

Mechanistic target of rapamycin complex 1 (mTORC1) is a multisubunit cellular kinase that coordinates protein synthesis with changing amino acid levels. During amino acid insufficiency, mTORC1 is repressed in uninfected cells, dampening protein synthesis and potentially restricting virus reproduction. Here, we establish that HSV-1 alters the responsiveness of mTORC1 to metabolic stress resulting from amino acid insufficiency. Unlike in uninfected cells, mTORC1 remains activated in HSV-1-infected cells deprived of amino acids. Synergistic action of the HSV-1 UL46 gene product, which stimulates PI 3-kinase, and the Us3 kinase supports virus reproduction during amino acid withdrawal. These results define how HSV-1, a medically important human pathogen associated with a range of diseases, uncouples mTORC1 activation from amino acid availability. Furthermore, they help explain how the virus reproduces during physiological stress. Reproduction triggered by physiological stress is characteristic of herpesvirus infections, where lifelong latency is punctuated by episodic reactivation events.
KEYWORDS: HSV-1 replication, amino acid withdrawal, mTORC1, translation control
ABSTRACT
By sensing fundamental parameters, including nutrient availability, activated mechanistic target of rapamycin complex 1 (mTORC1) suppresses catabolic outcomes and promotes anabolic processes needed for herpes simplex virus 1 (HSV-1) productive growth. While the virus-encoded Us3 Ser/Thr kinase is required to activate mTORC1, whether stress associated with amino acid insufficiency impacts mTORC1 activation in infected cells and virus reproduction was unknown. In contrast to uninfected cells, where amino acid withdrawal inhibits mTORC1 activation, we demonstrate that mTORC1 activity is sustained in HSV-1-infected cells during amino acid insufficiency. We show that in the absence of Us3, the insensitivity of mTORC1 to amino acid withdrawal in infected cells was dependent on the host kinase Akt and establish a role for the HSV-1 UL46 gene product, which stimulates phosphatidylinositol (PI) 3-kinase signaling. Significantly, virus reproduction during amino acid insufficiency was stimulated by the viral UL46 gene product. By synergizing with Us3, UL46 reprograms mTORC1 such that it is insensitive to amino acid withdrawal and supports sustained mTORC1 activation and virus reproduction during amino acid insufficiency. This identifies an unexpected function for UL46 in supporting virus reproduction during physiological stress and identifies a new class of virus-encoded mTORC1 regulators that selectively uncouple mTORC1 activation from amino acid sufficiency.
IMPORTANCE Mechanistic target of rapamycin complex 1 (mTORC1) is a multisubunit cellular kinase that coordinates protein synthesis with changing amino acid levels. During amino acid insufficiency, mTORC1 is repressed in uninfected cells, dampening protein synthesis and potentially restricting virus reproduction. Here, we establish that HSV-1 alters the responsiveness of mTORC1 to metabolic stress resulting from amino acid insufficiency. Unlike in uninfected cells, mTORC1 remains activated in HSV-1-infected cells deprived of amino acids. Synergistic action of the HSV-1 UL46 gene product, which stimulates PI 3-kinase, and the Us3 kinase supports virus reproduction during amino acid withdrawal. These results define how HSV-1, a medically important human pathogen associated with a range of diseases, uncouples mTORC1 activation from amino acid availability. Furthermore, they help explain how the virus reproduces during physiological stress. Reproduction triggered by physiological stress is characteristic of herpesvirus infections, where lifelong latency is punctuated by episodic reactivation events.
INTRODUCTION
In addition to its fundamental role maintaining homeostasis in living cells, tissues, and organisms, amino acid (AA) availability can exert a powerful influence on virus reproduction by regulating protein production (1). To coordinate fluctuating environmental AA levels with protein synthesis, cells rely in part upon mechanistic target of rapamycin complex 1 (mTORC1), a multisubunit kinase whose activation is dependent upon AA sufficiency (2). By phosphorylating the translational repressor 4E-BP1 and ribosomal protein p70S6-kinase (S6K1), activated mTORC1 stimulates cap-dependent mRNA translation (3, 4). While viruses often activate mTORC1 signaling, how this pathway responds to AA insufficiency, which normally restricts mTORC1 activation, in virus-infected cells is not well understood (4, 5). Significantly, monitoring amino acid sufficiency offers viruses a window into host cell fitness. During acute infections, metabolic stress responses associated with nutrient insufficiency or induced by infection could limit virus replication and function as a cell-intrinsic host defense (1, 6–10). Virus infection, however, often remodels host stress responses, impacting how fundamental parameters of cellular homeostasis, including energy and AA availability, are sensed. Subverting metabolic stress responses thereby facilitates completion of the viral replicative cycle. For example, mTORC1 activation is sustained during energy insufficiency in cells infected by herpes simplex virus 1 (HSV-1) (11) or human cytomegalovirus (HCMV) (12, 13). In addition, HCMV infection maintains mTORC1 activity during AA deprivation (14, 15). Although specific viral gene products required to remodel mTORC1 responses to AA insufficiency have not been identified, they may also enable replication of latent viruses, like herpesviruses, whose reproductive growth program is triggered by physiological stress (16, 17).
