

Pseudorabies virus (PRV) is an alphaherpesvirus related to human pathogens such as herpes simplex virus 1 and varicella-zoster virus (VZV). The natural host of PRV is the swine, but it can infect most mammals. In susceptible animals other than pigs, PRV infection always causes a characteristic lethal pruritus known as the “mad itch.” The role of the immune response in the clinical outcome of PRV infection is still poorly understood. Here, we show that a systemic host inflammatory response is responsible for the severe pruritus and acute death of mice infected with virulent PRV-Becker but not mice infected with attenuated strain PRV-Bartha. In addition, we identified IL-6 and G-CSF as two main cytokines that play crucial roles in the regulation of this process. Our findings give new insights into neuroinflammatory diseases and strengthen further the similarities between VZV and PRV infections at the level of innate immunity.
KEYWORDS: pseudorabies virus, cytokines, nervous system, mice, immunopathogenesis, inflammation
ABSTRACT
Pseudorabies virus (PRV) is an alphaherpesvirus that infects the peripheral nervous system (PNS). The natural host of PRV is the swine, but it can infect most mammals, including cattle, rodents, and dogs. In these nonnatural hosts, PRV always causes a severe acute and lethal neuropathy called the “mad itch,” which is uncommon in swine. Thus far, the pathophysiological and immunological processes leading to the development of the neuropathic itch and the death of the animal are unclear. Using a footpad inoculation model, we established that mice inoculated with PRV-Becker (virulent strain) develop a severe pruritus in the foot and become moribund at 82 h postinoculation (hpi). We found necrosis and inflammation with a massive neutrophil infiltration only in the footpad and dorsal root ganglia (DRGs) by hematoxylin and eosin staining. PRV load was detected in the foot, PNS, and central nervous system tissues by quantitative reverse transcription-PCR. Infected mice had elevated plasma levels of proinflammatory cytokines (interleukin-6 [IL-6] and granulocyte colony-stimulating factor [G-CSF]) and chemokines (Gro-1 and monocyte chemoattractant protein 1). Significant IL-6 and G-CSF levels were detected in several tissues at 82 hpi. High plasma levels of C-reactive protein confirmed the acute inflammatory response to PRV-Becker infection. Moreover, mice inoculated with PRV-Bartha (attenuated, live vaccine strain) did not develop pruritus at 82 hpi. PRV-Bartha also replicated in the PNS, and the infection spread further in the brain than PRV-Becker. PRV-Bartha infection did not induce the specific and lethal systemic inflammatory response seen with PRV-Becker. Overall, we demonstrated the importance of inflammation in the clinical outcome of PRV infection in mice and provide new insights into the process of PRV-induced neuroinflammation.
IMPORTANCE Pseudorabies virus (PRV) is an alphaherpesvirus related to human pathogens such as herpes simplex virus 1 and varicella-zoster virus (VZV). The natural host of PRV is the swine, but it can infect most mammals. In susceptible animals other than pigs, PRV infection always causes a characteristic lethal pruritus known as the “mad itch.” The role of the immune response in the clinical outcome of PRV infection is still poorly understood. Here, we show that a systemic host inflammatory response is responsible for the severe pruritus and acute death of mice infected with virulent PRV-Becker but not mice infected with attenuated strain PRV-Bartha. In addition, we identified IL-6 and G-CSF as two main cytokines that play crucial roles in the regulation of this process. Our findings give new insights into neuroinflammatory diseases and strengthen further the similarities between VZV and PRV infections at the level of innate immunity.
INTRODUCTION
Pseudorabies virus (PRV) is the causative agent of Aujeszky’s disease in swine (1). The virus causes respiratory disease, abortion, and neurological disorders, resulting in serious economic losses for the pig industry worldwide (2). Recently, severe PRV outbreaks associated with highly virulent, antigenic-variant novel strains have occurred on several pig farms in both northern and southern China (3, 4). The virus is a member of the alphaherpesvirus subfamily, which also includes several human pathogens such as herpes simplex virus 1 (HSV-1), HSV-2, and varicella-zoster virus (VZV), which cause cold sores, genital lesions, and chicken pox, respectively (5). These viruses are all pantropic, able to infect many different cell types, but all of them exhibit a marked neurotropism by invasion of the peripheral nervous system (PNS) and occasionally the central nervous system (CNS) of their host species.
PRV infection in swine usually starts by viral replication in the epithelial cells of the nasal and oropharyngeal mucosa (6). Then, the infection spreads to PNS neurons, innervating the infected epithelium, and viral particles travel via retrograde transport to the sensory and autonomic peripheral ganglia, where a latent lifelong infection is established (7, 8). Upon reactivation, viral replication occurs, and particles spread in the anterograde direction along the sensory nerves back to the mucosal surfaces where the infection initiated. Adult pigs typically exhibit symptoms of respiratory disease with a low mortality rate. In contrast, in younger swine, PRV infection causes an acute neurological disease, such as encephalitis, with a high fatality rate often suggested to be due to productive viral replication in the CNS (9, 10). PRV infection can also spread via a cell-associated viremia in peripheral blood mononuclear cells from the primary replication site to target organs such as pregnant uterus (11). There, secondary replication ensues in the endothelial cells of the pregnant uterus that can result in vasculitis and multifocal thrombosis, usually leading to abortion (12, 13).
While domesticated and feral swine are the natural hosts of PRV, the virus can infect most mammals, including rodents, dogs, cattle, and horses (14–17). Clinical manifestations of PRV infection in these hosts differ from those in adult swine. PRV infection causes a severe pruritus called the “mad itch,” followed by peracute death (18). By the use of a mouse flank infection model of PRV pathogenesis, it was demonstrated that mice infected with wild-type virulent PRV (PRV-Becker) develop a severe pruritus only in the skin of the infected dermatome, resulting in self-mutilation and rapid death of the animals with no detectable behavioral CNS pathology (19). The virus replicated in the skin, PNS neurons, and spinal cord, but few infectious virus particles were detected in the brain. In contrast, mice infected with an attenuated, live vaccine PRV strain (PRV-Bartha) lived more than twice as long as PRV-Becker-infected animals and did not develop pruritus, but late in infection these mice exhibited severe CNS abnormalities due to widespread infection in the brain. Interestingly, they also found that when PRV-Becker-infected mice were anesthetized with ketamine at the time the pruritus began, no lesions were observed; however, the mice still died at the same time as nonanesthetized mice. These data indicated that scratching and self-mutilation alone were not the cause of death. Brittle’s comparative study suggested that a peripheral host response to PRV infection of the PNS might be a determining factor in the death of the infected animals rather than viral replication in the CNS.
While the innate immune system acts as a first line of defense by initiating an inflammatory response to prevent spread of viral infections, the main challenge is to ensure that the inflammation is resolved, homeostasis restored, and viral replication contained with little damage to the host. However, control is not always obtained and an uncontrolled inflammatory response often leads to a damaging systemic inflammation also known as a “cytokine storm” that can be fatal for the host (20). The best example is the cytokine storm caused by influenza virus infection in humans and animal models where excessive plasma levels of proinflammatory cytokines (such as interleukin-1 [IL-1], IL-6, and tumor necrosis factor alpha [TNF-α]) and chemokines (such as monocyte chemoattractant protein 1 [MCP-1], interferon gamma-induced protein 10 [IP-10], and macrophage inflammatory protein 1 [MIP-1]) increase acute-phase signaling and trafficking of innate immune cells to the primary site of infection (21, 22). This acute-phase response usually results in widespread tissue damage and systemic inflammation. The latter is often responsible for organ dysfunction and failure, particularly of the lungs, kidneys, and circulatory system, and ultimately death (20, 23). Similar lethal systemic inflammatory responses have also been reported during dengue virus, Sindbis virus, and SARS-CoV infections (24–26). Interestingly, a recent study also demonstrated that VZV infection induces high levels of IL-6 transcription and protein production in human skin explant cultures (27).
Here, we sought to determine whether infection of PNS neurons with virulent PRV-Becker strain induces a similar cytokine storm that may be responsible for the severe pruritus and acute death of the infected mice. We also sought to determine whether a more encephalitic pathogenesis is associated with attenuated PRV-Bartha infection. We used the footpad inoculation model that has been widely used to study alphaherpesvirus infection of PNS separately from CNS infection (14, 28, 29). Infection in this model travels a greater distance from the periphery to the PNS and CNS neurons via the sciatic nerve. As a result, it is possible to obtain a more accurate assessment of the kinetics of the immunopathological processes associated with development of pruritus.
