

Abstract
Oligodeoxynucleotide (ODN) synthesis, which avoids the formation of side products, is of great importance to biochemistry-based technology development. One side reaction of ODN synthesis is the cyanoethylation of the nucleobases. We suppressed this reaction by synthesizing ODNs using fully protected deoxynucleoside 3′-phosphoramidite building blocks, where the remaining reactive nucleobase residues were completely protected with acyl-, diacyl-, and acyl-oxyethylene-type groups. The detailed analysis of cyanoethylation at the nucleobase site showed that N3-protection of the thymine base efficiently suppressed the Michael addition of acrylonitrile. An ODN incorporating N3-cyanoethylthymine was synthesized using the phosphoramidite method, and primer extension reactions involving this ODN template were examined. As a result, the modified thymine produced has been proven to serve as a chain terminator.
Keywords: protecting group, oligodeoxynucleotide synthesis, cyanoethylation
1. Introduction
Much attention has been focused recently on chemically synthesized oligodeoxyribonucleotides (ODNs) for use in biochemical and therapeutic studies [1,2,3]. Many modified ODNs have, therefore, been synthesized to improve their original sequence specificity, stability, hybridization ability, enzyme resistance, and various functional properties. Demand for effective syntheses of longer ODNs has also increased since the total synthesis of a bacterial gene of Mycoplasma genitalium was achieved by Gibson et al. [4,5], who utilized extensive stepwise ligations of medium-size ODNs. At present, the chemical synthesis of ODNs seems limited to a one hundred nucleotide-length level. This limitation is due to the fact that side reactions of the synthesis increase as the ODN chain length increases. Among the most serious side reactions observed is the cyanoethylation of the base residues during the deprotection step. Several improved procedures to avoid this side reaction have been reported. From a different viewpoint, however, this side reaction could be avoided altogether through the full protection of all reactive functional groups on the base moieties. To date, no such example of a full protection strategy with the phosphoramidite method has been reported, although as reported by our group, such strategies were employed in the phosphotriester approach. In this paper, we report the synthesis of ODNs using the full-protection mode and discuss the problems associated with this strategy.
The phosphoramidite approach for the chemical synthesis of ODNs is an established procedure [6,7,8,9] that has achieved high reliability in coupling efficiency, practicality, and versatility. Through the use of appropriately protected monomer building blocks, side reactions on the nucleobases have been minimized. The 2-cyanoethyl group has been commonly utilized for the protection of internucleotidic phosphate groups because of the ease of deprotection with the use of concentrated NH3 [10,11,12]. However, this protecting group generates the toxic and potentially carcinogenic compound acrylonitrile through β-elimination in the deprotection step [13,14]. The generated acrylonitrile has also been proven to be reactive towards the amino or imido groups on the nucleobases [14,15,16,17,18] to give addition products. It has also been reported that the imido NH group of the thymine residue is predominantly cyanoethylated over the other nucleobases (A, G, and C) [19]. In fact, Ravikumar has reported that ODN derivatives, cyanoethylated at the thymine residue, were isolated as side products when concentrated NH3 (aq) was used for the deprotection step [20]. Some researchers have recently studied the synthesis of ODNs without cyanoethylation at the nucleobase site because oligonucleotide products contaminated with cyanoethylated species would affect the chemical and pharmaceutical properties when used as DNA probes and nucleic acid drugs, respectively. Beaucage et al. has used the 4-[N-(2,2,2-trifluoroacetyl)amino]butyl group in place of the 2-cyanoethyl group for protection of the phosphate group [21]. The 4-[N-(2,2,2-trifluoroacetyl)amino]butyl group has no addition ability after the deprotection step because the protecting group is converted to a cycloaminoalkane derivative through the deprotection process. Wada et al. reported that the formation of N3-cyanoethylthymidine was efficiently suppressed by using nitromethane as a scavenger under the deprotection conditions [22]. Chang et al. indicated that the use of t-butylamine was efficient for avoidance of alkylation by acrylonitrile at the deprotection step [23].
In this study, we synthesized ODNs with the focus of preventing the cyanoethylation at the nucleobase site through the use of fully protected monomers. The three kinds of deoxynucleoside 3′-phosphoramidite building blocks (C, A, G) were completely protected by acyl type protecting groups at the amino group of the nucleobases, as shown in Figure 1. In the thymine residue, the benzoyl (Bz) group was used to block the addition reaction. In the cytosine residue, the phthaloyl (phth) group was used to completely mask the two protons of the 4-amino substituent, because it was known that 4-N-benzoylcytidine is only slightly reactive toward acrylonitrile [21]. On the other hand, the adenine and guanine residues were comparatively non-reactive towards cyanoethylation in comparison to the thymine residue. The adenine and guanine moieties, however, were protected with phthaloyl and isobutyryloxyethylene (iBu·dibe) groups, respectively, to entirely eliminate the possibility of the cyanoethylation side reaction.
Figure 1.

Fully protected monomers.
In this paper, we report the synthesis of ODNs using these fully protected monomers. In addition, the stabilities of the fully protected base derivatives under various conditions were also examined with the focus of whether these protecting groups could be effective in avoiding the acrylonitrile-mediated Michael addition reaction. Lastly, we report the base recognition ability of N3-cyanoethylated T, when introduced into DNA, based on Tm experiments and DNA polymerase-mediated insertion reactions.
2. Results and Discussion
2.1. Synthesis of Fully Protected Deoxynucleoside 3′-Phosphoramidite Building Blocks
To evaluate the deactivation of the fully protected nucleobases towards cyanoethylation with acrylonitrile, we synthesized fully protected phosphoramidite units, as previously reported [24,25,26,27,28,29,30,31,32,33,34]. The benzoyl group was selected as a full protecting group for the thymine residue [24,25]. The N3-protection of 2′-deoxythymidine by reaction with benzoyl chloride gave compound 2 quantitatively via the transient O-trimethylsilylation. The tritylation of compound 2 with DMTrCl, as per standard procedure, produced the 5′-protected product 3 in 98% yield. The phosphitylation of 3 with bis(diisopropylamino)(2-cyanoethoxy)phosphine produced the fully protected phosphoramidite unit 4 in 73% yield (Scheme 1).
Scheme 1.

Synthesis of fully protected 2′-deoxythymidine derivative 4.
For the full protection of the cytosine and adenine residues, the phthaloyl group was utilized [26,27,28,29,30]. This protecting group enabled us to protect the two protons of the amino group of the C or A base simultaneously via formation of a five-membered ring. It was found that 2′-deoxyadenosine derivatives protected with the phthaloyl group exhibited significantly greater suppression of depurination under acid conditions than when protected with the simple benzoyl group. The phthaloyl group also possesses a unique property that, even when ring opening occurred, re-cyclization could be performed during the capping reaction with acetic anhydride. In this study, we developed a new route to the amidite units using selective deacylation of the commercially available 4-benzoyldeoxycytidine 3′-phosphoramidite derivative, followed by acylation of the resulting N-free species 5 with phthaloyl chloride [31]. Thus, the desired phosphoramidite building block 6 was obtained in 83% yield (Scheme 2). In a similar manner, the 6-N-phthaloyldeoxyadenosine 3′-phosphoramidite derivative 8 was synthesized in 61% yield by treatment of the N-free derivative 7 with phthaloyl chloride in the presence of diisopropylethylamine in THF.