HSV-1 establishes a permanent, latent infection in peripheral nervous system neurons (18, 19). While viral genes required for reproduction are repressed during latency, episodic virus productive growth in response to environmental and physiological stress results in infectious virus production and shedding from mucosal surfaces (20). Significantly, physiological stress that suppresses mTORC1 signaling in latently infected neurons promotes virus reproductive growth (16, 17). In contrast, mTORC1 activation is enforced during the HSV-1 productive growth cycle by the virus-encoded Us3 gene product, an alphaherpesvirus subfamily-specific Ser/Thr kinase (Fig. 1A) (21–23). Despite lacking primary sequence homology, other than an ATP-binding motif, with the host kinase Akt, Us3 directly phosphorylates tuberous sclerosis complex subunit 2 (TSC2) and other Akt substrates on sites targeted by Akt (21). TSC2 S939/T1462 phosphorylation catalyzed by Us3 stimulates Rheb-GTP accumulation and constitutive mTORC1 activation, which subsequently stimulate viral protein synthesis and reproduction (21). Inhibiting mTORC1 reduced viral protein production and virus reproduction; furthermore, virus replication was restricted by TSC2 in the absence of Us3 (21). This exposed a potential paradox whereby inhibiting mTORC1 triggers virus reproduction, yet mTORC1 is needed for efficient virus protein production and replication.
FIG 1.

Sensitivity of mTORC1 activation to AA insufficiency in uninfected and HSV-1-infected cells. (A) Cartoon illustrating how HSV-1 manipulates mTORC1 activation in virus-infected cells. The HSV-1-encoded Ser/Thr kinase Us3 enforces mTORC1 activation during infection by phosphorylating TSC2 residues S939 and T1462, the same residues targeted by Akt. This phosphorylation event inhibits TSC Rheb-GAP activity, allowing Rheb-GTP to activate mTORC1. VP11/12, which is encoded by the HSV-1 UL46 gene, stimulates PI 3-kinase (PI3K) to activate Akt. Akt inhibitor VIII (AKTVIII) is a small molecule that specifically inhibits Akt. Activation of mTORC1 in uninfected cells requires amino acid sufficiency. By promoting assembly of GTP-bound RagA/B with GDP-bound RagC/D, amino acids stimulate binding of mTORC1 to the RAG complex on the lysosomal membrane surface and position mTORC1 proximal to its activator Rheb. Once activated, mTORC1 phosphorylates substrates, including 4E-BP1 and p70S6K1, to stimulate productive virus replication. (B) NHDFs growth arrested by serum deprivation were mock infected (M) or infected with the WT HSV-1 F strain (WT-F). At 9 hpi, cells were incubated in AA-free RPMI 1640 for 50 min (−), incubated in AA-free RPMI 1640 for 50 min followed by a 30-min restimulation in AA-replete RPMI 1640 (±), or left in AA-replete DMEM for 50 min (+). Total protein was isolated and analyzed by immunoblotting using the indicated antibodies. Migration of hyperphosphorylated (Hyper-) and hypophosphorylated (Hypo-) 4E-BP1 is indicated to the left of the panel. HSC70 serves as a loading control. pS6K1, S6K1 phosphorylated at Thr389.
To reconcile these findings, the possibility that a viral function expressed during the reproductive cycle might modify how stress regulates mTORC1 activity in infected cells was considered. While we recently showed that Us3 subverts the host energy-sensing program to support replication during energy insufficiency (11), the responsiveness of mTORC1 signaling to AA availability in cells productively infected with HSV-1 was unknown. Here, we demonstrate that HSV-1 infection alters the responsiveness of mTORC1 to metabolic stress resulting from AA insufficiency. Unlike in uninfected cells, where mTORC1 is inhibited by AA withdrawal, mTORC1 activation is unexpectedly sustained in HSV-1-infected cells deprived of AAs. We show that the HSV-1 UL46 gene product, which stimulates phosphatidylinositol (PI) 3-kinase signaling, supports virus reproduction during AA insufficiency and synergizes with Us3 to facilitate productive replication during AA withdrawal. UL46 and Us3 define a new class of virus-encoded mTORC1 regulators that can selectively uncouple mTORC1 activation from AA sufficiency to support virus replication during metabolic stress resulting from limited AA availability.
RESULTS
Insensitivity of mTORC1 to AA insufficiency in HSV-1-infected cells.
To determine how mTORC1 responds to AA insufficiency in HSV-1-infected cells, phosphorylation of mTORC1 substrates S6K1 and 4E-BP1 was evaluated in the presence and absence of AA in the growth medium. Triplicate normal human dermal fibroblast (NHDF) cultures were mock infected or infected with wild-type (WT) HSV-1. While one replicate remained in AA-replete medium for the duration of the experiment, the remaining duplicate cultures were challenged with AA-free medium (AA withdrawal) at 9 h postinfection (hpi). After 50 min, one culture was subsequently stimulated with AA-replete medium for an additional 30 min (AA stimulation). Analysis of S6K1 and 4E-BP1 phosphorylation by immunoblotting revealed that AA withdrawal decreased the abundance of phosphorylated S6K1 and stimulated the accumulation of hypophosphorylated 4E-BP1 in mock-infected cells (Fig. 1B, compare lane 1 to lane 2). As expected, AA stimulation resulted in phospho-S6K1 accumulation and increased hyperphosphorylated 4E-BP1 levels (Fig. 1B, compare lanes 2 to lane 3). In agreement with previously published results, HSV-1 infection stimulated mTORC1, resulting in increased phospho-S6K1 and 4E-BP1 hyperphosphorylation (Fig. 1B, compare lane 1 to lane 4). However, phosphorylated S6K1 and 4E-BP1 persisted following AA withdrawal in HSV-1-infected cells compared to mock-infected cells (Fig. 1B, compare lane 2 to lane 5). Moreover, AA stimulation of infected cultures after AA withdrawal did not increase S6K1 phosphorylation or 4E-BP1 hyperphosphorylation to the extent observed in mock-infected cells under these conditions (Fig. 1B, compare lanes 2 and 3 to lanes 5 and 6). This establishes that mTORC1 signaling in HSV-1-infected cells is relatively insensitive to changes in the AA concentration and resists stress induced by AA insufficiency compared to mock-infected cells. This further suggested that virus-encoded functions responsible for controlling mTORC1 in HSV-1-infected cells might influence the responsiveness of mTORC1 signaling to AA levels.