In this study, we measured and compared the level of proinflammatory cytokines and chemokines in the plasma and tissues of PRV-infected and control animals. In addition, we performed histological examination and viral load quantification on several tissues to establish correlations between the development of clinical, virological, and inflammatory processes in the pathogenesis of PRV in mice.
RESULTS
PRV-Becker infection induces a severe pruritus and inflammation of the footpad in mice.
Mice either were mock inoculated or inoculated with PRV-Becker (8 × 106 PFU) or PRV-Bartha (108 PFU), and body temperature and weight were monitored daily. These titers ensured that all animals were infected. At the start of the experiment, mice weighed an average of 24.6 ± 2.2 g and had a mean body temperature of 37.1 ± 0.6°C. At 82 h postinoculation (hpi), PRV-Becker-infected mice showed a significant 12% decrease in body weight and had a mean body temperature of 36.3 ± 1.4°C compared to PRV-Bartha-infected and control mice (P < 0.05) (Fig. 1A). These were the signs of imminent death and indicated that the animals had reached a moribund state and should be euthanized. PRV-Becker-infected mice began showing clinical signs at around 70 to 72 hpi, characterized by swelling of the inoculated foot and frequent tremors (Fig. 1B). By 82 hpi, all PRV-Becker-inoculated mice showed constant tremors in the inoculated leg and distinctive PRV symptoms, such as intense scratching and biting of the foot, resulting in a severe inflammation of the footpad (Fig. 1C). In contrast, all PRV-Bartha-inoculated animals remained asymptomatic at 82 hpi, and the inoculated footpad was indistinguishable from the control group with no signs of inflammation. By 200 hpi, the animals showed significant decreases in body weight (P < 0.05) and displayed behavioral CNS symptoms (the humane endpoint). Taken together, these results confirmed the two distinct pathogeneses of PRV-Becker and PRV-Bartha infections in mice using another model of PRV inoculation, the footpad, and highlighted the importance of the inflammatory response in the clinical presentation of PRV-Becker infection (19).
FIG 1.

Clinical observations of PRV infection in mice. (A and B) Body weight and temperature (A) and clinical score (B) for B57BL/6 mice following PRV infection with the Becker strain (8 × 106 PFU) (red), the Bartha strain (108 PFU) (blue), or controls (black). (n = 10 per group). *, P < 0.05. (C) Representative images of mouse right hind paws at 82 hpi after PRV inoculation. Black arrows indicate the sites of abrasion.
PRV-Becker infection induces a massive neutrophil infiltration in the inoculated footpad and inflammation in the DRGs.
Histopathological examination of several tissues (brain, heart, lungs, spleen, pancreas, liver, bladder, foot, kidneys, spinal cord, and dorsal root ganglia [DRGs]) was performed after hematoxylin and eosin (H&E) staining. Samples were taken from three mice in each experimental and control group.
At 82 hpi, all tissues, except the footpad and DRGs, showed no histopathological changes. PRV-Becker-infected foot sections revealed epidermal necrosis and severe dermal inflammation (edema and fibrin) (Fig. 2A, panels b). A massive infiltration of neutrophils (identified by a multilobed nucleus) was also observed in the epidermis, dermis, and connective tissues of all three PRV-Becker-infected mice. All footpads of the three control and three PRV-Bartha-infected mice were normal (Fig. 2A, panels a and c).
FIG 2.

PRV-Becker-infected mice showed signs of severe inflammation in the footpad and DRGs 82 h after footpad inoculation. (A and B) H&E staining of mouse inoculated footpads (A) and ipsilateral DRGs (B) from control (a), PRV-Becker-infected (b), and PRV-Bartha-infected (c) mice at 82 hpi. Histopathological manifestations observed in PRV-Becker-infected animal tissues (epidermal and neuronal necrosis and neutrophil infiltration) were absent from all examined mock-infected and PRV-Bartha-infected mice. The results are representative of three biological replicates for a given type of tissue. Black arrows indicate representative areas of inflammation with immune cell infiltration. Scale bars (50 μm) are indicated for each image.
PRV-Becker-infected DRGs showed minimal neuronal necrosis and mixed inflammation in all three infected mice (Fig. 2B, panels b). The mixed inflammation infiltrate consisted mainly of neutrophils and lymphocytes. The inflammatory cells were associated with neuronal necrosis that was characterized by shrunken, eosinophilic neurons with pyknotic nuclei. All DRGs of the three control and three PRV-Bartha-infected mice were normal (Fig. 2B, panels a and c). Overall, PRV-Becker infection induced severe inflammation in the footpad and DRGs 82 h after footpad inoculation.
PRV replication was detected only in the foot, PNS, and CNS tissues.
Next, we determined whether the fatal clinical outcome and distinct pathology of PRV-Becker-infected mice could be attributed to viral replication in a specific tissue other than the footpad and DRG.
Tissues, as listed above, were collected after euthanasia for quantitative real-time PCR (qRT-PCR) analysis to determine PRV DNA loads. DNA concentration was then converted to PFU as previously described (30). As shown in Fig. 3A, PRV-Becker DNA was detected only in the footpad (approximately 4 × 103 PFU/mg of tissue), DRGs (2.82 × 105 PFU/mg of tissue), spinal cord (1.2 × 104 PFU/mg of tissue), and brain (102 PFU/mg of tissue) at 82 hpi. Interestingly, detectable PRV-Bartha DNA was found only in the footpad (4 × 102 PFU/mg of tissue) and DRGs (5 × 102 PFU/mg of tissue) at that time (Fig. 3B). However, at 200 hpi, a significantly higher PRV-Bartha load was detected in the DRGs (8.4 × 103 PFU/mg of tissue), spinal cord (1.57 × 104 PFU/mg of tissue), and brain (1.80 × 104 PFU/mg of tissue) (Fig. 3C). The PRV-Bartha loads were comparable in the footpad at 82 and 200 hpi. No detectable PRV genomes were found in other tissues of PRV-Becker- or PRV-Bartha-infected mice and in any tissues of the control animals. Overall, we found that both PRV-Becker and PRV-Bartha DNA could be detected only in the mouse footpad and nervous system, indicating that widespread viral replication in vital organs was not correlated with the rapid death of the animals.
FIG 3.

Quantitation of the PRV genome in mouse tissues after footpad inoculation. At the indicated time points, PRV DNA was quantitated in mouse tissues by qRT-PCR using UL54 primers. PRV-Becker (red) and PRV-Bartha (blue) loads are expressed as PFU per mg of tissue. PRV-Becker and PRV-Bartha loads were detected only in the foot, DRGs, spinal cord, and brain (n = 10 per group). Dotted lines indicate the detection limit.
PRV-Becker infection, but not PRV-Bartha infection, induces a specific systemic inflammatory response in mice.
To ascertain whether the inflammatory response to PRV-Becker infection could be correlated with the acute death of the infected animals, we measured and compared the levels of 12 proinflammatory cytokines and 6 proinflammatory chemokines in the plasma of PRV-Becker-infected, PRV-Bartha-infected, and control mice at 82 hpi.
Among 12 proinflammatory cytokines tested, we found a significant 4-fold increase in both IL-6 and granulocyte colony-stimulating factor (G-CSF) plasma levels of PRV-Becker-infected mice compared to PRV-Bartha-infected and control groups at 82 hpi (Fig. 4A). The plasma levels of 2 proinflammatory chemokines (Gro-1 and MCP-1) were also significantly higher in PRV-Becker infected mice compared to control and PRV-Bartha infected animals (P < 0.01) (Fig. 4B). Other chemokines (Rantes, IP-10, TLSF [CXCL12], and MIP-1α) were not increased in the plasma of PRV-Becker, PRV-Bartha, and control groups at 82 hpi. At 200 hpi, no detectable amount of any of the cytokines and chemokines tested above was observed in the plasma of PRV-Bartha-infected mice (data not shown).
FIG 4.