Scheme 2.

Synthesis of fully protected 2′-deoxycytidine derivative 6 and 2′-deoxyadenosine derivative 8.
For full protection of the guanine base, we used a 1,2-bis(isobutyryloxy)ethylene (iBu·dibe) group as an additional protecting group with the widely used isobutyryl group on the 2-amino substituent. This double protecting mode was used for the synthesis of G-rich oligodeoxynucleotides in the phosphotriester approach, and proved to effectively avoid the side reactions associated with the guanine base [32,33,34]. Moreover, the iBu·dibe group could be simultaneously removed under the deprotection conditions of the other N-acyl protecting groups at T, C, and A. In this study, we synthesized the phosphoramidite unit of deoxyguanosine, protected with the iBu·dibe group (Scheme 3). 3′,5′-Bis(t-butyldimethylsilyl)-2-isobutyryl-2′-deoxyguanine derivative 9, having an isobutyryl group at the 2-N-amino group, was allowed to react with excess glyoxal and isobutyric anhydride in pyridine, which formed the fully protected derivative 10 in 98% yield. When the N-unprotected 2′-deoxyguanosine derivative was used as the starting material in this reaction, the desired compound 10 could not be obtained, but a complex mixture containing various glyoxal adducts was formed. Treatment of compound 10 with Et3N·3HF in THF gave the desilylated product 11. Tritylation of 11 with DMTrCl, as per usual conditions, produced the 5′-protected product 12 in 60% yield. The phosphitylation of 12 with bis(diisopropylamino)(2-cyanoethoxy)phosphine produced the fully protected phosphoramidite unit 13 in 93% yield.
Scheme 3.

Synthesis of fully protected 2′-deoxyguanosine derivative 13.
2.2. Evaluation of the Stability of the Fully Protected Nucleobases toward Cyanoethylation
We introduced the fully protected phosphoramidite units of TBz, dCphth, dAphth, and dGiBu·dibe into a resin linked to UnyLinker™ in order to evaluate the resistance of the fully protected base moieties to the addition reaction of acrylonitrile under the conditions required for removal of the cyanoethyl group in the solid phase. Subsequently, each monomer loaded resin was treated with 100 equiv of acrylonitrile in DBU/CH3CN, which corresponded to the conditions estimated when a 100mer ODN was synthesized. To compare the stability of the fully protected monomers, stability of the common protected monomers [T, 4-N-acetyl-dC (dCac), 6-N-phenoxyacetyl-dA (dApac), and 2-N-(4-isopropyl)phenoxyacetyl-dG (dGiPrpac)] was also examined under the same conditions.
Cyanoethylation for common protected T, which is the most reactive compared to the other bases, was observed after 1 min by HPLC analysis. The N3-cyanoethylated derivative (TCE) of thymidine increased as time passed and was formed in 52% yield after 24 h. The time course of the remaining T monomer in the reaction of T or TBz with acrylonitrile is shown in Figure 2. Results indicated that T decreased linearly in a time-dependent manner. In contrast, when TBz was used in this evaluation, TCE was not detected by HPLC analysis within the 24 h period. Therefore, protection of T at the N3-amino group proved to be very effective for suppression of cyanoethylation of T.
Figure 2.

The remaining amount (%) of T or TBz in its reaction with acrylonitrile. Open and filled circles refer to the remaining amounts (%) of T and TBz in use of T and TBz, respectively. Conditions: 100 equiv of acrylonitrile in DBU/CH3CN.
Cyanoethylation of the other fully protected monomers (Cphth, Aphth, and GiBu·dibe) did not occur within 24 h under the same conditions as those used for the ODN synthesis using the common protected monomers (Figure 3i). In the case of Cphth and Aphth, the minor product (peak a) was phthalamide resulted from the ammonolysis of the phthaloyl group. In the case of GiBu·dibe, the side product (peak b) was observed ahead of the peak of dG. This product is not an alkylated product but an incompletely deprotected G residue because the product (peak b) was decreased by prolonged treatment with concentrated NH3. To examine the stability of GiBu·dibe in DBU/CH3CN in detail, we carried out the reaction of the GiBu·dibe derivative 10 with DBU/CH3CN. After the reaction for 5 min, the decomposition of the GiBu·dibe derivative 10 was observed by TLC analysis. The decomposed compounds did not involve 3´,5´-O-bis(tert-butyldimethylsilyl)-2´-deoxyguanosine. The side product (peak b) might be an incompletely deprotected dG derivative or a compound having a moiety derived from the iBu·dibe group.
Figure 3.

Analysis of cyanoethylation of (i) the fully protected monomers (Cphth, Aphth, and GiBu·dibe) and (ii) the common protected monomers (Cac, Apac, and GiPrpac) by reverse-phase HPLC; (iii) Analysis of deprotection of the fully protected monomers by concentrated NH3 at 55 °C for 6 h.
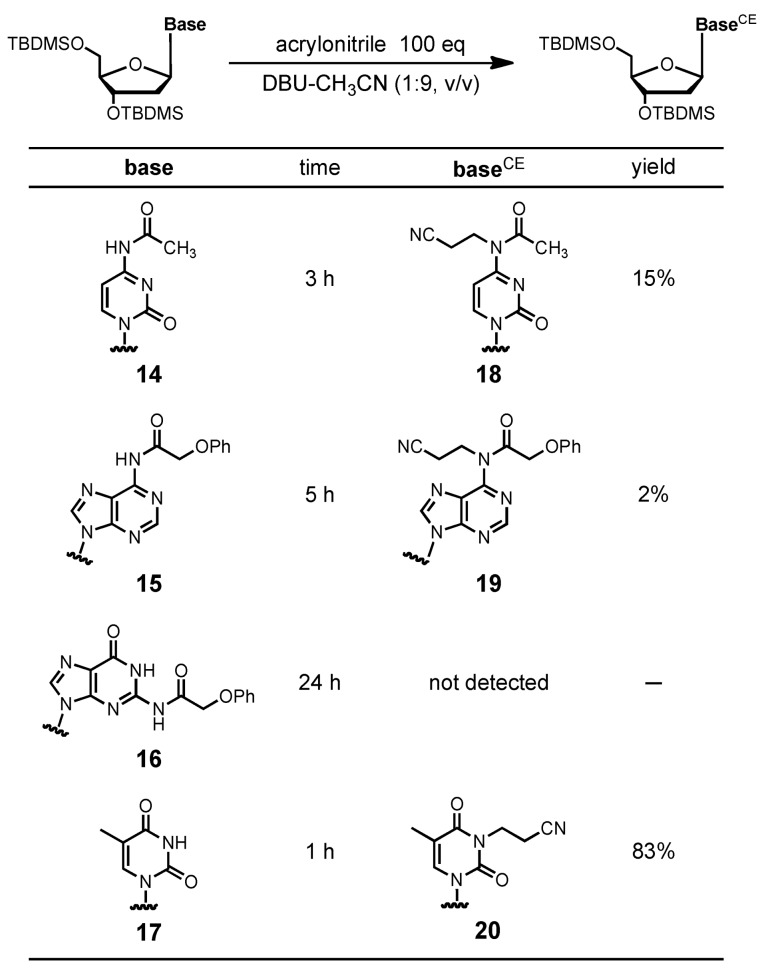
On the other hand, cyanoethylation for the common protected monomers (Cac, Apac, and GiPrpac) was not observed (Figure 3ii). The results indicated that the reactivity of the N-monoacylated C, A, and G residues toward acrylonitrile is lower than that of the T residue. However, we found that N-monoacylated C and A residues were alkylated by acrylonitrile in DBU/CH3CN in solution phase, as shown in Table 1. The reaction of 4-N-acetyl-2′-deoxycytidine derivative 14 with acrylonitrile gave the 4-N-cyanoethylated derivative 18 in 15% yield. 6-N-Phenoxyacetyl-2′-deoxyadenosine derivative 15 was slightly alkylated under the same conditions to give the 6-N-cyanoethylated derivative 19 in 2% yield. Therefore, there is a possibility that the common protected monomers Cac and Apac are alkylated by acrylonitrile on the ODN synthesis even if the C and A residues are protected by the monoacylated protecting group when DBU was used. It seems that the fully protected approach is important for the C and A residues to avoid the cyanoethylation. Cyanoethylation of a 2-N-phenoxyacetyl-2′-deoxyguanosine derivative was not detected by TLC and mass analysis after 24 h. It was found that the N-monoacylated G residue could suppress the cyanoethylation by acrylonitrile.