Responsiveness of mTORC1 to AA withdrawal is antagonized by multiple viral functions.
To investigate if the HSV-1 Us3 Ser/Thr kinase influenced mTORC1 activation in infected cells during AA insufficiency, NHDFs were either mock infected or infected with WT or Us3-deficient (ΔUs3) HSV-1. At 9 hpi, duplicate cultures were exposed to AA-free medium prior to harvesting or restimulated with AA-replete medium. A control, unmanipulated culture remained in AA-replete medium until collection at 10 hpi. Cell-free lysates were fractionated by SDS-PAGE, and phosphorylation of mTORC1 substrates 4E-BP1 and S6K1 was evaluated by immunoblotting. In contrast to mock-infected cells, phospho-S6K1 and hyperphosphorylated 4E-BP1 remained detectable during AA withdrawal in cells infected with either WT HSV-1 or ΔUs3 (Fig. 2A, compare lanes 5 and 8 to lane 2). Unexpectedly, levels of S6K1 and 4E-BP1 phosphorylation during AA withdrawal and restimulation were similar in cells infected with WT and Us3-deficient viruses (Fig. 2A, compare lanes 5 and 6 to lanes 8 and 9). This raised the possibility that the insensitivity of mTORC1 in HSV-1-infected cells to AA insufficiency is not solely dependent upon Us3.
FIG 2.
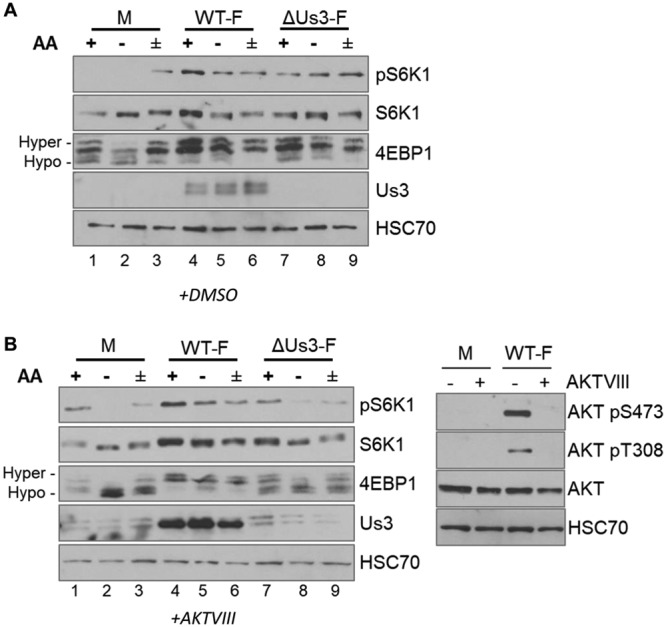
Responsiveness of mTORC1 to AA withdrawal in infected cells is regulated by the HSV-1 Us3 and host AKT Ser/Thr kinases. NHDFs growth arrested by serum deprivation were mock infected or infected with the WT HSV-1 F strain (WT-F) or a Us3-deficient virus (ΔUs3-F). At 4 hpi, cultures were treated with dimethyl sulfoxide (DMSO) (A) or the AKT inhibitor Akt VIII (B, left). At 9 hpi, cultures were either incubated in AA-free RPMI 1640 for 50 min (−), incubated in AA-free RPMI 1640 for 50 min followed by a 30-min restimulation in AA-replete RPMI 1640 (±), or left in AA-replete DMEM for 50 min (+). Total protein was isolated and analyzed by immunoblotting using the indicated antibodies. Migration of hyperphosphorylated (Hyper-) and hypophosphorylated (Hypo-) 4E-BP1 is indicated to the left. HSC70 serves as a loading control. pS6K1, S6K1 phosphorylated at Thr389. (B, right) Control showing the efficacy of Akt inhibitor VIII treatment. Total protein isolated from NHDFs mock infected (M) or infected with WT HSV-1 strain F in the presence (+) or absence (−) of Akt inhibitor VIII (AKTVIII) was analyzed by immunoblotting using the indicated phosphospecific anti-Akt antibodies (AKTpS473 and AKTpT308) or an antibody recognizing total Akt (AKT). HSC70 is a loading control.
To separate any contribution of the host kinase Akt, which can also activate mTORC1, from the HSV-1 kinase Us3, the experiment was repeated in the presence of an Akt inhibitor (Akt inhibitor VIII [Akt VIII]). Phosphorylation of S6K1 and 4E-BP1 in Akt VIII-treated cells infected with WT HSV-1 was relatively insensitive to AA withdrawal and restimulation (Fig. 2B, lanes 4 to 6). However, compared to cells infected with WT HSV-1, phosphorylation of S6K1 was reduced and higher levels of hypophosphorylated 4E-BP1 were detected in cells infected with ΔUs3 (Fig. 2B, compare lane 4 to lane 7). Furthermore, phosphorylation of both mTORC1 substrates was sensitive to AA withdrawal/restimulation in ΔUs3-infected cells (Fig. 2B, compare lanes 4, 5, and 6 to lanes 7, 8, and 9) compared to cells infected with WT HSV-1. These results were consistent with a model where Us3 and/or a host Akt-dependent function was required to sustain mTORC1 signaling during AA insufficiency in HSV-1-infected cells.