Plasma cytokine and chemokine levels in PRV-infected and control mice. (A) Relative levels of 12 proinflammatory cytokines measured in the plasma of mock-infected (black), PRV-Becker-infected (red), and PRV-Bartha-infected (blue) mice at 82 hpi as determined by ELISA. (B) Concentrations of six proinflammatory chemokines measured in the plasma of these three groups. The concentration is expressed in pg per ml (n = 10 per group). ****, P < 0.0001; ns, not significant. (C) Kinetics of IL-6, G-CSF, Gro-1, and MCP-1 expression in the plasma of mock-infected, PRV-Becker-infected, and PRV-Bartha-infected mice. The concentration is expressed in pg per ml (n = 5 per group). *, P < 0.05; **, P < 0.01; ****, P < 0.0001; ns, not significant.
In addition, we quantified and compared the plasma levels of IL-6, G-CSF, Gro-1, and MCP-1 at 24 and 48 hpi between PRV-infected and control mice to establish the kinetics of cytokine/chemokine production and to determine the peak of production. As shown in Fig. 4C, a significant increase in all four inflammatory markers was detected only at 82 hpi in the plasma of PRV-Becker-infected mice compared to controls. No significant differences in plasma levels of these four inflammatory markers were observed between PRV-Bartha-infected and control animals at all time points. Taken together, these results demonstrated that PRV-Becker induces a specific systemic inflammatory response at the time the animals are moribund, which correlates with the fatal outcome of the infection in mice.
PRV-Becker infection induces local cytokine production in uninfected tissues at 82 hpi.
To determine whether the systemic increase of IL-6, G-CSF, Gro-1, and MCP-1 could be responsible for the death of PRV-Becker-infected mice, the levels of these inflammatory markers were quantified in several tissue samples and compared to control groups at 82 hpi. Since local cytokine production is a significant indicator for disease pathogenesis or progression, its assessment in tissues should provide additional information for monitoring the pathological events in a target tissue, rather than the evaluation of systemic cytokine levels alone (31, 32).
Interestingly, we found that IL-6 levels were significantly increased in all tissues compared to controls at 82 hpi (P < 0.05) (Fig. 5A). Significant G-CSF levels were detected only in the footpad, DRGs, spinal cord, brain, heart and liver of PRV-Becker-infected mice (P < 0.01) (Fig. 5B). Significant Gro-1 and MCP-1 levels were detected only in the inoculated footpad at 82 hpi (P < 0.05 and P < 0.01, respectively) (Fig. 5C and D).
FIG 5.

Local cytokine and chemokine protein levels in PRV-Becker-infected and control mouse tissues. IL-6 (A), G-CSF (B), Gro-1 (C), and MCP-1 (D) levels were significantly upregulated in PRV-Becker-infected (red) and control (black) mouse tissues at 82 hpi. Protein levels were quantified by ELISA and are expressed as pg per mg of tissue (n = 10 per group). *, P < 0.05. The dotted line indicates the detection limit.
PRV-Becker infection induces a high level of CRP in the plasma.
The short time window (<12 h) between the appearance of the clinical symptoms and the moribund state of the animals, the absence of histopathological changes in nearly all tissues, and the presence of local and systemic inflammation, demonstrate that PRV-Becker-induced inflammation in mice is a hyperacute process. It is well known that uncontrolled acute inflammation due to bacterial or viral infections often culminates in organ failure and death (33–35). To investigate the potential relationship between the systemic inflammation and acute deaths of PRV-infected mice, we measured and compared the plasma level of C-reactive protein (CRP) between PRV-Becker-infected, PRV-Bartha-infected, and control mice at 82 hpi. CRP is an acute-phase protein synthesized by hepatocytes in response to proinflammatory cytokines, IL-6 in particular (36). This specific biomarker of acute inflammation is studied both as a causal factor in the prediction of coronary heart disease and as a measurement of disease severity during influenza infections (37–39). At 82 hpi, we found that PRV-Becker-infected mice had significantly higher plasma levels of CRP (10.3 ± 2.4 mg/liter) compared to PRV-Bartha-infected or control animals (1.8 ± 0.3 and 2.7 ± 1.8 mg/liter, respectively) (Fig. 6A). This result suggests that PRV-induced systemic inflammation is a hyperacute process in mice.
FIG 6.
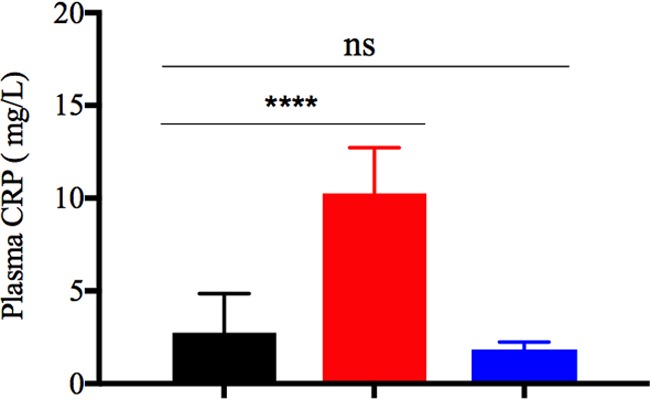
Plasma level of CRP in PRV-infected and control mice. The plasma level of CRP was measured by ELISA in PRV-Becker-infected (red), PRV-Bartha-infected (blue), and control (black) mice at 82 hpi. Protein levels are expressed in mg per liter. ****, P < 0.0001 (n = 10 per group).
DISCUSSION
PRV infection in swine has been widely studied; however, little information is available on the causes of the distinctive pathogenesis of PRV in nonnatural hosts. In the present study, we demonstrate that infection with a virulent PRV strain (PRV-Becker) induces a systemic and lethal inflammatory response in mice.
First, we showed that PRV-Becker infection induces high plasma levels of the proinflammatory cytokines IL-6 and G-CSF, as well as the proinflammatory chemokines Gro-1 and MCP-1, at 82 hpi (moribund stage). As expected, these inflammatory markers were not found elevated in the plasma of attenuated PRV-Bartha-infected animals at that time (82 hpi). Control IL-6, G-CSF, Gro-1, and MCP-1 values were within the range of baseline values previously described in literature (40–42). These results are consistent with the hypothesis that PRV-Becker infection induces a systemic inflammatory response when the animals are moribund. Second, the fact that little PRV-Becker DNA was detected in the brain at 82 hpi clearly suggests that this uncontrolled inflammatory response is responsible for the severe pruritus and acute death of the animals rather than viral replication in the brain. We suggest that the absence of the markers of a systemic inflammatory response in PRV-Bartha-infected mice explains why these infected animals live longer, giving time for the infection to spread unabated to the brain. Ultimately, if animals live long enough, widespread infection in the brain may lead to fatal viral encephalitis as previously described (43, 44). This conclusion is consistent with our findings in which high concentrations of PRV-Bartha DNA were detected in the brain in animals with CNS abnormalities at 200 hpi.
The presence of IL-6 and G-CSF in the plasma of PRV-Becker-infected animals is striking. Both are inflammatory cytokines produced by various cells, including immune cells (neutrophils, macrophages, and T lymphocytes), neurons, and endothelial cells. IL-6 has pleiotropic effects on inflammation, immune response, hematopoiesis, and neurogenesis (45, 46). Typically, IL-6 is produced following pathogen stimulation in the initial stage of the inflammation and is the main inducer of an acute-phase response in the liver (47). G-CSF is a key regulator in neutrophil production. This cytokine influences the survival, proliferation, and differentiation of all cells in the neutrophil lineage, from hematopoietic stem cells to mature neutrophils (48). G-CSF also influences the migration of neutrophils across the vascular endothelium (49). Moreover, G-CSF has a neuroprotective effect in several murine models of acute spinal cord injury. For instance, this cytokine is able to rapidly induce autophagy after spinal cord injury to inhibit neuronal apoptosis and is considered an effective auxiliary therapeutic intervention for spinal cord injury (50). However, sustained levels of IL-6 and G-CSF can overwhelm the immune response and can be detrimental for the host. Elevated serum levels of IL-6 are positively correlated with disease severity in bacterial sepsis, as well as in Sindbis virus, influenza virus A, enterovirus 71 (E71), and hepatitis B virus infections (25, 51–53). In this study, we detected high levels of IL-6 (∼600 pg/ml) in the blood of PRV-infected mice similar to those described during E71 and Sindbis infections. It is known that E71 infection induces a systemic lethal inflammation response in mice. E71-induced clinical manifestations and disease outcome are not a direct consequence of viral replication and spread in tissues but result from an uncontrolled inflammatory response. Here, we found that IL-6 and G-CSF were elevated in several tissues, suggesting that both local and systemic inflammatory responses may also contribute to the fatal outcome of PRV-Becker infection in mice.