Table 1.
Cyanoethylation of the common protected monomers in solution phase reaction.
|
In the C and A monomers, the phth group for complete protection of the amino groups was effective for avoidance of the cyanoethylation because N-monoacylated monomers proved to react with acrylonitrile. The iBu·dibe group on the GiBu·dibe base was rapidly decomposed within 5 min during the DBU/CH3CN treatment required for removal of the 2-cyanoethyl group at the internucleotidic phosphate group as mentioned before but cyanoethylation for the GiBu·dibe residue was not observed under the conditions of acrylonitrile-DBU/CH3CN (Figure 3i). However, the fully protected species of TBz, Cphth, Aphth, and GiBu·dibe were completely removed by treatment with concentrated NH3 at 55 °C for 6 h (Figure 3iii). Therefore, the full protection mode can be used when it is necessary to protect the base moieties to avoid any side reactions which are expected when the usual monoacylated base moieties are used for further modification of sugar or phosphate residues.
2.3. Synthesis of Oligonucleotides Using the Fully Protected Phosphoramidites
To evaluate if the full protection of the base residues was effective for the oligonucleotide synthesis, we synthesized an ODN with the fully protected monomers using crosslinked polystyrene beads (UnyLinker™ NittoPhase®). Selective removal of the 2-cyanoethyl groups at the internucleotidic phoshate groups was performed by treatment with DBU/CH3CN (1:9, v/v) for 1 min [35,36]. The successive release of the base-protected oligomer from the resin and complete deprotection of the other base-labile protecting groups were simultaneously conducted by treatment with concentrated ammonia at 55 °C for 14 h. The mixture thus obtained was analyzed by anion-exchange HPLC (Figure 4i). The result showed that the desired ODN appeared as the main product. In contrast, several minor products were also observed in this HPLC analysis. It was expected that treatment of dGiBu·dibe with DBU/CH3CN produced these byproducts, as indicated the previous section. To confirm this expectation, we prepared base-protected T10 and ATCATCATCG derivatives, which were synthesized on the same resins by use of TBz, Cphth, Aphth, and GiPrpac phosphoramidite building blocks. In the latter oligomer, the GiPrpac monomer unit was used in place of GiBu·dibe. These protected ODNs were treated with DBU-CH3CN under the same conditions followed by treatment with concentrated NH3. The analysis by HPLC suggested that the corresponding ODNs were obtained as single products and the minor products were not produced as observed in the reaction using ODN containing the GiBu·dibe monomer (Figures 4ii, iii). When a base-protected ATCATCATCG oligomer on the resin having fully protected monomers (TBz, Cphth, Aphth, and GiBu·dibe) was deprotected by treatment with concentrated NH3 at 55 °C for 6 h, the desired ODN was obtained without side products (Figure 4iv).
Figure 4.

Anion-exchange HPLC profiles of ODNs obtained by using (i) the fully protected monomers (TBz, Cphth, Aphth, and GiBu·dibe); (ii) only TBz monomer and (iii) TBz, Cphth, Aphth, and GiPrpac monomers after removal of the cyaonoethyl groups of the internucleotidic phosphate groups by treatment with DBU-CH3CN (1:9, v/v); (iv) Anion-exchange HPLC profile of deprotection of ODN having the fully protected monomers (TBz, Cphth, Aphth, and GiBu·dibe) by concentrated NH3.
When a longer ODN is synthesized in the solid phase, there are considerable concerns in addition to cyanoethylation at the nucleobases. One expected problem is the depurination of the A and G residues under acidic conditions, which is required for the repeated removal of the DMTr group during chain elongation. In the synthesis of a longer ODN, the total time of the repeated acidic treatments increases with chain length. Therefore, we examined the stability of the glycosyl bonds of the fully protected Aphth and GiBu·dibe bases in 3% TCA/CH2Cl2, which is used for removal of the DMTr group in the current DNA synthesis. To evaluate the stability of these nucleobases, we synthesized ODNs containing the fully protected monomer Aphth or GiBu·dibe at the central position. The synthesized ODNs on the resin were treated with 3% TCA in CH2Cl2 for 24 h. After the treatment with concentrated ammonia, the mixtures thus obtained were analyzed by reverse-phase HPLC (Figure 5). In the reaction mixture, the side products originating from the depurination of Aphth or GiBu·dibe were not detected. The results showed that the Aphth and GiBu·dibe residues were stable under acidic conditions for extended durations. These fully protecting groups on the purine bases will undoubtedly become useful for the synthesis of long chain DNAs.
Figure 5.

Analysis of depurination using ODNs containing (A) Aphth or (B) GiBu·dibe by reverse-phase HPLC.
2.4. Evaluation of Base Recognition Ability of Cyanoethylated T by Tm Experiments
As mentioned above, cyanoethylation of the nucleobases, especially the T residue, is well known as a side reaction in ODN synthesis. However, little attention has been devoted to study the properties of ODNs containing TCE. We investigated the base recognition ability of TCE using Tm experiments and DNA polymerase-mediated insertion reactions.
For the synthesis of ODN containing TCE, we synthesized the TCE phosphoramidite unit 23 from a T derivative 21, as shown in Scheme 4. N3-Cyanoethylation of 21 with acrylonitrile gave the cyanoethylated derivative 22 after acetylation of the 3′-hydroxy group. The phosphitylation of 22 with bis(diisopropylamino)(2-cyanoethoxy)phosphine gave the cyanoethylated phosphoramidite unit 23 in 58% yield. Subsequently, we synthesized an ODN containing TCE using the phosphoramidite unit 23, as shown in Table 2. At the deprotection step of the ODN synthesis, the N3-cyaoethyl moiety of T was found to be very stable in concentrated NH3.