As both Us3 and Akt inhibit TSC GAP activity by phosphorylating the TSC2 subunit, whether TSC controls mTORC1 responsiveness to AAs in HSV-1-infected cells was investigated. To address this, NHDFs treated with control, nonsilencing (NS) small interfering RNA (siRNA) or siRNA specific for TSC2 (21) were mock infected or infected with ΔUs3. Akt inhibitor VIII was added at 5 hpi to exclude any contribution of Akt. Figure 3A shows that mTORC1 activation in cells infected with ΔUs3 and treated with NS siRNA remained sensitive to AA withdrawal, as evidenced by reduced S6K1 phosphorylation and accumulation of hypophosphorylated 4E-BP1 (compare lane 1 to lane 2). In addition, increased S6K1 and 4E-BP1 phosphorylation following AA addition indicated that they were likewise responsive to restimulation by AAs (Fig. 3A, compare lane 2 to lane 3). In contrast, S6K1 and 4E-BP1 phosphorylation mostly persisted in TSC2-depleted, ΔUs3-infected NHDFs upon AA withdrawal and restimulation, indicating that mTORC1 activity was insensitive to AA insufficiency (Fig. 3A, compare lane 4 to lanes 5 and 6). Significantly, TSC2 depletion in mock-infected cells, however, did not detectably change mTORC1 responsiveness to AA insufficiency (Fig. 3B, compare lane 1 to lanes 2 and 3 and compare lane 4 to lanes 5 and 6). This demonstrates that the insensitivity of mTORC1 to AA withdrawal in HSV-1-infected cells is regulated by TSC2. That TSC2 depletion in HSV-1-infected, but not uninfected, cells is sufficient to alter mTORC1 responsiveness to AAs is consistent with the possibility that additional viral functions regulate this pathway.
FIG 3.
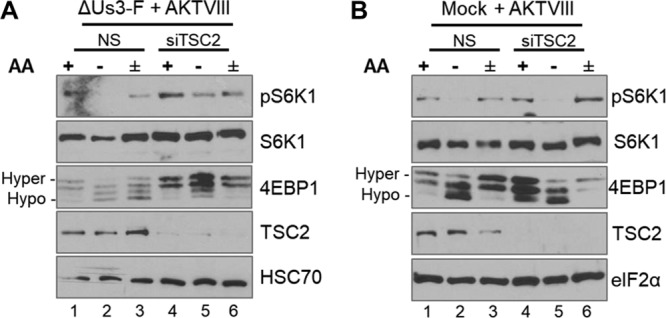
Insensitivity of mTORC1 to AA withdrawal in HSV-1-infected cells is controlled in part by TSC. Growth-arrested NHDFs treated with TSC2 siRNA (siTSC2) or control, nonsilencing siRNA (NS) were infected with the F strain ΔUs3 mutant (ΔUs3-F) (A) or mock infected (B). At 12 hpi, cultures were treated with Akt VIII, and at 15 hpi, they were either incubated in AA-free RPMI 1640 (−) for 50 min, incubated in AA-free RPMI 1640 for 50 min followed by a 30-min stimulation in AA-replete RPMI 1640 (±), or left in AA-replete DMEM for 50 min (+). Total protein was isolated, fractionated by SDS-PAGE, and analyzed by immunoblotting using the indicated antibodies. The α subunit of eukaryotic initiation factor 2 (eIF2α) and HSC70 served as loading controls for panels A and B, respectively.
While Us3 stimulates TSC2 phosphorylation to activate mTORC1 (21), the HSV-1 UL46 gene product VP11/12 stimulates PI 3-kinase and can activate Akt (22, 23). To investigate the contribution of UL46 to mTORC1 signaling during AA insufficiency, experiments were performed in the absence of Akt inhibitor VIII. NHDFs were either mock infected or infected with the WT, ΔUs3, or a virus doubly deficient for both Us3 and UL46 (ΔUs3 ΔUL46). Analysis of cell-free lysates by immunoblotting revealed that while WT HSV-1 stimulated 4E-BP1 phosphorylation and S6K1 phosphorylation (Fig. 4A, compare lanes 1 and 2), less phosphorylated p70S6K1 and more hypophosphorylated 4E-BP1 accumulated in ΔUs3-infected cells (Fig. 4A, compare lanes 2 and 3). Phosphorylation of 4E-BP1 on T37/46 is a prerequisite for subsequent S65 phosphorylation, which subsequently results in eIF4E release and stimulates cap-dependent translation (24). Reduced 4E-BP1 phosphorylation in ΔUs3-infected cells primarily reflected reductions in levels of both phospho-T37/46 and phospho-S65 (Fig. 4A). In contrast, phospo-S6K1, 4E-BP1 phospho-S65, T37/46, and hyperphosphorylated 4E-BP1 were not detectable in cells infected with a virus doubly deficient for Us3 and UL46 (Fig. 4A, lane 4). Thus, while mTORC1 activity is reduced in cells infected with ΔUs3 compared to the WT, mTORC1 activity is greater in cells infected with ΔUs3 than in cells infected with ΔUs3 ΔUL46. Stimulation of Akt by UL46 likely accounts for mTORC1 activation in ΔUs3-infected cells.
FIG 4.
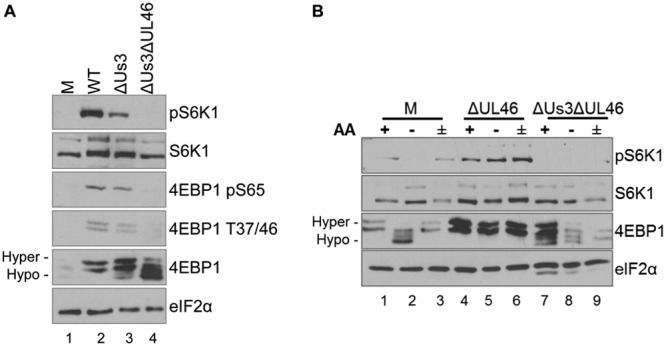
Regulation of mTORC1 sensitivity to AA insufficiency by UL46 and Us3. (A) NHDFs growth arrested by serum deprivation were mock infected or infected with HSV-1 Kos37 strain-derived viruses (wild type [WT], Us3-deficient virus [ΔUs3], or Us3/UL46 doubly deficient virus [ΔUs3 ΔUL46]). At 18 hpi, the cell lysate was fractionated by SDS-PAGE and analyzed by immunoblotting with the indicated antibodies. (B) Growth-arrested NHDFs were mock infected or infected with ΔUL46 or ΔUs3 ΔUL46. At 17 hpi, cells were either incubated in either AA-free RPMI 1640 for 50 min (−), incubated in AA-free RPMI 1640 for 50 min followed by a 30-min restimulation in AA-replete RPMI 1640 (±), or left in AA-replete DMEM for 50 min (+). Total protein was isolated and analyzed by immunoblotting using the indicated antibodies. Migration of hyperphosphorylated (Hyper-) and hypophosphorylated (Hypo-) 4E-BP1 is indicated to the left. pS6K1, S6K1 phosphorylated at Thr389.
