channel. J Am Chem Soc 138:8143–8155. doi: 10.1021/jacs.6b03142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dobler M. 1981. Ionophores and their structures. John Wiley and Sons Ltd, New York, NY. [Google Scholar]
- 17.Kevin Ii DA, Meujo DA, Hamann MT. 2009. Polyether ionophores: broad-spectrum and promising biologically active molecules for the control of drug-resistant bacteria and parasites. Expert Opin Drug Discov 4:109–146. doi: 10.1517/17460440802661443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huczynski A. 2012. Polyether ionophores—promising bioactive molecules for cancer therapy. Bioorg Med Chem Lett 22:7002–7010. doi: 10.1016/j.bmcl.2012.09.046. [DOI] [PubMed] [Google Scholar]
- 19.Krenn BM, Gaudernak E, Holzer B, Lanke K, Van Kuppeveld FJ, Seipelt J. 2009. Antiviral activity of the zinc ionophores pyrithione and hinokitiol against picornavirus infections. J Virol 83:58–64. doi: 10.1128/JVI.01543-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qiu M, Chen Y, Chu Y, Song S, Yang N, Gao J, Wu Z. 2013. Zinc ionophores pyrithione inhibits herpes simplex virus replication through interfering with proteasome function and NF-kappaB activation. Antiviral Res 100:44–53. doi: 10.1016/j.antiviral.2013.07.001. [DOI] [PubMed] [Google Scholar]
- 21.Te Velthuis AJ, van den Worm SH, Sims AC, Baric RS, Snijder EJ, van Hemert MJ. 2010. Zn(2+) inhibits coronavirus and arterivirus RNA polymerase activity in vitro and zinc ionophores block the replication of these viruses in cell culture. PLoS Pathog 6:e1001176. doi: 10.1371/journal.ppat.1001176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iacoangeli A, Melucci-Vigo G, Risuleo G. 2000. The ionophore monensin inhibits mouse polyomavirus DNA replication and destabilizes viral early mRNAs. Biochimie 82:35–39. doi: 10.1016/S0300-9084(00)00358-8. [DOI] [PubMed] [Google Scholar]
- 23.Edwardson JM. 1984. Effects of monensin on the processing and intracellular transport of influenza virus haemagglutinin in infected MDCK cells. J Cell Sci 65:209–221. [DOI] [PubMed] [Google Scholar]
- 24.Bron R, Kendal AP, Klenk HD, Wilschut J. 1993. Role of the M2 protein in influenza virus membrane fusion: effects of amantadine and monensin on fusion kinetics. Virology 195:808–811. doi: 10.1006/viro.1993.1435. [DOI] [PubMed] [Google Scholar]
- 25.Kim M, Kim SY, Lee HW, Shin JS, Kim P, Jung YS, Jeong HS, Hyun JK, Lee CK. 2013. Inhibition of influenza virus internalization by (-)-epigallocatechin-3-gallate. Antiviral Res 100:460–472. doi: 10.1016/j.antiviral.2013.08.002. [DOI] [PubMed] [Google Scholar]
- 26.Shin JS, Ku KB, Jang Y, Yoon YS, Shin D, Kwon OS, Go YY, Kim SS, Bae MA, Kim M. 2017. Comparison of anti-influenza virus activity and pharmacokinetics of oseltamivir free base and oseltamivir phosphate. J Microbiol 55:979–983. doi: 10.1007/s12275-017-7371-x. [DOI] [PubMed] [Google Scholar]
- 27.Lu D, Choi MY, Yu J, Castro JE, Kipps TJ, Carson DA. 2011. Salinomycin inhibits Wnt signaling and selectively induces apoptosis in chronic lymphocytic leukemia cells. Proc Natl Acad Sci U S A 108:13253–13257. doi: 10.1073/pnas.1110431108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mitani M, Yamanishi T, Miyazaki Y. 1975. Salinomycin: a new monovalent cation ionophore. Biochem Biophys Res Commun 66:1231–1236. doi: 10.1016/0006-291X(75)90490-8. [DOI] [PubMed] [Google Scholar]