Scheme 4.

Synthesis of TCE phosphoramidite unit 23.
Table 2.
Sequences and isolated yields of ODNs containing TCE.
| ODN | Sequence | Yield (%) | MALDI-TOF mass [M+H]+ | |
|---|---|---|---|---|
| calcd. | found | |||
| 1 | 5′-d(CGTCTTCETCTGC)-3′ | 48 | 3326.6 | 3329.4 |
| 2 | 5′-d(TTAGACTCEGTAACCGGTCTTCGCGCG)-3′ | 33 | 7699.3 | 7691.4 |
To clarify the base pairing properties of TCE in DNA duplexes, Tm experiments were performed using duplexes formed between modified ODNs and their complementary ODNs, with matched or single-mismatched sequences at the central position, as shown in Table 3. A duplex with a matched natural T-A base pair showed a Tm value of 58.1 °C. In contrast, the Tm value of a duplex with a N3-cyanoethylated TCE-A base pair decreased to 36.0 °C, which is 22.1 °C lower than that of the natural T-A matched base pair. In addition, the Tm values of DNA duplexes containing the mismatched pairs (for T, C, and G) also decreased, showing a ΔTm of −7.2 °C, −6.8 °C, and −8.4 °C, respectively. These results indicated that the base pairs having TCE were unstable and the Tm values (for all bases) of DNA duplexes containing TCE became similar to each other. Therefore, it was found that the sequence dependence of ODN having TCE was completely lacking. A significant drop of the thermal stability of the duplex especially was observed when X was the matched A base. This is apparent because the hydrogen bonding ability of TCE was lost by addition of acrylonitrile to the N3-NH group, which is an accepter site of a hydrogen bond for the A-T base pair.
Table 3.
Tm values for DNA–DNA 11-mer duplexes containing TCE.
| DNA–DNA duplex | d(GCA GAX AGA CG)-5′ | ||||
|---|---|---|---|---|---|
| 5′-d(CGT CTY TCT GC) | |||||
| Y = T | Y = TCE | ||||
| X | Tm (°C)a | Tm (°C)a | ΔTm (°C)b | ||
| A | 58.1 | 36.0 | −22.1 | ||
| T | 43.6 | 36.4 | −7.2 | ||
| C | 42.0 | 35.2 | −6.8 | ||
| G | 44.0 | 35.6 | −8.4 | ||
aThe Tm values are accurate within ±0.5 °C. The Tm measurements were performed in a buffer containing 10 mM sodium phosphate (pH 7.0), 1 M NaCl, 0.1 mM EDTA, and 2 µM duplex. bΔTm is the difference in the Tm values between the unmodified and modified ODNs.
2.5. Evaluation of the Base Recognition Ability of Cyanoethylated T by DNA Polymerase-Mediated Insertion Reactions
To study the template specific incorporation of dNTPs using DNA polymerase, we examined the single insertion reaction of dNTPs toward the opposite TCE in templates using Taq polymerase [37], a well-known thermostable enzyme used in PCR reactions [38,39]. The template incorporating T or TCE at position X was annealed with a 5′-FAM-labeled 18-nt primer in the presence of DNA polymerase. Following the enzymatic reaction, the products were analyzed by PAGE, as shown in Figure 6B. Consequently, the incorporation of dATP was observed using the template containing a natural T base. In contrast, dNTPs were not incorporated into the opposite site of TCE in the template by Taq DNA polymerase. It was found that N3-cyanoethylation of the T residue caused the inhibition of the base recognition by DNA polymerase and TCE in the template terminated the primer extension to give no incorporation of dNTPs at the opposite site. Therefore, if synthesized ODNs including cyanoethylated species would be used as PCR templates, PCR would be stopped at the modified base to produce shorter duplicated products. Since PCR is a system that enables amplification of a trace amount of DNA, a minor contamination of N3-cyanoethylated T does not affect the whole PCR process. TCE, therefore, could be used as a new type of chain terminator in DNA duplications.
Figure 6.

Single dNTP insertion reactions using Taq DNA polymerases. (A) Sequences of 5′-FAM labeled 18-nt primer and 25-nt templates; (B) PAGE analysis of single-insertion reactions.
3. Experimental
General
1H-, 13C-, and 31P-NMR spectra were recorded at 500, 126 and 203 MHz, respectively. The chemical shifts were measured from tetramethylsilane for 1H NMR spectra, CDCl3 (77 ppm) for 13C-NMR spectra and 85% phosphoric acid (0 ppm) for 31P NMR spectra. UV spectra were recorded on a U-2000 spectrometer. Column chromatography was performed with silica gel C-200 and a minipump for a goldfish bowl was conveniently used to attain sufficient pressure for rapid chromatographic separation. HPLC was performed using the following systems. Reversed-phase HPLC was done on a system with a 3D UV detector and a C18 column (4.6 x 150 mm). A linear gradient (0-30%) of solvent I [0.03 M ammonium acetate buffer (pH 7.0)] in solvent II (CH3CN) was used at 30 °C at a rate of 1.0 mL/min for 30 min. Anion-exchange HPLC was done on an apparatus with a 3D UV detector and a FAX column (Waters, 4.6 × 100 mm). A linear gradient (0-60%) of Solvent III [1 M NaCl and 25 mM phosphate buffer, 10% CH3CN (v/v)] in solvent IV [25 mM phosphate buffer, 10% CH3CN (v/v)] was used at 50 °C at a flow rate of 1.0 mL/min for 45 min. ESI mass was performed by use of MarinerTM (PerSeptive Biosystems Inc.). MALDI-TOF mass was performed by use of Bruker Daltonics [Matrix: 3-hydroxypicolinic acid (100 mg/ml) in H2O-diammonium hydrogen citrate (100 mg/ml) in H2O (10:1, v/v)]. Compounds 1 and 21 were purchased from GeneACT, Inc. Compounds 5, 7 [31], 9 [40], 14 [26],and 16 [41] were prepared according to the published procedure. Common protected phosphoramidites were purchased from Glen Research Corporation.
N3-Benzoyl-5´-O-(4, 4'-dimethoxytrityl)thymidine (3). Compound 2 (2.42 g, 7.0 mmol) was rendered anhydrous by repeated coevaporation with dry pyridine (3 mL × 1) and dissolved in dry pyridine (70 mL). To the solution was added DMTrCl (3.08 g, 9.1 mmol) and the mixture was stirred at room temperature for 5 h. The reaction was quenched by addition of saturated aqueous NaHCO3. The mixture was partitioned between CHCl3 and H2O. The organic phase was collected, dried over Na2SO4, filtered, and evaporated under reduced pressure. The residue was chromatographed on a column of silica gel with CHCl3-MeOH (100:0–98:2, v/v) containing 1% Et3N to give the fractions containing 3. The fractions were collected and evaporated under reduced pressure. The residue was finally evaporated by repeated coevaporation three times each with toluene and CHCl3 to remove the last traces of Et3N to give 3 (4.45 g, 98%). 1H-NMR (CDCl3) δ 1.50 (s, 3H), 2.36–2.42 (m, 2H), 3.40 (dd, 1H, J = 2.2 Hz, J = 9.5 Hz), 3.52 (dd, 1H, J = 2.7 Hz, J = 9.2 Hz), 3.81 (s, 6H), 4.05–4.06 (m, 1H), 4.60 (m, 1H), 6.39 (t, 1H, J = 6.6 Hz), 6.85–6.87 (m, 4H), 7.27–7.32 (m, 7H), 7.41 (d, 2H, J = 7.6 Hz), 7.49 (t, 2H, J = 7.6 Hz), 7.64 (t, 1H, J = 7.6 Hz), 7.70 (s, 1H), 7.94 (d, 1H, J = 7.6 Hz); 13C-NMR (CDCl3) δ 12.1, 41.4, 55.5, 63.7, 72.7, 85.2, 86.4, 87.3, 111.5, 113,57, 113.59, 127.5, 128.30, 128.37, 129.3, 130.3, 130.7, 131.9, 135.2, 135.50, 135.57, 135.63, 144.5, 149.5, 159.0, 163.1, 169.3. HRMS (ESI) m/z (M+Na)+: calcd for C38H36N2NaO8+ 671.2364; found, 671.2364.