To determine whether UL46 contributes to mTORC1 activation during AA insensitivity, NHDFs were either mock infected or infected with a UL46-deficient virus or a ΔUs3 ΔUL46. At 17 hpi, duplicate cultures were exposed to AA-free medium prior to harvesting, one of which was restimulated with AA-replete medium. A control, unmanipulated culture remained in AA-replete medium. Cell-free lysates were fractionated by SDS-PAGE, and phosphorylation of mTORC1 substrates 4E-BP1 and S6K1 was evaluated by immunoblotting. As expected, while AA withdrawal in mock-infected NHDFs reduced S6K1 phosphorylation and 4E-BP1 hyperphosphorylation, the addition of AAs stimulated both S6K1 and 4E-BP1 phosphorylation (Fig. 4B, lanes 1 to 3). However, differences in S6K1 and 4E-BP1 phosphorylation in ΔUL46-infected cells were not detected irrespective of whether AAs were withdrawn or added back to depleted cultures (Fig. 4B, lanes 4 to 6). In contrast, hypophosphorylated 4E-BP1 was more abundant in cells infected with ΔUs3 ΔUL46 (Fig. 4B, compare lanes 4 and 7), and this was reduced even further by AA withdrawal (Fig. 4B, compare lanes 7 and 8). Moreover, AA stimulation did not detectably increase 4E-BP1 phosphorylation in ΔUs3 ΔUL46-infected cells (Fig. 4B, compare lanes 8 and 9). Phosphorylated S6K1 was not detected in ΔUs3 ΔUL46-infected cells under any conditions, preventing any assessment of how this mTORC1 substrate responded to AA withdrawal and stimulation.
Synergy between Us3 and UL46 supports HSV-1 productive replication during AA insufficiency.
To evaluate the relative contributions of Us3 and UL46 to HSV-1 reproduction under conditions of AA insufficiency, NHDFs were infected with WT HSV-1, the Us3 deletion mutant ΔUs3, the UL46 deletion mutant ΔUL46, or the doubly deficient mutant ΔUs3 ΔUL46 (multiplicity of infection [MOI] = 10−2). At 5.5 hpi, the culture medium was replaced with AA-replete or AA-free RPMI 1640. The cell-free lysate was prepared at 48 hpi, and titers were determined by a plaque assay using permissive Vero cells. Us3-deficient viruses ΔUs3 and ΔUs3 ΔUL46 showed an ∼10-fold reduction in yield compared to Us3-expressing WT HSV-1 and ΔUL46 in AA-replete medium (Fig. 5). Replication of all tested viruses declined in response to AA withdrawal. While the growth of WT HSV-1 and ΔUs3 was reduced between 13- and 17-fold by AA withdrawal, the growth of ΔUL46 was reduced 32-fold, and that of the ΔUs3 ΔUL46 doubly deficient virus was reduced by 72-fold (Fig. 5A). Under these conditions, the reductions in growth of WT and Us3-deficient HSV-1 upon AA withdrawal were similar, whereas the yield of UL46-deficient virus was markedly reduced. Significantly, a deficiency for both Us3 and UL46 displayed a synthetic genetic interaction and resulted in the greatest reduction of virus reproduction in response to AA withdrawal (Fig. 5A). Thus, HSV-1 deficient in both ΔUs3 and ΔUL46 is more sensitive to AA insufficiency than either single mutant virus or than WT HSV-1. This synthetic genetic interaction between Us3 and UL46 is consistent with their acting within a common pathway to support virus reproduction during AA insufficiency.
FIG 5.
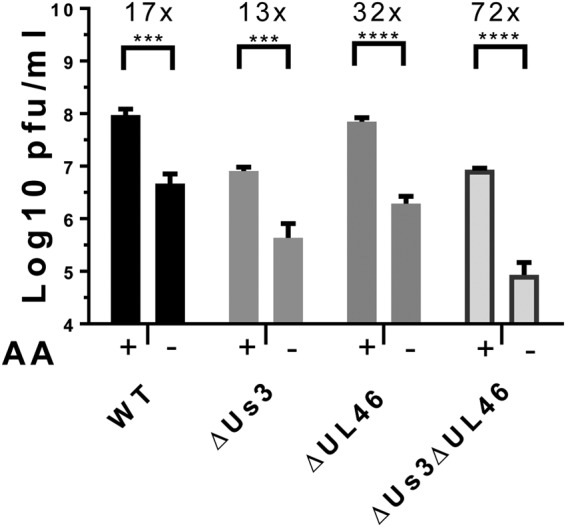
Us3 and UL46 synergistically promote HSV-1 reproduction during AA insufficiency. NHDFs were infected with HSV-1 Kos37 strain-derived viruses (wild type [WT], a Us3 deletion mutant [ΔUs3], a UL46 deletion mutant [ΔUL46]), or HSV-1 doubly deficient in Us3 and UL46 [ΔUs3 ΔUL46] at an MOI of 10−2. At 5.5 hpi, culture medium was replaced with AA-replete (+) or AA-free (−) RPMI 1640. At 48 hpi, cell-free lysates were prepared by freeze-thawing, and infectious virus was quantified by a plaque assay on Vero cells. (n = 4) (***, P < 0.001; ****, P < 0.0001 [as determined by 2-way analysis of variance {ANOVA} with multiple comparisons]). Error bars indicate standard errors of the means (SEM).