Interestingly, we demonstrated a very high IL-6 protein levels (∼30,000 pg/ml) in the footpad of PRV-Becker-infected mice. This result strongly correlates with a recent study, which demonstrated the same exact concentration of IL-6 in human skin explant cultures after VZV infection (27). Thus, these findings support the fact that VZV and PRV have very similar innate immune responses to infection of skin.
Moreover, at 82 hpi after PRV-Becker infection, both IL-6 and G-CSF were significantly increased in the heart, and high plasma levels of CRP were detected. Taken together, these findings suggest that PRV-induced systemic inflammation is a hyperacute process that could possibly lead to heart failure in mice. Indeed, a previous study showed that PRV infection causes peripheral nervous system pathology and cardiac injury in infected dogs (54). These researchers proposed that excessive sympathetic cardiac stimulation, associated with neuritis, ganglioneuritis, and cardiac injury, is associated with heart failure.
While high levels of IL-6 and G-CSF were detected in the majority of the tissues examined, Gro-1 and MCP-1 were elevated only in the plasma and footpads of PRV-Becker-infected mice. It is certainly relevant to note that Gro-1 and MCP-1 are known to regulate the migration of neutrophils and monocyte/macrophages, respectively, to the site of injury (55, 56). We suggest that both chemokines remained elevated in the footpad because of the intense scratching and resulting damage and inflammation of the foot. Ultimately, soluble Gro-1 and MCP-1 leaked from the footpad to the blood circulation and thus were detected at high levels in the plasma. However, the fact that both Gro-1 and MCP-1 were not produced in most of the tissues examined suggests that they are not directly responsible for the lethal systemic inflammatory response induced by PRV-Becker infection. These cytokines rather represent collateral damage of the inflammation caused by the direct recruitment of neutrophils to the site of infection.
Consistent with elevated levels of local and systemic inflammatory markers, we also found an aberrant infiltration of neutrophils in the footpad and ipsilateral DRGs of PRV-Becker-infected animals at 82 hpi. This infiltration was accompanied by epidermal and neuronal necrosis, respectively. All of these signs were absent in the tissues of PRV-Bartha-infected mice. Neutrophils represent around 60 to 70% of the circulating white blood cells in most mammals. These cells are essential cellular components of the innate immune system and represent the first line of defense against invading pathogens, such as bacteria, viruses, and fungi (57). Upon infection, neutrophils are first recruited from the bloodstream and migrate to the inflammation site. Their presence is usually one hallmark of acute inflammation. Once at the inflammation site, they become activated and mediate a variety of effector functions, including phagocytosis and killing of pathogens through the production of reactive oxygen species, degranulation, and the generation of neutrophil extracellular traps (58). These neutrophils also orchestrate the direct recruitment of other immune cells, such as monocytes, from the circulation or indirectly via the production of cytokines and chemokines. This phase amplifies the neutrophil response at the site of injury. Finally, neutrophils are usually removed from the area either by phagocytosis via macrophages, apoptosis, or reverse migration (59). This last phase is crucial to ensure the local resolution of the acute inflammation and prevent tissue damage.
Recently, neutrophils have been also shown to play an essential role in the pathogenesis of viral respiratory diseases, including influenza A virus infection (60). Moreover, neutrophils have been shown to promote neurotoxicity on DRG neurons and are considered responsible for hypersensitivity and/or neuropathic pain after peripheral nerve injury (61). Here, the migration of neutrophils to the infected footpad and DRG neurons and their subsequent production of IL-6 and G-CSF are likely to increase and propagate the inflammation and cause tissue damage. Future experiments will focus on the early phases of this inflammatory process, in particular the kinetics of neutrophil recruitment to the infected footpad and DRG neurons. It would be interesting to assess the therapeutic efficacy of inhibitors of neutrophil recruitment, such as fucoidan, on the immunopathology of PRV in mice (62).
Surprisingly, only 4 of 12 inflammatory markers tested were found elevated in plasma samples, indicating that the PRV-induced systemic inflammatory response does not resemble the traditional cytokine storm observed during influenza or Sindbis virus infections. One explanation is that influenza and Sindbis viruses are both RNA viruses and are detected by different pathogen-recognition receptors on host cells compared to DNA viruses. While retinoic acid-inducible gene I (RIG-I)-like receptors play essential roles in the recognition of RNA viruses in various cells, DNA viruses are mainly detected by Toll-like receptors (TLRs), IFI16, and cGAS sensors (63, 64). Detection of viral components by these pathogen recognition receptors in host cells activates distinct intracellular signaling cascades, leading to the secretion of type I IFNs, proinflammatory cytokines, and chemokines. Interestingly, TLR-2 has been shown to play an important role in recognition of HSV-1 and to contribute to encephalitis in infected mice (65). TLR-2 knockout mice (KO) inoculated intraperitoneally with HSV-1 survived, while the wild-type animals died by 6 days postinfection. In addition, this study showed that TLR-2 KO mice had significantly lower serum levels of IL-6 compared to the wild type. Future studies will test whether PRV similarly interacts with TLR2 on epidermal cells and DRG neurons in vivo and whether this interaction is responsible for the initiation of the uncontrolled inflammatory response in infected mice.
We detected PRV-Becker DNA in the footpad, DRG neurons, spinal cord, and brain at the latest time after infection just before death (82 hpi), while only low concentrations of PRV-Bartha DNA were detected in the footpad and DRG neurons at that time. These results suggest that PRV-Becker replicates more efficiently in the foot and DRG neurons than PRV-Bartha. There is a strong correlation of viral gene expression in the footpad and DRG neurons and inflammation in PRV-Becker-infected mice. We also found inclusion bodies indicative of viral replication in pockets of inflammation in DRGs (data not shown). Interestingly, the difference in replication may reflect the fact that PRV-Becker can suppress the interferon (IFN) response and PRV-Bartha cannot. As a result, more virus particles may invade axon terminals of the DRG neurons and replicate in the cell bodies. A recent study showed that PRV-Becker inhibits the IFN response in swine plasmacytoid DCs, while PRV-Bartha elicits a much more robust type I IFN response in these cells (66). This difference was attributed to a deletion of the glycoprotein gE/gI gene complex in the genome of Bartha.
Continued replication of PRV-Becker in DRG neurons is known to be responsible for the severe pruritus observed in infected mice. By using several PRV mutants, it was demonstrated that the pruritus stimulus was mediated, at least in part, by gE, gI, and US9 proteins (19). By infecting the salivary gland and imaging the submandibular ganglia (SMG), it was also shown that PRV-Becker infection of PNS neurons induces electrical coupling in axons, which gives rise to a synchronous and cyclical activity in neuronal cell bodies in the SMG (67). In addition, it was observed that newly made PRV particles in infected SMG neurons were transported back in axons to the glands where the infection was initiated. Therefore, they developed the concept of “round-trip” reseeding and amplification of infection in the ganglia. This reseeding phenomenon occurs only after PRV-Becker infection but not after PRV Bartha infection, primarily because this strain lacks the Us9 protein required for sorting virion proteins into axons. The ability to reseed the gland increases the infection of the innervating PNS ganglia and the involvement of more axons in electrical coupling, thus directly contributing to the pruritus. Our results are consistent with this concept. We found that the PRV-Becker-inoculated footpad had epidermal necrosis with a massive immune cell infiltration at 82 hpi. The epidermis layer did not regenerate to support efficient viral replication at that time. This finding suggests that the large amount of infectious PRV-Becker virus detected in the footpad likely resulted from virions that originally infected the DRG, replicated, and returned to the footpad rather than from local viral replication in the epidermis. Also, reseeding may stimulate a powerful inflammatory response in the primary infected tissue. We observed an effective healing of the PRV-Bartha-inoculated footpad, suggesting that the regenerated epidermis layer might support viral replication. Still, PRV-Bartha DNA was barely detected because particles could not be transported back from the DRG neurons to the initial site of infection. Interestingly, we found that mice inoculated with UV-inactivated PRV-Becker remained asymptomatic and were still alive at 82 hpi (data not shown). This finding confirms that infectious virus and active viral replication in the DRG neurons are necessary to cause the pruritus and subsequently to kill the infected animals.