N3-Benzoyl-5´-O-(4,4'-dimethoxytrityl)thymidine 3'-(2-cyanoethyl N,N-diisopropylphosphor-amidite) (4). Compound 3 (380 mg, 0.59 mmol) was rendered anhydrous by repeated coevaporation with dry pyridine (1 mL × 1), dry toluene (1 mL × 1), dry CH3CN (1 mL × 1) and dissolved in dry CH2Cl2 (6 mL). To the solution was added bis(diisopropylamino)(2-cyanoethoxy)phosphine (205 g, 0.65 mmol), 1H-tetrazole (25 mg, 0.35 mmol), diisopropylamine (50 μL, 0.35 mmol) and the mixture was stirred at room temperature for 3 h. The reaction was quenched by addition of saturated aqueous NaHCO3. The mixture was partitioned between CHCl3 and aqueous NaHCO3. The organic phase was collected, dried over Na2SO4, filtered, and evaporated under reduced pressure. The residue was chromatographed on a column of silica gel with CHCl3-MeOH (100:0–98:2, v/v) containing 1% Et3N to give the fractions containing 4. The fractions were collected and evaporated under reduced pressure. The residue was finally evaporated by repeated coevaporation three times each with toluene and CHCl3 to remove the last traces of Et3N to give 4 (338 mg, 73%). 1H-NMR (CDCl3) δ 1.07 (d, 3H, J = 6.5 Hz), 1.16 (d, 9H, J = 5.6 Hz), 1.48 (s, 3H), 2.39 (t, 1H, J = 6.0 Hz), 2.48 (m, 1H), 2.55 (t, 1H, J = 6.0 Hz), 2.61 (m, 1H), 3.40 (t, 1H, J = 10.1 Hz), 3.54–3.71 (m, 5H), 3.76 (s, 6H), 4.18, 4.24 (2s, 1H), 4.75 (m, 1H), 6.42–6.45 (m, 1H), 6.86–6.89 (m, 4H), 7.24 (m, 1H), 7.32–7.37 (m, 6H), 7.43–7.48 (m, 4H), 7.59 (t, 1H, J = 7.3 Hz), 7.79, 7.84 (2s, 1H), 7.94 (d, 1H, J = 7.3 Hz); 13C-NMR (CDCl3) δ 12.1, 12.1, 20.4, 20.5, 20.6, 20.6, 24.8, 40.5, 43.4, 43.5, 43.5, 43.6, 55.5, 55.5, 55.5, 58.3, 58.4, 58.6, 63.3, 63.4, 73.7, 74.0, 85.2, 85.8, 86.1, 87.3, 111.4, 111.4, 113.5, 113.6, 117.9, 118.1, 127.5, 128.3, 128.5, 128.5, 129.4, 130.4, 130.7, 131.9, 135.3, 135.5, 135.5, 135.6, 135.6, 135.9, 144.6, 144.6, 149.6, 149.7, 159.1, 163.1, 169.5; 31P-NMR (CDCl3) δ 149.5, 149.9 (2s). HRMS (ESI) m/z (M+Na)+: calcd for C47H53N4NaO9P+ 871.3442; found, 871.3441.
4-N-Phthaloyl-5´-O-(4,4'-dimethoxytrityl)-2´-deoxycytidine 3'-(2-cyanoethyl N,N-diisopropylphos-phoramidite) (6). Compound 5 (869 mg, 1.19 mmol) was rendered anhydrous by repeated coevaporation with dry pyridine (1 mL × 1), dry toluene (1 mL × 1), dry CH3CN (1 mL × 1), and dissolved in dry THF (12 mL). To the solution was added phthaloyl chloride (189 μL, 1.31 mmol), diisopropylethylamine (456 μL, 2.62 mmol) at 0 °C and the mixture was stirred at room temperature for 2 h. The reaction was quenched by addition of H2O. The mixture was partitioned between ethyl acetate and H2O. The organic phase was collected, dried over Na2SO4, filtered, and evaporated under reduced pressure. The residue was chromatographed on a column of silica gel with hexane-ethyl acetate (40:60–30:70, v/v) containing 1% pyridine to give the fractions containing 6. The fractions were collected and evaporated under reduced pressure. The residue was finally evaporated by repeated coevaporation three times each with toluene and CHCl3 to remove the last traces of pyridine to give 6 (846 mg, 83%). 1H-NMR (CDCl3) δ 1.04–1.17 (m, 12H), 2.40–2.45 (m, 2H), 2.62 (t, 1H, J = 6.3 Hz), 2.72–2.82 (m, 1H), 3.40–3.61 (m, 6H), 3.78 (s, 6H), 4.20–4.21 (m, 1H), 4.59–4.69 (m, 1H), 6.22–6.32 (m, 2H), 6.81–6.86 (m, 4H), 7.26–7.39 (m, 9H), 7.79–7.82 (m, 2H), 7.94–7.97 (m, 2H), 8.51, 8.60 (2d, 1H, J = 7.3 Hz); 13C-NMR (CDCl3) δ 20.1, 20.2, 20.4, 24.4, 24.5, 24.6, 40.7, 41.1, 43.1, 43.2, 43.3, 53.4, 55.2, 58.1, 58.4, 61.5, 61.9, 70.9, 71.2, 71.8, 72.1, 85.8, 86.9, 87.5, 87.6, 100.5, 113.2, 117.3, 117.5, 124.3, 127.1, 128.0, 128.1, 128.2, 130.07, 130.13, 131.4, 135.1, 135.3, 144.0, 145.2, 154.6, 158.6, 159.1, 164.9; 31P-NMR (CDCl3) δ 149.6, 150.2 (2s). HRMS (ESI) m/z (M+H)+: calcd for C47H51N5O9P+ 860.3419; found, 860.3411.