DISCUSSION
Perturbations in homeostasis resulting from infection often trigger cell-intrinsic stress responses that can restrict virus reproduction (1, 25). By sensing fundamental parameters, including nutrient availability, activated mTORC1 suppresses catabolic outcomes and promotes anabolic processes needed for virus growth (4, 5, 25, 26). In HSV-1-infected cells, the virus-encoded Us3 Ser/Thr kinase is required to activate mTORC1 (21). Here, we establish that the inhibition of mTORC1 in response to AA withdrawal is remodeled by productive HSV-1 infection. Unexpectedly, virus reproduction during AA insufficiency was stimulated by the viral UL46 gene product, which stimulates PI 3-kinase signaling and synergizes with the Us3 Ser/Thr kinase. By working together, Us3 and UL46 undermine the host response to suppress mTORC1 activation by AA insufficiency and reprogram mTORC1 such that it is insensitive to AA withdrawal. This identifies a new class of virus-encoded mTORC1 regulators that selectively uncouple mTORC1 activation from AA sufficiency to support virus replication during metabolic stress resulting from limited AA availability.
Normally, physiological stress associated with nutrient and energy insufficiency limits anabolic responses and restricts mTORC1 activation (27–29). In HSV-1-infected cells, the constitutively active Us3 Ser/Thr kinase enforces mTORC1 activation by targeting TSC2 to sustain virus reproduction during energy insufficiency (11). While Us3 in part contributes similarly to mTORC1 activation during AA insufficiency, virus reproduction is augmented by the UL46 gene product, a second virus-encoded function that synergizes with Us3 to sustain mTORC1 activation during AA withdrawal. Importantly, HSV-1-enforced mTORC1 activation during energy insufficiency required Us3 kinase activity but was not detectably dependent upon UL46 (11). This suggests that UL46 plays a specific role in maintaining mTORC1 activation in response to a discrete stress resulting from AA but not energy insufficiency. Although the exact reason underlying the differential requirement for UL46 in response to AA but not energy insufficiency is unknown, it might reflect the fact that energy insufficiency is sensed exclusively through a TSC-dependent pathway effectively countered by Us3, whereas AA sensing requires TSC-dependent and -independent inputs, with the latter likely involving the Ras-related GTP binding (RAG) proteins. Besides stimulating PI 3-kinase-dependent Akt activation in the absence of Us3, UL46 might conceivably influence the TSC-independent pathway via either PI 3-kinase or an additional presently uncharacterized mechanism. This could include targeting the cellular RAG proteins, which transduce AA-dependent signals to mTORC1, or interfering with TSC subcellular localization on Rheb-containing cytoplasmic membrane surfaces, which impairs mTORC1 inactivation in response to AA withdrawal (30, 31). Nevertheless, the synthetic genetic interaction between Us3 and UL46 supports their action within a common pathway to support virus reproduction during AA insufficiency. The molecular basis for the observed synergy between UL46 and Us3 in sustaining mTORC1 activation during AA withdrawal, however, remains unknown and requires further investigation.
The capacity to reproduce during physiological stress is an important characteristic of herpesvirus infection biology. Indeed, sustained mTORC1 activation during AA withdrawal has been documented in HCMV-infected fibroblasts (14, 15). By manipulating mTORC1 subcellular localization and redistributing it to the perinuclear virus assembly compartment in infected cells, HCMV removes mTORC1 from host membranes containing the RAG proteins, which regulate mTORC1 activity in response to AA (14). While the assembly compartment is a subcellular structure not found in alphaherpesvirus-infected cells, a related mechanism involving altering mTORC1 subcellular distribution in relation to its regulatory molecules remains possible.
Amino acid starvation can induce a panoply of responses that have a profound impact on infection biology, including activation of innate cell-intrinsic responses, and trigger reproduction of latent viruses (32, 33). In the case of HSV-1, continuous mTORC1 signaling is required to maintain latency in neurons (16). By interfering with mTORC1 signaling, physiological stress signals to relieve epigenetic repression of latent genomes stimulate lytic virus gene expression and promote virus reproduction (16, 20). Synergy among multiple viral functions, including Us3 and UL46, likely plays critical roles, enabling HSV-1 to complete the reproductive growth cycle during episodes of physiological stress, like AA and/or nutrient insufficiency, which if unchecked would restrict mTORC1 activation, virus protein synthesis, and virus replication.
MATERIALS AND METHODS
Cell culture, viruses, and chemicals.
Vero cells (ATCC) were grown in Dulbecco’s modified Eagle medium (DMEM) supplemented with 5% calf serum. Fibroblasts (NHDFs) (purchased from Lonza, Walkersville, MD) were grown in DMEM supplemented with 5% fetal bovine serum (FBS), and growth was arrested by serum deprivation in 0.2% fetal bovine serum as described previously (34). The WT HSV-1 Kos37 strain and ΔUs3, ΔUL46, and ΔUs3 ΔUL46 viruses were described previously (23). F strain HSV1 wild type (WT) and HSV1ΔUs3 (R7041) were described previously (35, 36). RPMI 1640 (catalog number R8999-04A) was purchased from U.S. Biological. Commercially dialyzed FBS (catalog number 26400-036), minimum essential medium (MEM) nonessential amino acid solution (catalog number 11140-50), and MEM amino acid solution (catalog number 11130-051) were purchased from Gibco. Akt VIII (catalog number 124018) was purchased from Calbiochem/Millipore.