Furthermore, we demonstrated that infection by PRV-Becker spreads faster from the footpad to the brain than does infection by PRV-Bartha. This difference is due primarily to the two distinct modes of neuroinvasion of these strains and the function of the UL21 protein, which is compromised in PRV-Bartha (68). It is well known that PRV-Becker can spread both in the retrograde and anterograde directions, whereas PRV-Bartha only spreads in a retrograde direction. The mouse footpad mainly consists of sensory nerve fibers with a few sympathetic nerve fibers (sweat glands) that are innervating the epidermis and dermis layers. Accordingly, we suggest that PRV-Becker can travel in axons in the retrograde direction via the sensory route to reach DRG cell bodies. Newly replicated particles can exit DRG cell bodies and not only travel back to the footpad but also travel anterograde to the dorsal horn of the spinal cord and from there to the CNS. PRV-Becker also can enter sympathetic nerve fibers in the footpad and reach the ventral horn of the spinal cord by retrograde transport (motor route). In the footpad model, the course of infection was slightly longer than in the flank inoculation model. In the footpad model, PRV-Becker-infected mice showed symptoms around 70 hpi and died at around 82 hpi compared to 40 and 72 hpi, respectively, in the flank model. This time delay reflects the different kinetics of viral replication and spread between the two models: PRV particles must travel longer distances through the sciatic nerve from the footpad to reach PNS neurons than the distance to DRG cell bodies from the skin flank. While PRV-Bartha can reach the DRG cell bodies via the peripheral axons of DRG neurons, newly replicated virions cannot spread to the dorsal horn of the spinal cord from the infected DRG because there is no anterograde sorting and spread (19). Thus, PRV-Bartha infection can only spread to the CNS by invading sympathetic motor nerve fibers in the footpad. Most importantly, we demonstrated that both PRV-Becker and Bartha DNA could be detected only in the mouse footpad and nervous system and not in other organs, confirming that widespread viral replication in vital organs was not responsible for the rapid death of the animals.
In conclusion, our data show that a systemic host inflammatory response is responsible for the severe pruritus and acute death of mice infected with virulent PRV-Becker but not with attenuated strain PRV-Bartha. We identified IL-6 and G-CSF as two cytokines that play crucial roles in the regulation of this process. Further research on modulating the inflammatory response could help to limit the severity of the disease in susceptible hosts and broaden our understanding of PRV-induced neuroinflammation.
MATERIALS AND METHODS
Viruses.
A wild-type virulent PRV strain (PRV-Becker) and a live-attenuated PRV strain (PRV-Bartha) were used in this study. PRV-Becker is a virulent field isolate from dog, originally isolated at Iowa State University, with subsequent laboratory passage (69). PRV-Bartha is a highly passaged vaccine strain, derived from the original Aujeszky strain, which was isolated in Hungary (70). The attenuated live Bartha strain has mutations in the glycoprotein C (gC), gM, and UL21 genes and a deletion in the unique short region spanning the gI, gE, Us9, and Us2 genes (71–74). All viral stocks were grown and titers were determined on monolayers of PK-15 pig kidney cells (American Type Culture Collection).
Mouse footpad inoculation model.
Male C57BL/6 mice between 5 and 7 weeks old were purchased from The Jackson Laboratory (Bar Harbor, ME). C57BL/6 mice are sensitive to PRV infection, as previously described (19, 75). The protocol used for the footpad inoculation experiments was adapted from a previously described protocol (28). Mice were anesthetized with 1 to 3% isoflurane gas, and the right hind footpads, between the heel and walking pads, were gently abraded about 20 times with an emery board until the stratum corneum was removed in order not to induce bleeding and to ensure that virus was inoculated onto the stratum spinosum of the epidermis. A 20-μl droplet of virus inoculum containing 8.106 PFU of PRV-Becker or 108 PFU of PRV-Bartha, resuspended in medium (Dulbecco modified Eagle medium, 2% fetal calf serum, and antibiotics; HyClone [GE Healthcare Life Sciences]), was applied onto the abraded area of the skin. We used a higher virus inoculum for PRV-Bartha than PRV-Becker because previous study demonstrated that the 50% lethal dose for PRV-Bartha was about twice that determined for PRV-Becker (19). These viral doses were optimized in a preliminary experiment. Briefly, we inoculated mice with several virus doses (105, 106, 107, and 108 PFU) for each PRV strain. We selected the appropriate PRV-Becker and PRV-Bartha doses that ensure all inoculated mice to show symptoms at 82 and 200 hpi, respectively. Mock inoculations (medium only) were carried out in parallel. The inoculum was gently rubbed 5 to 10 times with the shaft of an 18-gauge hypodermic needle to facilitate adsorption of the virus. The mice were kept under anesthesia for 30 min until the abraded footpad was dry, and then the animals were placed in separate cages for further analysis.
Mice were weighed daily, and temperatures were measured using a rectal probe. For clinical evaluation, mice were scored daily for symptoms of virulent PRV infection using the following 3-point system: 0 = posture normal, footpad red at abrasion/inoculation site; 1 = foot swollen and frequent tremors (∼10 per min) in leg upon attempt to scratch; 2 = foot and hip swollen, constant tremors in leg upon attempt to scratch, scratching and licking of the foot, lack of weight bearing when walking and hunched posture; and 3 = same as score 2 but with biting and bleeding of the footpad. Mice were euthanized when scored as 2+. PRV-Bartha-infected animals were euthanized when animals displayed behavioral CNS abnormalities, such as ataxia or gait disturbances, as previously described (19). PRV-Becker- and PRV-Bartha-infected animals were euthanized at 82 and 200 hpi, respectively. A set of controls was euthanized at both time points.
Tissue collection and homogenization.
Mice were euthanized by CO2 asphyxiation at the moribund state. Fresh tissues, including the footpad, dorsal root ganglia (DRGs), spinal cord, brain, heart, lungs, spleen, pancreas, liver, kidneys, and bladder, were collected, flash-frozen in liquid nitrogen, and stored at –80°C. Portions (100 mg) of tissue were weighed and placed in a 2-ml microcentrifuge tube containing a sterile steel bead (Qiagen) and 500 μl of modified radioimmunoprecipitation assay buffer containing 0.5 M EDTA (pH 8.0), 1 M Tris-HCl (pH 8.0), 5 M NaCl, 10% sodium dodecyl sulfate, and protease cocktail inhibitor tablets (cOmplete Mini EDTA-free; Roche Diagnostics). Tissues were disrupted using a TissueLyser (Qiagen) using 20 cycles/s for 2 min, followed by a 1-min wait, and 20 cycles/s for 2 min and then centrifuged at high speed (17,900 × g) for 10 min. Tissues were stored at –20°C until use in enzyme-linked immunosorbent assay (ELISA) and qRT-PCR.
Blood collection.
Whole blood (∼200 μl) was collected through submandibular bleeding daily by alternating sides of the face and transferred into 1.5-ml EDTA capillary collection tubes (BD Vacutainer). After collection, the samples were first centrifuged for 10 min at 1,500 × g and 4°C to separate cells from plasma and then for 15 min at 2,000 × g and 4°C to deplete platelets. Samples were stored at –80°C until ELISA analysis.
Histology.
At the humane endpoint, mock-infected or PRV-infected mice were sacrificed and exsanguinated by transcardiac perfusion with a phosphate-buffered saline/10% formalin, neutral buffered solution (Sigma). Tissue samples were carefully dissected and placed in 10% formalin at 4°C for 24 h. The ipsilateral DRGs of lower lumbar and sacral levels were collected for histopathological analyses. Samples were processed and embedded in paraffin, and 4- to 6-μm sections were prepared and stained with H&E by Charles River Histopathology Services. H&E-stained sections were evaluated for signs of inflammation and epidermal and neuronal necrosis.
Quantification of inflammatory markers by ELISA.
We performed multi-analyte profiling of 12 cytokines and 4 chemokines in plasma samples using the mouse inflammatory cytokines multi-analyte ELISArray kit (Qiagen) and the multiplex immunoassays with Firefly particle technology (Abcam), respectively. Quantitation of single analyte IL-6, G-CSF, Gro-1, MCP-1, TLSF, and MIP-1α levels in plasma and/or tissue homogenates were performed using commercial ELISA kits from Thermo Fischer, Qiagen, and Abcam. Mouse cardiac marker C-reactive protein (CRP) plasma levels were also analyzed by commercial ELISA kits from R&D Systems, LSBio, and Abcam, respectively. The assays were conducted according to the manufacturer’s recommendations. All samples were measured in duplicate.