6-N-Phthaloyl-5´-O-(4,4'-dimethoxytrityl)-2´-deoxyadenosine 3'-(2-cyanoethyl N,N-diisopropyl-phosphoramidite) (8). Compound 7 (1.06 g, 1.40 mmol) was rendered anhydrous by repeated coevaporation with dry pyridine (1 mL × 1), dry toluene (1 mL × 1), dry CH3CN (1 mL × 1) and dissolved in dry THF (14 mL). To the solution was added phthaloyl chloride (221.7 μL, 1.54 mmol), diisopropylethylamine (1.22 mL, 7.0 mmol) at 0 °C and the mixture was stirred at room temperature for 2 h. The reaction was quenched by addition of H2O. The mixture was partitioned between CHCl3 and H2O. The organic phase was collected, dried over Na2SO4, filtered, and evaporated under reduced pressure. The residue was chromatographed on a column of silica gel with hexane-ethyl acetate (40:60–30:70, v/v) containing 1% pyridine to give the fractions containing 8. The fractions were collected and evaporated under reduced pressure. The residue was finally evaporated by repeated coevaporation three times each with toluene and CHCl3 to remove the last traces of pyridine to give 8 (761 mg, 61%). 1H-NMR (CDCl3) δ 1.13–1.22 (m, 12H), 2.48 (t, 1H, J = 6.3 Hz), 2.62–2.68 (m, 3H), 2.72–2.75 (m, 1H), 3.35–3.37 (m, 1H), 3.59–3.77 (m, 9H), 4.33–4.35 (m, 1H), 4.77–4.78 (m, 1H), 6.55 (t, 1H, J = 5.7 Hz), 6.79–6.81 (m, 4H), 7.18–7.30 (m, 7H), 7.40 (d, 2H, J = 7.3 Hz), 7.82–7.83 (m, 2H), 8.01–8.02 (m, 2H), 8.34, 8.36 (2s, 1H), 8.96 (d, 1H, J = 3.7 Hz); 13C-NMR (CDCl3) δ 20.4, 20.6, 24.65, 24.70, 24.76, 24.8, 39.7, 39.8, 43.4, 43.5, 55.35, 55.36, 58.4, 58.5, 63.3, 63.5, 73.6, 73.7, 74.3, 74.4, 85.11, 85.13, 86.7, 113.3, 117.5, 117.6, 124.5, 127.08, 127.11, 128.0, 128.2, 128.3, 130.11, 130.15, 130.19, 130.23, 130.27, 132.1, 134.9, 135.6, 135.7, 135.8, 144.44, 144.48, 144.5, 152.5, 152.5, 153.5, 153.6, 158.7, 165.7; 31P-NMR (CDCl3) δ 149.9, 150.2 (2s). HRMS (ESI) m/z (M+H)+: calcd for C48H51N7O8P+ 884.3531; found, 884.3533.
2-N-Isobutyryl-1,N2-[1,2-di(isobutyryloxy)ethylene]-3´,5´-O-bis(tert-butyldimethylsilyl)-2´-deoxy-guanosine (10). 37% Glyoxal (5 mL, 40 mmol) was added to compound 9 (1.26 g, 2.0 mmol) at room temperature. The mixture was rendered anhydrous by repeated coevaporation with dry pyridine (1 mL x1) and dissolved in dry pyridine (20 mL). After the solution was stirred at room temperature for 3 h, isobutyric anhydride (6.5 mL, 40 mmol) was added, and the mixture was stirred at room temperature for 1 h. The reaction was quenched by addition of saturated aqueous NaHCO3. The mixture was partitioned between CHCl3 and H2O. The organic phase was collected, dried over Na2SO4, filtered, and evaporated under reduced pressure. The residue was chromatographed on a column of silica gel with hexane-ethyl acetate (90:10–70:30, v/v) to give the fractions containing 10. The fractions were collected and evaporated under reduced pressure to give 10 (1.51 g, 98%). 1H-NMR (CDCl3) δ 0.11 (s, 12H), 0.92 (s, 18H), 1.15–1.19 (m, 12H), 1.25–1.31 (m, 6H), 2.35–2.38 (m, 2H), 2.54–2.59 (m, 2H), 3.80 (m, 2H), 4.00–4.04 (m, 2H), 4.51 (m, 1H), 6.29 (t, 1H, J = 6.5 Hz), 6.78 (d, 1H, J = 10.0 Hz), 6.83 (d, 1H, J = 6.3 Hz), 8.016, 8.021 (2s, 1H); 13C-NMR (CDCl3) δ -5.4, -5.3, -4.65, -4.63, 18.0, 18.46, 18.50, 18.53, 18.55, 18.57, 18.6, 18.7, 18.80, 18.83, 18.9, 19.0, 25.8, 26.1, 33.85, 33.94, 33.97, 42.6, 63.1, 72.3, 72.4, 78.6, 81.6, 84.3, 88.3, 120.6, 137.1, 148.1, 148.3, 153.7, 174.2, 174.4, 175.6. HRMS (ESI) m/z (M+H)+: calcd for C36H62N5O9Si2+ 764.4081; found, 764.4021.
2-N-Isobutyryl-1,N2-[1,2-di(isobutyryloxy)ethylene]-2´-deoxyguanosine (11). Compound 10 (1.0 g, 1.3 mmol) was dissolved in dry THF (6 mL). To the solution was added triethylamine (544 μL, 3.9 mmol) and Et3N·3HF (635 μL, 3.9 mmol) and the mixture was stirred at room temperature for 12 h. The mixture was partitioned between CHCl3-iPrOH (3:1, v/v) and aqueous NaHCO3. The organic phase was collected, dried over Na2SO4, filtered, and evaporated under reduced pressure. The residue was chromatographed on a column of silica gel with CHCl3-MeOH (100:0–95:5, v/v) to give the fractions containing 11. The fractions were collected and evaporated under reduced pressure to give 11 quantitatively. 1H-NMR (DMSO) δ1.05–1.11 (m, 12H), 1.17–1.22 (m, 6H), 2.36–2.40 (m, 1H), 2.59–2.64 (m, 3H), 3.52–3.57 (m, 2H), 3.87 (m, 1H), 3.96 (m, 1H), 4.36 (m, 1H), 4.95–4.96 (m, 1H), 5.36 (d, 1H, J = 4.2 Hz), 6.23–6.24 (m, 1H), 6.70 (s, 1H), 6.77 (s, 1H), 8.29, 8.30, 8.31 (3s, 1H); 13C-NMR (DMSO) δ 18.7, 18.8, 18.8, 19.1, 19.1, 19.4, 19.5, 33.7, 33.8, 40.8, 61.9, 62.0, 70.9, 71.0, 78.6, 79.7, 82.2, 84.6, 84.6, 88.4, 120.4, 120.5, 139.2, 139.4, 148.2, 148.3, 148.7, 153.8, 174.8, 175.0, 175.0, 176.0. HRMS (ESI) m/z (M+H)+: calcd for C24H34N5O9+ 536.2351; found, 536.2348.