Antibodies and siRNAs.
S6K1-pThr389 (catalog number 9234), S6K1 (catalog number 9202), Akt (catalog number 9272), phospho-Akt (catalog number 9271), and TSC2 (catalog number 3612) were purchased from Cell Signaling Technology. The HSC70 antibody (catalog number 10011384) was obtained from Cayman, anti-4E-BP1 (catalog number A300-501A) was obtained from Bethyl Laboratories, and the tubulin antibody (catalog number T5168) was obtained from Sigma. The Us3 antibody was a gift from B. Roizman (University of Chicago). AllStars negative-control siRNA was purchased from Qiagen, and the TSC2 siRNA (CCAAUGUCCUCUUGUCUUU) was chemically synthesized by Sigma. TSC2 was depleted as described previously (21). In summary, NHDFs were seeded in a 12-well dish and then transfected with siRNA using Lipofectamine RNAi Max (Invitrogen) according to the manufacturer’s instructions. The following day, the transfection protocol was repeated. Approximately 24 h later, cells were growth arrested by serum deprivation in 0.2% FBS for 72 h and then infected with HSV-1.
Amino acid starvation.
Commercially available dialyzed fetal bovine serum was subjected to extensive further dialysis against phosphate-buffered saline (PBS) to ensure removal of AAs as described previously (14). NHDFs were growth arrested in 0.2% dialyzed FBS for 72 h and then infected at an MOI of 5. At the indicated time points postinfection, cells were washed twice in PBS and then incubated in AA-free RPMI 1640 supplemented with dialyzed FBS for 50 min. A subset of these AA-starved cells was restimulated with RPMI 1640 supplemented with dialyzed FBS, essential amino acids, nonessential amino acids, and l-glutamine for an additional 30 min before collection.
Multicycle virus growth assay.
NHDFs were seeded in a 12-well tissue culture dish. Upon reaching confluence, cells were infected at an MOI of 10−2 in a volume of 0.3 ml per well for 1.5 h, after which the virus inoculum was removed and replaced with fresh medium. At 5.5 hpi, cells were washed twice and incubated in RPMI 1640 supplemented with 5% dialyzed FBS or in RPMI 1640 supplemented with 5% dialyzed FBS, MEM nonessential amino acids, MEM essential amino acids, and l-glutamine. At 48 hpi, cultures were collected and freeze-thawed three times, and infectious virus was quantified by a plaque assay on Vero cells.
ACKNOWLEDGMENTS
We thank members of the Mohr laboratory and Angus Wilson for stimulating discussions.
This work was supported by grants from the NIH to I.M. (R01GM056927 and AI073898) and grants from the Canadian Institutes for Health Research (FRN 12172) and Alberta Innovates-Health Solutions to J.R.S. J.R.S. holds a Canada research chair in molecular virology (tier I). E.I.V. was supported in part by NIH grant 5T32AI7647 and an American Cancer Society postdoctoral fellowship (grant PF-16-048-01-MPC).
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Tsalikis J, Croitoru DO, Philpott DJ, Girardin SE. 2013. Nutrient sensing and metabolic stress pathways in innate immunity. Cell Microbiol 15:1632–1641. doi: 10.1111/cmi.12165. [DOI] [PubMed] [Google Scholar]
- 2.Wolfson R, Sabatini DM. 2017. The dawn of the age of amino acid sensors for the mTORC1 pathway. Cell Metab 26:301–309. doi: 10.1016/j.cmet.2017.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Proud CG. 2018. Phosphorylation and signal transduction pathways in translational control. Cold Spring Harb Perspect Biol 29:a033050. doi: 10.1101/cshperspect.a03305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stern-Ginossar N, Thompson SR, Mathews MB, Mohr I. 2018. Translational control in virus-infected cells. Cold Spring Harb Perspect Biol 11:a033001. doi: 10.1101/cshperspect.a033001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mohr I, Sonenberg N. 2012. Host translation at the nexus of infection and immunity. Cell Host Microbe 12:470–483. doi: 10.1016/j.chom.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alwine JC. 2008. Modulation of host cell stress responses by human cytomegalovirus. Curr Top Microbiol Immunol 325:263–279. [DOI] [PubMed] [Google Scholar]
- 7.Shenk T, Alwine JC. 2014. Human cytomegalovirus: coordinating cellular stress, signaling, and metabolic pathways. Annu Rev Virol 1:355–374. doi: 10.1146/annurev-virology-031413-085425. [DOI] [PubMed] [Google Scholar]
- 8.Russell RC, Yuan HX, Guan KL. 2014. Autophagy regulation by nutrient signaling. Cell Res 24:42–57. doi: 10.1038/cr.2013.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choi Y, Bowman JW, Jung JU. 2018. Autophagy during viral infection—a double-edged sword. Nat Rev Microbiol 16:341–354. doi: 10.1038/s41579-018-0003-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hardie DG, Schaffer BE, Brunet A. 2016. AMPK: an energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol 26:190–201. doi: 10.1016/j.tcb.2015.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vink EI, Smiley JR, Mohr I. 2017. Subversion of host responses to energy insufficiency by Us3 supports herpes simplex virus 1 replication during stress. J Virol 91:e00295-17. doi: 10.1128/JVI.00295-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kudchodkar SB, Del Prete GQ, Maguire TG, Alwine JC. 2007. AMPK-mediated inhibition of mTOR kinase is circumvented during immediate-early times of human cytomegalovirus infection. J Virol 81:3649–3651. doi: 10.1128/JVI.02079-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moorman NJ, Cristea IM, Terhune SS, Rout MP, Chait BT, Shenk T. 2008. Human cytomegalovirus protein UL38 inhibits host cell stress responses by antagonizing the tuberous sclerosis protein complex. Cell Host Microbe 3:253–262. doi: 10.1016/j.chom.