Quantitative real-time PCR assay.
Homogenized tissues were digested with proteinase K (New England Biolabs) in Tween 20 for 60 min at 55°C, followed by inactivation for 10 min at 95°C prior to qRT-PCR run. Viral genomic DNA was quantified by using UL54-specific primers as previously published (76). This set of primers (5′-TGC-AGC-TAC-ACC-CTC-GTC-C-3′ and 5′-TCA-AAA-CAG-GTG-GTT-GCA-GTA-AA-3′; Integrated DNA Technologies) generated a 65-bp fragment of the viral gene UL54 after amplification. qRT-PCR was performed with an Eppendorf RealPlex Mastercycler. The reaction mixture was prepared using a Kapa Syber Fast qPCR kit, and samples were prepared as triplicates. Each experiment was performed in duplicates. The amplification reactions were carried out in a total volume of 10 µl, containing 2 µl of template DNA, 5 μl of 2× SYBR Fast qPCR Master Mix Universal (Kapa Biosystems), 0.4-µl portions of each forward and reverse primer (2.5 µM), and 2.2 µl of RNase-free water. The amplification conditions consisted of preincubation at 95° for 2 min and 40 cycles of denaturation (5 s at 95°C), annealing (20 s at 55°C), and extension (10 s at 72°C). The quantification cycle (CT) was calculated as the cycle number at which the concentration increase became exponential. The specific target amplification was analyzed by melting-curve analysis using Mastercycler EP RealPlex 2.2 software.
To quantitate viral DNA, a standard curve was obtained for each experiment by coamplification of known amounts of PRV DNA. Five consecutive 10-fold dilution of PRV stocks was prepared containing from 105 to 101 PFU. The amounts of PRV DNA in samples were obtained by plotting CT values onto the standard curve and are expressed as PFU/mg of tissue (30).
Statistical analyses.
Significant differences (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001) between mock-, PRV-Becker-, and PRV-Bartha-inoculated animals were identified by using one-way analysis of variance (ANOVA), followed by either a Tukey’s post hoc test or a two-sided Dunnett’s post hoc test. If homoscedasticity of the variables was not met as assessed by Levene’s test, the data were log transformed prior to ANOVA. The normality of the residuals was verified by the use of the Shapiro-Wilk test. If the variables remained heteroscedastic or normality was not met after log transformation, a Kruskal-Wallis test, followed by a Mann-Whitney post hoc test, was performed. All analyses were conducted in GraphPad Prism v7.0d (GraphPad Software, La Jolla, CA). Values in the text, graphs, and figure legends throughout the manuscript are means ± the standard deviations.
Ethics statement.
All animal experiments were performed in accordance with protocol 208316 and reviewed and approved by the Institution Animal Care and Use Committee of Princeton University. Princeton personnel are required to adhere to applicable federal, state, local, and institutional laws and policies governing animal research, including the Animal Welfare Act and Regulations; the Public Health Service Policy on Humane Care and Use of Laboratory Animals; the Principles for the Utilization and Care of Vertebrate Animals Used in Testing, Research, and Training; and the Health Research Extension Act of 1985.
ACKNOWLEDGMENTS
We acknowledge Halina Staniszewska Goraczniak and Charles River Laboratories for excellent technical support. We also thank Christina DeCoste and Katherine Rittenbach and the Molecular Biology Flow Cytometry Resource Facility, which is partially supported by a Cancer Institute of New Jersey Cancer Center Support Grant (P30CA072720).
J.V.C. is a researcher funded by the Research Foundation Flanders (FWO 11Y5415N). L.W.E. is supported by a U.S. National Institutes of Health grant (R37NS033506). We declare that we have no competing interests.
K.L. designed and performed all the experiments, statistically evaluated the results, designed the figures, and wrote the first draft of the manuscript. J.B.V. helped to perform experiments. J.V.C. helped with the interpretation of the results and statistical analyses. O.O.K. helped with the design of qRT-PCR experiments. L.W.E. helped in conceptualization of the project, provided funding and resources, and was involved in supervision. All authors reviewed and edited the manuscript.
REFERENCES
- 1.Gustafson DP. 1986. Pseudorabies, p 209–223. In Leman AD, Glock RD, Mengeling WL, Penny RHC, Scholl E, Straw B (ed), Diseases of swine, 6th ed Iowa State University Press, Ames, IA. [Google Scholar]
- 2.Wittmann G, Rziha H-J. 1989. Aujeszky’s disease (pseudorabies) in pigs, p 230–325. In Knipe DM, Howley PM (ed), Herpesvirus diseases of cattle, horses, and pigs, vol 9 Kluwer Academic Publishers, Boston, MA. [Google Scholar]
- 3.Gu Z, Hou C, Sun H, Yang W, Dong J, Bai J, Jiang P. 2015. Emergence of highly virulent pseudorabies virus in southern China. Can J Vet Res 79:221–228. [PMC free article] [PubMed] [Google Scholar]
- 4.Wu R, Bai C, Sun J, Chang S, Zhang X. 2013. Emergence of virulent pseudorabies virus infection in Northern China. J Vet Sci 14:363–365. doi: 10.4142/jvs.2013.14.3.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steiner I, Kennedy PGE, Pachner AR. 2007. The neurotropic herpes viruses: herpes simplex and varicella-zoster. Lancet Neurol 6:1015–1028. doi: 10.1016/S1474-4422(07)70267-3. [DOI] [PubMed] [Google Scholar]
- 6.Masic M, Ercegan M, Petrovic M. 1965. The significance of the tonsils in the pathogenesis and diagnosis of Aujeszyk’s disease in pigs. Zentralbl Veterinarmed B 12:398–405. [PubMed] [Google Scholar]
- 7.Tirabassi RS, Townley RA, Eldridge MG, Enquist LW. 1998. Molecular mechanisms of neurotropic herpesvirus invasion and spread in the CNS. Neurosci Biobehav Rev 22:709–720. doi: 10.1016/S0149-7634(98)00009-8. [DOI] [PubMed] [Google Scholar]
- 8.Gutekunst DE, Pirtle EC, Miller LD, Stewart WC. 1980. Isolation of pseudorabies virus from trigeminal ganglia of a latently infected sow. Am J Vet Res 41:1315–1316. [PubMed] [Google Scholar]
- 9.Kluge JP, Beran GW, Hill HT, Platt KB. 1999. Pseudorabies (Aujeszky’s disease), p 233–246. In Straw BE, D’Allaire S, Mengeling WL, Taylor TJ (ed), Diseases of swine, 8th ed Iowa State University Press, Ames, IA. [Google Scholar]
- 10.Verpoest S, Cay B, Favoreel H, De Regge N. 2017. Age-dependent differences in pseudorabies virus neuropathogenesis and associated cytokine expression. J Virol 91:e02058-16. doi: 10.1128/JVI.02058-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nawynck HJ, Pensaert MB. 1995. Cell-free and cell-associated viremia in pigs after oronasal infection with Aujeszky’s disease virus. Vet Microbiol 43:307–314. doi: 10.1016/0378-1135(94)00103-4. [DOI] [PubMed] [Google Scholar]
- 12.Nauwynck HJ, Pensaert MB. 1992. Abortion induced by cell-associated pseudorabies virus in vaccinated sows. Am J Vet Res 53:489–493. [PubMed] [Google Scholar]
- 13.Kluge JP, Mare CJ. 1974. Swine pseudorabies: abortion, clinical disease, and lesions in pregnant gilts infected with pseudorabies virus (Aujeszky’s disease). Am J Vet Res 35:991–995. [PubMed] [Google Scholar]
- 14.Field HJ, Hill TJ. 1974. The pathogenesis of pseudorabies in mice following peripheral inoculation. J Gen Virol 23:145–157. doi: 10.1099/0022-1317-23-2-145. [DOI] [PubMed] [Google Scholar]
- 15.Crandell RA, Mesfin GM, Mock RE. 1982. Horizontal transmission of pseudorabies virus in cattle. Am J Vet Res 43:326–328. [PubMed] [Google Scholar]
- 16.Cramer SD, Campbell GA, Njaa BL, Morgan SE, Smith SK, McLin WR, Brodersen BW, Wise AG, Scherba G, Langohr IM, Maes RK. 2011. Pseudorabies virus infection in Oklahoma hunting dogs. J Vet Diagn Invest 23:915–923. doi: 10.1177/1040638711416628. [DOI] [PubMed] [Google Scholar]