2-N-Isobutyryl-1,N2-[1,2-di(isobutyryloxy)ethylene]-3´,5´-O-(4,4'-dimethoxytrityl)-2´-deoxy-guanosine (12). Compound 11 (696 mg, 1.3 mmol) was rendered anhydrous by repeated coevaporation with dry pyridine (1 mL × 1), and dissolved in dry pyridine (13 mL). To the solution was added DMTrCl (533 mg, 1.6 mmol), and the mixture was stirred at room temperature for 16 h. The reaction was quenched by addition of saturated aqueous NaHCO3. The mixture was partitioned between CHCl3 and H2O. The organic phase was collected, dried over Na2SO4, filtered, and evaporated under reduced pressure. The residue was chromatographed on a column of silica gel with CHCl3-MeOH (100:0–98:2, v/v) containing 1% Et3N to give the fractions containing 12. The fractions were collected and evaporated under reduced pressure. The residue was finally evaporated by repeated coevaporation three times each with toluene and CHCl3 to remove the last traces of Et3N to give 12 (657 mg, 60%). 1H-NMR (CDCl3) δ 1.14–1.19 (m, 12H), 1.26–1.31 (m, 6H), 2.55–2.59 (m, 4H), 3.80 (s, 6H), 3.92–4.03 (m, 3H), 4.74–4.75 (m, 1H), 6.28 (d, 1H, J = 5.4 Hz), 6.77, 6.78 (2s, 1H), 6.81–6.84 (m, 4H), 7.17 (d, 1H, J = 8.8 Hz), 7.22–7.32 (m, 9H), 8.60, 8.64 (2s, 1H); 13C -NMR (CDCl3) δ 18.6, 18.7, 18.9, 19.0, 19.1, 34.0, 34.1, 41.3, 41.6, 55.4, 64.2, 72.3, 78.4, 78.5, 81.8, 81.9, 84.5, 86.7, 86.8, 86.9, 113.4, 120.7, 120.8, 127.2, 128.1, 128.3, 130.2, 130.3, 130.3, 135.7, 135.8, 135.9, 137.7, 137.9, 144.7, 148.3, 148.4, 148.5, 153.9, 158.8, 174.4, 175.8, 175.9. HRMS (ESI) m/z (M+H)+: calcd for C45H52N5O11+ 838.3658; found, 838.3659.
2-N-Isobutyryl-1,N2-[1,2-di(isobutyryloxy)ethylene]-3´,5´-O-(4,4'-dimethoxytrityl)-2´-deoxy-guanosine 3'-(2-cyanoethyl N,N-diisopropylphosphramidite) (13). Compound 12 (210 mg, 0.25 mmol) was rendered anhydrous by repeated coevaporation with dry pyridine (1 mL × 1), dry toluene (1 mL × 1), dry CH3CN (1 mL × 1), and dissolved in dry CH2Cl2 (2.5 mL). To the solution was added bis(diisopropylamino)(2-cyanoethoxy)phosphine (89 μL, 0.28 mmol), 1H-tetrazole (11 mg, 0.15 mmol), diisopropylamine (21 μL, 0.15 mmol), and the mixture was stirred at room temperature for 14 h. The reaction was quenched by addition of saturated aqueous NaHCO3. The mixture was partitioned between CHCl3 and aqueous NaHCO3. The organic phase was collected, dried over Na2SO4, filtered, and evaporated under reduced pressure. The residue was chromatographed on a column of silica gel with CHCl3-MeOH (100:0–98:2, v/v) containing 1% Et3N to give the fractions containing 13. The fractions were collected and evaporated under reduced pressure. The residue was finally evaporated by repeated coevaporation three times each with toluene and CHCl3 to remove the last traces of Et3N to give 13 (239 mg, 93%). 1H-NMR (CDCl3) δ 1.11–1.29 (m, 30H), 2.42 (m, 1H), 2.53–2.62 (m, 5H), 3.32–3.35 (m, 2H), 3.57–3.76 (m, 8H), 3.81–3.99 (m, 2H), 4.29–4.32 (m, 1H), 4.64 (m, 1H), 6.28 (m, 1H), 6.79–6.84 (m, 6H), 7.18–7.20 (m, 1H), 7.24–7.30 (m, 6H), 7.38–7.40 (m, 2H), 7.85, 7.90 (2s, 1H); 13C-NMR (CDCl3) δ 18.6, 18.7, 18.9, 19.1, 19.2, 20.4, 20.6, 24.8, 24.9, 34.0, 40.7, 40.9, 41.2, 43.5, 43.6, 55.5, 58.3, 58.4, 63.6, 63.7, 74.2, 74.3, 74.6, 78.5, 78.6, 81.7, 81.8, 84.3, 84.4, 86.0, 86.4, 86.9, 113.5, 117.6, 117.7, 120.9, 121.0, 127.2, 128.2, 128.4, 130.2, 130.3, 135.6, 135.7, 135.8, 137.2, 137.3, 144.6, 148.4, 148.5, 153.8, 158.9, 174.3, 174.4, 175.7, 175.8; 31P-NMR (CDCl3) 150.0, 150.4, 150.5 (3s). HRMS (ESI) m/z (M+H)+: calcd for C54H69N7O12P+ 1038.4736; found, 1038.4064.
Evaluation of the resistance for addition of acrylonitrile to protected monomers. Fully protected monomers and common protected monomers were introduced into UnyLinker™ NittoPhase® by use of the ABI 392 DNA synthesizer. The protected monomer (125 nmol) loading UnyLinker™ NittoPhase® were treated with acrylonitrile (0.82 μL, 12.5 μmol) in 4 mM DBU-CH3CN (200 μL). After the reaction for appropriate time, the reaction solution was removed by filtration. The DMTr group was removed by treatment with 3% trichloroacetic acid in CH2Cl2 (1 mL) for 1 min, and the resin was washed with CH2Cl2 (1 mL × 3), and CH3CN (1 mL × 3). The monomer was deprotected and released from UnyLinker™ NittoPhase® by treatment with concentrated NH3 aq (500 µL) at 55 °C for 6 h. The polymer support was removed by filtration and washed with distilled water (1 mL × 3). The filtrate was evaporated and purified by reversed-phase HPLC.
6-N-Phenoxyacetyl-3´,5´-O-bis(tert-butyldimethylsilyl)-2´-deoxyadenosine (15). 3´,5´-O-Bis(tert-butyldimethylsilyl)-2´-deoxyadenosine (960 mg, 2.0 mmol) was rendered anhydrous by repeated coevaporation with dry pyridine (1 mL × 1) and dissolved in dry pyridine (10 mL). To the solution was added phenoxyacetic anhydride (1.37 mg, 4.8 mmol), and the mixture was stirred at room temperature for 3 h. To the reaction was added concentrated NH3 aq (5 mL), and the mixture was stirred at room temperature for 10 min. The mixture was partitioned between CHCl3 and aqueous NaHCO3. The organic phase was collected, dried over Na2SO4, filtered, and evaporated under reduced pressure. The residue was chromatographed on a column of silica gel with CHCl3-MeOH (100:0, v/v) to give the fractions containing 15. The fractions were collected and evaporated under reduced pressure to give 15 (831 mg, 68%). 1H NMR (CDCl3) δ 1H NMR (CDCl3) δ 0.10 (s, 12H), 0.91 (s, 18H), 2.45–2.49 (m, 1H), 2.65–2.70 (m, 1H), 3.78 (dd, 1H, J = 2.9 Hz, J = 9.8 Hz), 3.87 (dd, 1H, J = 4.2 Hz, J = 9.2 Hz), 4.037–4.043 (m, 1H), 4.62–4.63 (m, 1H), 4.87 (s, 2H), 6.50 (t, 1H, J = 6.3 Hz), 7.08 (d, 3H, J = 8.3 Hz), 7.35 (t, 2H, J = 7.9 Hz), 8.33 (s, 1H), 8.79 (s, 1H), 9.41 (s, 1H); 13C-NMR (CDCl3) δ -5.3, -5.2, -4.6, -4.5, 18.1, 18.5, 26.0, 26.1, 40.9, 62.8, 68.6, 72.1, 84.9, 88.1, 115.0, 117.6, 122.1, 123.0, 129.8, 142.5, 148.7, 151.5, 152.3, 157.5, 167.9. HRMS (ESI) m/z (M+H)+: calcd for C30H48N5O5Si2+ 614.3188; found, 614.3186.