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clippinger AJ, Alwine JC. 2012. Dynein mediates the localization and activation of mTOR in normal and human cytomegalovirus-infected cells. Genes Dev 26:2015–2026. doi: 10.1101/gad.196147.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clippinger AJ, Maguire TG, Alwine JC. 2011. Human cytomegalovirus infection maintains mTOR activity and its perinuclear localization during amino acid deprivation. J Virol 85:9369–9376. doi: 10.1128/JVI.05102-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kobayashi M, Wilson AC, Chao MV, Mohr I. 2012. Control of viral latency in neurons by axonal mTOR signaling and the 4E-BP translation repressor. Genes Dev 26:1527–1532. doi: 10.1101/gad.190157.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cliffe AR, Arbuckle JH, Vogel JL, Geden MJ, Rothbart SB, Cusack CL, Strahl BD, Kristie TM, Deshmukh M. 2015. Neuronal stress pathway mediating a histone methyl/phospho switch is required for herpes simplex virus reactivation. Cell Host Microbe 18:649–658. doi: 10.1016/j.chom.2015.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roizman B, Zhou G. 2015. The 3 facets of regulation of herpes simplex virus gene expression: a critical inquiry. Virology 479–480:562–567. doi: 10.1016/j.virol.2015.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bloom DC. 2016. Alphaherpesvirus latency: a dynamic state of transcription and reactivation. Adv Virus Res 94:53–80. doi: 10.1016/bs.aivir.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 20.Wilson AC, Mohr I. 2012. A cultured affair: HSV latency and reactivation in neurons. Trends Microbiol 20:604–611. doi: 10.1016/j.tim.2012.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chuluunbaatar U, Roller R, Feldman ME, Brown S, Shokat KM, Mohr I. 2010. Constitutive mTORC1 activation by a herpesvirus Akt surrogate stimulates mRNA translation and viral replication. Genes Dev 24:2627–2639. doi: 10.1101/gad.1978310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wagner MJ, Smiley JR. 2011. Herpes simplex virus requires VP11/12 to activate Src family kinase-phosphoinositide 3-kinase-Akt signaling. J Virol 85:2803–2812. doi: 10.1128/JVI.01877-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eaton HE, Saffran HA, Wu FW, Quach K, Smiley JR. 2014. Herpes simplex virus protein kinases US3 and UL13 modulate VP11/12 phosphorylation, virion packaging, and phosphatidylinositol 3-kinase/Akt signaling activity. J Virol 88:7379–7388. doi: 10.1128/JVI.00712-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gingras AC, Raught B, Gygi SP, Niedzwiecka A, Miron M, Burley SK, Polakiewicz RD, Wyslouch-Cieszynska A, Aebersold R, Sonenberg N. 2001. Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev 15:2852–2864. doi: 10.1101/gad.912401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buchkovich NJ, Yu Y, Zampieri CA, Alwine JC. 2008. The TORrid affairs of viruses: effects of mammalian DNA viruses on the PI3K-Akt-mTOR signalling pathway. Nat Rev Microbiol 6:266–275. doi: 10.1038/nrmicro1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dibble CC, Manning BD. 2013. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat Cell Biol 15:555–564. doi: 10.1038/ncb2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jewell JL, Guan KL. 2013. Nutrient signaling to mTOR and cell growth. Trends Biochem Sci 38:233–242. doi: 10.1016/j.tibs.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yuan HX, Xiong Y, Guan KL. 2013. Nutrient sensing, metabolism, and cell growth control. Mol Cell 49:379–387. doi: 10.1016/j.molcel.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saxton RA, Sabatini DM. 2017. mTOR signaling in growth, metabolism, and disease. Cell 168:960–976. doi: 10.1016/j.cell.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. 2008. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320:1496–1501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Demetriades C, Doumpas N, Teleman AA. 2014. Regulation of TORC1 in response to amino acid starvation via lysosomal recruitment of TSC2. Cell 156:786–799. doi: 10.1016/j.cell.2014.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tattoli I, Sorbara MT, Vuckovic D, Ling A, Soares F, Carneiro LA, Yang C, Emili A, Philpott DJ, Girardin SE. 2012. Amino acid starvation induced by invasive bacterial pathogens triggers an innate host defense program. Cell Host Microbe 11:563–575. doi: 10.1016/j.chom.2012.04.012. [DOI] [PubMed] [Google Scholar]
- 33.Palmisano I, Della Chiara G, D’Ambrosio RL, Huichalaf C, Brambilla P, Corbetta S, Riba M, Piccirillo R, Valente S, Casari G, Mai A, Martinelli Boneschi F, Gabellini D, Poli G, Schiaffino MV. 2012. Amino acid starvation induces reactivation of silenced transgenes and latent HIV-1 provirus via down-regulation of histone deacetylase 4 (HDAC4). Proc Natl Acad Sci U S A 109:E2284–E2293. doi: 10.1073/pnas.1202174109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Walsh D, Mohr I. 2004. Phosphorylation of eIF4E by Mnk-1 enhances HSV-1 translation and replication in quiescent cells. Genes Dev 18:660–672. doi: 10.1101/gad.1185304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ryckman BJ, Roller RJ. 2004. Herpes simplex virus type 1 primary envelopment: UL34 protein modification and the US3-UL34 catalytic relationship. J Virol 78:399–412. doi: 10.1128/JVI.78.1.399-412.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Purves FC, Longnecker RM, Leader DP, Roizman B. 1987. Herpes simplex virus 1 protein kinase is encoded by open reading frame US3 which is not essential for virus growth in cell culture. J Virol 61:2896–2901. [DOI] [PMC free article] [PubMed] [Google Scholar]