- 17.Kimman TG, Binkhorst GJ, van den Ingh TS, Pol JM, Gielkens AL, Roelvink ME. 1991. Aujeszky’s disease in horses fulfills Koch’s postulates. Vet Rec 128:103–106. doi: 10.1136/vr.128.5.103. [DOI] [PubMed] [Google Scholar]
- 18.Shope RE. 1931. An experimental study of ‘mad itch’ with especial reference to its relationship to pseudorabies. J Exp Med 54:233–248. doi: 10.1084/jem.54.2.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brittle EE, Reynolds AE, Enquist LW. 2004. Two modes of pseudorabies virus neuroinvasion and lethality in mice. J Virol 78:12951–12963. doi: 10.1128/JVI.78.23.12951-12963.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG. 2012. Into the eye of the cytokine storm. Microbiol Mol Biol Rev 76:16–32. doi: 10.1128/MMBR.05015-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Q, Zhou Y-h, Yang Z-q. 2016. The cytokine storm of severe influenza and development of immunomodulatory therapy. Cell Mol Immunol 13:3–10. doi: 10.1038/cmi.2015.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.La Gruta NL, Kedzierska K, Stambas J, Doherty PC. 2007. A question of self-preservation: immunopathology in influenza virus infection. Immunol Cell Biol 85:85–92. doi: 10.1038/sj.icb.7100026. [DOI] [PubMed] [Google Scholar]
- 23.Wang S, Le TQ, Kurihara N, Chida J, Cisse Y, Yano M, Kido H. 2010. Influenza virus-cytokine-protease cycle in the pathogenesis of vascular hyperpermeability in severe influenza. J Infect Dis 202:991–1001. doi: 10.1086/656044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Costa VV, Fagundes CT, Souza DG, Teixeira MM. 2013. Inflammatory and innate immune responses in dengue infection: protection versus disease induction. Am J Pathol 182:1950–1961. doi: 10.1016/j.ajpath.2013.02.027. [DOI] [PubMed] [Google Scholar]
- 25.Klimstra WB, Ryman KD, Bernard KA, Nguyen KB, Biron CA, Johnston RE. 1999. Infection of neonatal mice with Sindbis virus results in a systemic inflammatory response syndrome. J Virol 73:10387–10398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang KJ, Su IJ, Theron M, Wu YC, Lai SK, Liu CC, Lei HY. 2005. An interferon-gamma-related cytokine storm in SARS patients. J Med Virol 75:185–194. doi: 10.1002/jmv.20255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jarosinski KW, Carpenter JE, Buckingham EM, Jackson W, Knudtson K, Moffat JF, Kita H, Grose C. 2018. Cellular stress response to varicella-zoster virus infection of human skin includes highly elevated interleukin-6 expression. Open Forum Infect Dis 5:ofy118. doi: 10.1093/ofid/ofy118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Engel JP, Madigan TC, Peterson GM. 1997. The transneuronal spread phenotype of herpes simplex virus type 1 infection of the mouse hind footpad. J Virol 71:2425–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guedon JM, Yee MB, Zhang M, Harvey SA, Goins WF, Kinchington PR. 2015. Neuronal changes induced by varicella zoster virus in a rat model of postherpetic neuralgia. Virology 482:167–180. doi: 10.1016/j.virol.2015.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koyuncu OO, MacGibeny MA, Hogue IB, Enquist LW. 2017. Compartmented neuronal cultures reveal two distinct mechanisms for alpha herpesvirus escape from genome silencing. PLoS Pathog 13:e1006608. doi: 10.1371/journal.ppat.1006608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hayden FG, Fritz R, Lobo MC, Alvord W, Strober W, Straus SE. 1998. Local and systemic cytokine responses during experimental human influenza A virus infection: relation to symptom formation and host defense. J Clin Invest 101:643–649. doi: 10.1172/JCI1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bordon J, Aliberti S, Fernandez-Botran R, Uriarte SM, Rane MJ, Duvvuri P, Peyrani P, Morlacchi LC, Blasi F, Ramirez JA. 2013. Understanding the roles of cytokines and neutrophil activity and neutrophil apoptosis in the protective versus deleterious inflammatory response in pneumonia. International J Infect Dis 17:e76–e83. doi: 10.1016/j.ijid.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 33.Cai B, Deitch EA, Ulloa L. 2010. Novel insights for systemic inflammation in sepsis and hemorrhage. Mediators Inflamm 2010:642462. doi: 10.1155/2010/642462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taubenberger JK, Morens DM. 2008. The pathology of influenza virus infections. Annu Rev Pathol 3:499–522. doi: 10.1146/annurev.pathmechdis.3.121806.154316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ng WF, To KF. 2007. Pathology of human H5N1 infection: new findings. Lancet 370:1106–1108. doi: 10.1016/S0140-6736(07)61490-1. [DOI] [PubMed] [Google Scholar]
- 36.Darlington GJ, Wilson DR, Lachman LB. 1986. Monocyte-conditioned medium, interleukin-1, and tumor necrosis factor stimulate the acute phase response in human hepatoma cells in vitro. J Cell Physiol 103:787–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Calabro P, Golia E, Yeh ET. 2012. Role of C-reactive protein in acute myocardial infarction and stroke: possible therapeutic approaches. Curr Pharm Biotechnol 13:4–16. doi: 10.2174/138920112798868764. [DOI] [PubMed] [Google Scholar]
- 38.Shrivastava AK, Singh HV, Raizada A, Singh SK. 2015. C-reactive protein, inflammation and coronary heart disease. Egyptian Heart J 67:89–97. doi: 10.1016/j.ehj.2014.11.005. [DOI] [Google Scholar]
- 39.Wu W, Shi D, Fang D, Guo F, Guo J, Huang F, Chen Y, Lv L, Li L. 2016. A new perspective on C-reactive protein in H7N9 infections. Int J Infect Dis 44:31–36. doi: 10.1016/j.ijid.2016.01.009. [DOI] [PubMed] [Google Scholar]
- 40.Wildhagen KCAA, Schrijver R, Beckers L, ten Cate H, Reutelingsperger CPM, Lutgens E, Nicolaes GAF. 2014. Effects of exogenous recombinant APC in mouse models of ischemia reperfusion injury and of atherosclerosis. PLoS One 9:e101446. doi: 10.1371/journal.pone.0101446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shaked Y, Tang T, Woloszynek J, Daenen LG, Man S, Xu P, Cai S-R, Arbeit JM, Voest EE, Chaplin DJ, Smythe J, Harris A, Nathan P, Judson I, Rustin G, Bertolini F, Link DC, Kerbel RS. 2009. Contribution of granulocyte colony-stimulating factor to the acute mobilization of endothelial precursor cells by vascular disrupting agents. Cancer Res 69:7524–7528. doi: 10.1158/0008-5472.CAN-09-0381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang H, Knight JS, Hodgin JB, Wang J, Guo C, Kleiman K, Eitzman DT. 2016. Psgl-1 deficiency is protective against stroke in a murine model of lupus. Sci Rep 6:28997. doi: 10.1038/srep28997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rassnick S, Enquist LW, Sved AF, Card JP. 1998. Pseudorabies virus-induced leukocyte trafficking into the rat central nervous system. J Virol 72:9181–9191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rinaman L, Card JP, Enquist LW. 1993. Spatiotemporal responses of astrocytes, ramified microglia, and brain macrophages to central neuronal infection with pseudorabies virus. J Neurosci 13:685–702. doi: 10.1523/JNEUROSCI.13-02-00685.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tanaka T, Narazaki M, Kishimoto T. 2014. IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol 6:a016295. doi: 10.1101/cshperspect.a016295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang P, Wen H, Ou S, Cui J, Fan D. 2012. IL-6 promotes regeneration and functional recovery after cortical spinal tract injury by reactivating intrinsic growth program of neurons and enhancing synapse formation. Exp Neurol 236:19–27. doi: 10.1016/j.expneurol.2012.03.019. [DOI] [PubMed] [Google Scholar]