- 29.Sulli C, Banik SS, Schilling J, Moser A, Xiang X, Payne R, Wanless A, Willis SH, Paes C, Rucker JB, Doranz BJ. 2013. Detection of proton movement directly across viral membranes to identify novel influenza virus M2 inhibitors. J Virol 87:10679–10686. doi: 10.1128/JVI.01190-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park S, Il Kim J, Lee I, Bae JY, Yoo K, Nam M, Kim J, Sook Park M, Song KJ, Song JW, Kee SH, Park MS. 2017. Adaptive mutations of neuraminidase stalk truncation and deglycosylation confer enhanced pathogenicity of influenza A viruses. Sci Rep 7:10928. doi: 10.1038/s41598-017-11348-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stauffer S, Feng Y, Nebioglu F, Heilig R, Picotti P, Helenius A. 2014. Stepwise priming by acidic pH and a high K+ concentration is required for efficient uncoating of influenza A virus cores after penetration. J Virol 88:13029–13046. doi: 10.1128/JVI.01430-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Amorim MJ, Bruce EA, Read EK, Foeglein A, Mahen R, Stuart AD, Digard P. 2011. A Rab11- and microtubule-dependent mechanism for cytoplasmic transport of influenza A virus viral RNA. J Virol 85:4143–4156. doi: 10.1128/JVI.02606-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smee DF, Hurst BL, Wong MH, Bailey KW, Tarbet EB, Morrey JD, Furuta Y. 2010. Effects of the combination of favipiravir (T-705) and oseltamivir on influenza A virus infections in mice. Antimicrob Agents Chemother 54:126–133. doi: 10.1128/AAC.00933-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nguyen JT, Hoopes JD, Smee DF, Prichard MN, Driebe EM, Engelthaler DM, Le MH, Keim PS, Spence RP, Went GT. 2009. Triple combination of oseltamivir, amantadine, and ribavirin displays synergistic activity against multiple influenza virus strains in vitro. Antimicrob Agents Chemother 53:4115–4126. doi: 10.1128/AAC.00476-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nguyen JT, Hoopes JD, Le MH, Smee DF, Patick AK, Faix DJ, Blair PJ, de Jong MD, Prichard MN, Went GT. 2010. Triple combination of amantadine, ribavirin, and oseltamivir is highly active and synergistic against drug resistant influenza virus strains in vitro. PLoS One 5:e9332. doi: 10.1371/journal.pone.0009332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoopes JD, Driebe EM, Kelley E, Engelthaler DM, Keim PS, Perelson AS, Rong L, Went GT, Nguyen JT. 2011. Triple combination antiviral drug (TCAD) composed of amantadine, oseltamivir, and ribavirin impedes the selection of drug-resistant influenza A virus. PLoS One 6:e29778. doi: 10.1371/journal.pone.0029778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Folz SD, Lee BL, Nowakowski LH, Conder GA. 1988. Anticoccidial evaluation of halofuginone, lasalocid, maduramicin, monensin and salinomycin. Vet Parasitol 28:1–9. doi: 10.1016/0304-4017(88)90013-1. [DOI] [PubMed] [Google Scholar]
- 38.Kadykalo S, Roberts T, Thompson M, Wilson J, Lang M, Espeisse O. 2017. The value of anticoccidials for sustainable global poultry production. Int J Antimicrob Agents doi: 10.1016/j.ijantimicag.2017.09.004. [DOI] [PubMed] [Google Scholar]
- 39.Rochdi M, Delort AM, Guyot J, Sancelme M, Gibot S, Gourcy JG, Dauphin G, Gumila C, Vial H, Jeminet G. 1996. Ionophore properties of monensin derivatives studied on human erythrocytes by 23Na NMR and K+ and H+ potentiometry: relationship with antimicrobial and antimalarial activities. J Med Chem 39:588–595. doi: 10.1021/jm9505829. [DOI] [PubMed] [Google Scholar]
- 40.Kapoor A, He R, Venkatadri R, Forman M, Arav-Boger R. 2013. Wnt modulating agents inhibit human cytomegalovirus replication. Antimicrob Agents Chemother 57:2761–2767. doi: 10.1128/AAC.00029-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dewangan J, Srivastava S, Rath SK. 2017. Salinomycin: a new paradigm in cancer therapy. Tumour Biol 39:1010428317695035. doi: 10.1177/1010428317695035. [DOI] [PubMed] [Google Scholar]