- 47.Schmidt-Arras D, Rose-John S. 2016. IL-6 pathway in the liver: from physiopathology to therapy. J Hepatol 64:1403–1415. doi: 10.1016/j.jhep.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 48.Bendall LJ, Bradstock KF. 2014. G-CSF: From granulopoietic stimulant to bone marrow stem cell mobilizing agent. Cytokine Growth Factor Rev 25:355–367. doi: 10.1016/j.cytogfr.2014.07.011. [DOI] [PubMed] [Google Scholar]
- 49.Yong KL. 1996. Granulocyte colony-stimulating factor (G-CSF) increases neutrophil migration across vascular endothelium independent of an effect on adhesion: comparison with granulocyte-macrophage colony-stimulating factor (GM-CSF). Br J Haematol 94:40–47. doi: 10.1046/j.1365-2141.1996.d01-1752.x. [DOI] [PubMed] [Google Scholar]
- 50.Guo Y, Liu S, Zhang X, Wang L, Gao J, Han A, Hao A. 2015. G-CSF promotes autophagy and reduces neural tissue damage after spinal cord injury in mice. Lab Invest 95:1439–1449. doi: 10.1038/labinvest.2015.120. [DOI] [PubMed] [Google Scholar]
- 51.Kosai K, Seki M, Yanagihara K, Nakamura S, Kurihara S, Izumikawa K, Kakeya H, Yamamoto Y, Tashiro T, Kohno S. 2008. Gabexate mesilate suppresses influenza pneumonia in mice through inhibition of cytokines. J Int Med Res 36:322–328. doi: 10.1177/147323000803600215. [DOI] [PubMed] [Google Scholar]
- 52.Khong WX, Foo DG, Trasti SL, Tan EL, Alonso S. 2011. Sustained high levels of interleukin-6 contribute to the pathogenesis of enterovirus 71 in a neonate mouse model. J Virol 85:3067–3076. doi: 10.1128/JVI.01779-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lan T, Chang L, Wu L, Yuan Y-F. 2015. IL-6 plays a crucial role in HBV infection. J Clin Transl Hepatol 3:271–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang L, Zhong C, Wang J, Lu Z, Liu L, Yang W, Lyu Y. 2015. Pathogenesis of natural and experimental pseudorabies virus infections in dogs. Virol J 12:44. doi: 10.1186/s12985-015-0274-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.De Filippo K, Dudeck A, Hasenberg M, Nye E, van Rooijen N, Hartmann K, Gunzer M, Roers A, Hogg N. 2013. Mast cell and macrophage chemokines CXCL1/CXCL2 control the early stage of neutrophil recruitment during tissue inflammation. Blood 121:4930–4937. doi: 10.1182/blood-2013-02-486217. [DOI] [PubMed] [Google Scholar]
- 56.Deshmane SL, Kremlev S, Amini S, Sawaya BE. 2009. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res 29:313–326. doi: 10.1089/jir.2008.0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Selders GS, Fetz AE, Radic MZ, Bowlin GL. 2017. An overview of the role of neutrophils in innate immunity, inflammation and host-biomaterial integration. Regen Biomater 4:55–68. doi: 10.1093/rb/rbw041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Papayannopoulos V. 2018. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol 18:134–147. doi: 10.1038/nri.2017.105. [DOI] [PubMed] [Google Scholar]
- 59.de Oliveira S, Rosowski EE, Huttenlocher A. 2016. Neutrophil migration in infection and wound repair: going forward in reverse. Nat Rev Immunol 16:378–391. doi: 10.1038/nri.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Camp JV, Jonsson CB. 2017. A role for neutrophils in viral respiratory disease. Front Immunol 8:550. doi: 10.3389/fimmu.2017.00550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shaw SK, Owolabi SA, Bagley J, Morin N, Cheng E, LeBlanc BW, Kim M, Harty P, Waxman SG, Saab CY. 2008. Activated polymorphonuclear cells promote injury and excitability of dorsal root ganglia neurons. Exp Neurol 210:286–294. doi: 10.1016/j.expneurol.2007.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Park J-H, Choi S-H, Park S-J, Lee YJ, Park JH, Song PH, Cho C-M, Ku S-K, Song C-H. 2017. Promoting wound healing using low molecular weight Fucoidan in a full-thickness dermal excision rat model. Marine Drugs 15:112. doi: 10.3390/md15040112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kawai T, Akira S. 2009. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int Immunol 21:317–337. doi: 10.1093/intimm/dxp017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Diner BA, Lum KK, Toettcher JE, Cristea IM. 2016. Viral DNA sensors IFI16 and cyclic GMP-AMP synthase possess distinct functions in regulating viral gene expression, immune defenses, and apoptotic responses during herpesvirus infection. mBio 7:e01553-16. doi: 10.1128/mBio.01553-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kurt-Jones EA, Chan M, Zhou S, Wang J, Reed G, Bronson R, Arnold MM, Knipe DM, Finberg RW. 2004. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc Natl Acad Sci U S A 101:1315–1320. doi: 10.1073/pnas.0308057100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lamote JA, Kestens M, Van Waesberghe C, Delva J, De Pelsmaeker S, Devriendt B, Favoreel HW. 2017. The pseudorabies virus glycoprotein gE/gI complex suppresses type I interferon production by plasmacytoid dendritic cells. J Virol 91:e02276-16. doi: 10.1128/JVI.02276-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Granstedt AE, Bosse JB, Thiberge SY, Enquist LW. 2013. In vivo imaging of alphaherpesvirus infection reveals synchronized activity dependent on axonal sorting of viral proteins. Proc Natl Acad Sci U S A 110:E3516–E3525. doi: 10.1073/pnas.1311062110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Curanovic D, Lyman MG, Bou-Abboud C, Card JP, Enquist LW. 2009. Repair of the UL21 locus in pseudorabies virus Bartha enhances the kinetics of retrograde, transneuronal infection in vitro and in vivo. J Virol 83:1173–1183. doi: 10.1128/JVI.02102-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Platt KB, Mare CJ, Hinz PN. 1979. Differentiation of vaccine strains and field isolates of pseudorabies (Aujeszky’s disease) virus: thermal sensitivity and rabbit virulence markers. Arch Virol 60:13–23. doi: 10.1007/BF01318093. [DOI] [PubMed] [Google Scholar]
- 70.Bartha A. 1961. Experimental reduction of virulence of Aujeszky’s disease virus. Magy Allatorv Lapja 16:42–45. [Google Scholar]
- 71.Dijkstra JM, Gerdts V, Klupp BG, Mettenleiter TC. 1997. Deletion of glycoprotein gM of pseudorabies virus results in attenuation for the natural host. J Gen Virol 78:2147–2151. doi: 10.1099/0022-1317-78-9-2147. [DOI] [PubMed] [Google Scholar]
- 72.Klupp BG, Lomniczi B, Visser N, Fuchs W, Mettenleiter TC. 1995. Mutations affecting the UL21 gene contribute to avirulence of pseudorabies virus vaccine strain Bartha. Virology 212:466–473. doi: 10.1006/viro.1995.1504. [DOI] [PubMed] [Google Scholar]
- 73.Lomniczi B, Watanabe S, Ben-Porat T, Kaplan AS. 1987. Genome location and identification of functions defective in the Bartha vaccine strain of pseudorabies virus. J Virol 61:796–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Robbins AK, Ryan JP, Whealy ME, Enquist LW. 1989. The gene encoding the gIII envelope protein of pseudorabies virus vaccine strain Bartha contains a mutation affecting protein localization. J Virol 63:250–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ono E, Tasaki T, Kobayashi T, Taharaguchi S, Nikami H, Miyoshi I, Kasai N, Arikawa J, Kida H, Shimizu Y. 1999. Resistance to pseudorabies virus infection in transgenic mice expressing the chimeric transgene that represses the immediate-early gene transcription. Virology 262:72–78. doi: 10.1006/viro.1999.9899. [DOI] [PubMed] [Google Scholar]
- 76.Tombácz D, Tóth JS, Petrovszki P, Boldogkoi Z. 2009. Whole-genome analysis of pseudorabies virus gene expression by real-time quantitative RT-PCR assay. BMC Genomics 10:491. doi: 10.1186/1471-2164-10-491. [DOI] [PMC free article] [PubMed] [Google Scholar]