- 42.Steverding D, Antoszczak M, Huczynski A. 2016. In vitro activity of salinomycin and monensin derivatives against Trypanosoma brucei. Parasit Vectors 9:409. doi: 10.1186/s13071-016-1698-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Borgström B, Huang X, Hegardt C, Oredsson S, Strand D. 2017. Structure-activity relationships in salinomycin: cytotoxicity and phenotype selectivity of semi-synthetic derivatives. Chemistry 23:2077–2083. doi: 10.1002/chem.201603621. [DOI] [PubMed] [Google Scholar]
- 44.Parry J. 2005. Use of antiviral drug in poultry is blamed for drug resistant strains of avian flu. BMJ 331:10. doi: 10.1136/bmj.331.7507.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perelson AS, Rong L, Hayden FG. 2012. Combination antiviral therapy for influenza: predictions from modeling of human infections. J Infect Dis 205:1642–1645. doi: 10.1093/infdis/jis265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rosenberg MR, Casarotto MG. 2010. Coexistence of two adamantane binding sites in the influenza A M2 ion channel. Proc Natl Acad Sci U S A 107:13866–13871. doi: 10.1073/pnas.1002051107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tran TT, Kim M, Jang Y, Lee HW, Nguyen HT, Nguyen TN, Park HW, Le Dang Q, Kim JC. 2017. Characterization and mechanisms of anti-influenza virus metabolites isolated from the Vietnamese medicinal plant Polygonum chinense. BMC Complement Altern Med 17:162. doi: 10.1186/s12906-017-1675-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jang YJ, Achary R, Lee HW, Lee HJ, Lee CK, Han SB, Jung YS, Kang NS, Kim P, Kim M. 2014. Synthesis and anti-influenza virus activity of 4-oxo- or thioxo-4,5-dihydrofuro[3,4-c]pyridin-3(1H)-ones. Antiviral Res 107:66–75. doi: 10.1016/j.antiviral.2014.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Christodoulopoulos I, Droniou-Bonzom ME, Oldenburg JE, Cannon PM. 2010. Vpu-dependent block to incorporation of GaLV Env into lentiviral vectors. Retrovirology 7:4. doi: 10.1186/1742-4690-7-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jang Y, Lee HW, Shin JS, Go YY, Kim C, Shin D, Malpani Y, Han SB, Jung YS, Kim M. 2016. Antiviral activity of KR-23502 targeting nuclear export of influenza B virus ribonucleoproteins. Antiviral Res 134:77–88. doi: 10.1016/j.antiviral.2016.07.024. [DOI] [PubMed] [Google Scholar]
- 51.Vanderlinden E, Vanstreels E, Boons E, ter Veer W, Huckriede A, Daelemans D, Van Lommel A, Roth E, Sztaricskai F, Herczegh P, Naesens L. 2012. Intracytoplasmic trapping of influenza virus by a lipophilic derivative of aglycoristocetin. J Virol 86:9416–9431. doi: 10.1128/JVI.07032-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cotter CR, Jin H, Chen Z. 2014. A single amino acid in the stalk region of the H1N1pdm influenza virus HA protein affects viral fusion, stability and infectivity. PLoS Pathog 10:e1003831. doi: 10.1371/journal.ppat.1003831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vanderlinden E, Goktas F, Cesur Z, Froeyen M, Reed ML, Russell CJ, Cesur N, Naesens L. 2010. Novel inhibitors of influenza virus fusion: structure-activity relationship and interaction with the viral hemagglutinin. J Virol 84:4277–4288. doi: 10.1128/JVI.02325-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fivelman QL, Adagu IS, Warhurst DC. 2004. Modified fixed-ratio isobologram method for studying in vitro interactions between atovaquone and proguanil or dihydroartemisinin against drug-resistant strains of Plasmodium falciparum. Antimicrob Agents Chemother 48:4097–4102. doi: 10.1128/AAC.48.11.4097-4102.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Forkuo AD, Ansah C, Boadu KM, Boampong JN, Ameyaw EO, Gyan BA, Arku AT, Ofori MF. 2016. Synergistic anti-malarial action of cryptolepine and artemisinins. Malar J 15:89. doi: 10.1186/s12936-016-1137-5. [DOI] [PMC free article] [PubMed] [Google Scholar]