General procedure for evaluation of cyanoethylation of compounds 14-17. An appropriate compound (0.30 mmol) was dissolved in DBU-CH3CN [3 mL, (1:9, v/v)]. To the solution was added acrylonitrile (1.96 mL, 30 mmol). After being stirred at room temperature for appropriate time, as shown in Table 1, the mixture was partitioned between CHCl3 and H2O. The organic phase was collected, dried over Na2SO4, filtered, and evaporated under reduced pressure. The residue was chromatographed on a column of silica gel with CHCl3-MeOH (100:0–99:1, v/v) to give the fractions containing the cyanoethylated compound. The fractions were collected and evaporated under reduced pressure to give the cyanoethylated compound. Thus, the following compounds 18–20 were obtained.
4-N-Acetyl-4-N-2-cyanoethyl-3´,5´-O-bis(tert-butyldimethylsilyl)-2´-deoxycytidine (18). The compound 18 was obtained (25.3 mg, 15%). 1H NMR (CDCl3) δ 0.11 (s, 12H), 0.90 (s, 18H), 2.13–2.18 (m, 1H), 2.47 (s, 3H), 2.52–2.57 (m, 1H), 2.84–2.93 (m, 2H), 3.78 (dd, 1H, J = 2.6 Hz, J = 10.8 Hz), 3.95–3.97 (m, 2H), 4.26 (t, 2H, J = 6.6 Hz), 4.38–4.41 (m, 1H), 6.61–6.23 (m, 1H), 6.88 (d, 1H, J = 7.3 Hz), 8.39 (d, 1H, J = 7.6 Hz); 13C-NMR (CDCl3) δ -5.4, -5.3, -4.8, -4.4, 17.5, 18.1, 18.5, 22.6, 25.8, 26.0, 42.3, 42.4, 61.8, 70.0, 87.1, 88.0, 99.3, 144.0, 154.9, 164.2, 172.1. HRMS (ESI) m/z (M+H)+: calcd for C26H47N4O5Si2+ 551.3080; found, 551.3079.
6-N-2-Cyanoethyl-6-N-phenoxyacetyl-3´,5´-O-bis(tert-butyldimethylsilyl)-2´-deoxyadenosine (19). The compound 19 was obtained (3.4 mg, 2%). 1H NMR (CDCl3) δ 0.12 (s, 12H), 0.92 (s, 18H), 2.46–2.51 (m, 1H), 2.57–2.62 (m, 1H), 2.93 (t, 2H, J = 7.3 Hz), 3.79 (dd, 1H, J = 2.7 Hz, J = 9.9 Hz), 3.89 (dd, 1H, J = 3.9 Hz, J = 9.3 Hz), 4.05–4.06 (m, 1H), 4.57–4.60 (m, 2H), 4.62–4.64 (m, 1H), 5.17 (d, 2H, J = 3.9 Hz), 6.52 (t, 1H, J = 6.2 Hz), 6.67 (d, 2H, J = 8.1 Hz), 6.93 (t, 1H, J = 7.3 Hz), 7.20 (t, 2H, J = 7.9 Hz), 8.42 (s, 1H), 8.70 (s, 1H); 13C-NMR (CDCl3) δ -5.3, -5.2, -4.6, -4.5, 17.3, 18.2, 18.6, 25.9, 26.1, 41.7, 43.6, 62.8, 68.9, 71.9, 84.9, 88.3, 114.6, 117.6, 121.7, 125.5, 129.6, 142.7, 151.4, 151.6, 152.7, 157.7, 170.8. HRMS (ESI) m/z (M+H)+: calcd for C33H51N6O5Si2+ 667.3454; found, 667.3454.
N3-(2-Cyanoethyl)-3´,5´-O-bis(tert-butyldimethylsilyl)thymidine (20). The compound 20 was obtained (127.3 mg, 83%). 1H NMR (CDCl3) δ 0.11 (s, 12H), 0.90 (s, 18H), 1.93 (s, 3H), 1.95–2.03 (m, 1H), 2.25–2.28 (m, 1H), 2.73–2.76 (m, 2H), 3.76 (d, 1H, J = 11.0 Hz), 3.86 (d, 1H, J = 11.5 Hz), 3.95 (m, 1H), 4.24–4.29 (m, 2H), 4.39 (m, 1H), 6.34 (t, 1H, J = 6.8 Hz), 7.50 (s, 1H); 13C-NMR (CDCl3) δ -5.3, -5.2, -4.7, -4.5, 13.3, 16.2, 18.1, 18.5, 25.9, 26.1, 36.8, 41.6, 63.1, 72.4, 85.8, 88.1, 110.2, 117.3, 134.3, 150.6, 163.1. HRMS (ESI) m/z (M+H)+: calcd for C25H46N3O5Si2+ 524.2971; found, 524.2913.
Synthesis of ODN using fully protected monomers: The synthesis of ODN d[ATCATCATCG] by use of an ABI 392 DNA synthesizer was carried out. After chain elongation was finished, the fully protected ODN (125 nmol) on UnyLinker™ NittoPhase® beads was treated with DBU-CH3CN [200 mL, (1:9, v/v)] for 1 min to remove the 2-cyanoethyl groups from the phosphate moieties. The ODN was deprotected and released from the resin by treatment with concentrated NH3 aq (500 µL) at 55 °C for 14 h. The polymer support was removed by filtration and washed with distilled water (1 mL × 3). The filtrate was evaporated and purified by anion-exchange HPLC. MALDI-TOF Mass (M+H)+: calcd for C97H125N35O58P9+ 2986.55; found 2987.91.
Evaluation of depurination using ODNs containing Aphth or GiBu·dibe. ODN d[TTTTXTTTT] (X = Aphth or GiBu·dibe) (0.5 µmol) on UnyLinker™ NittoPhase® beads was treated with 3% trichloroacetic acid in CH2Cl2 (1 mL) for 24 h. After the filtration of the reaction solution, the resin was washed with CH2Cl2 (1 mL × 3) and CH3CN (1 mL × 3). The ODN was deprotected and released from UnyLinker™ NittoPhase® by treatment with concentrated NH3 aq (500 µL) at 55 °C for 6 h. The polymer support was removed by filtration and washed with distilled water (1 mL × 3). The filtrate was evaporated and analyzed by reversed-phase HPLC. d[TTTTAphthTTTT]: MALDI-TOF Mass (M+H)+ calcd for C90H118N21O59P8+ 2684.48; found 2686.58. d[TTTTGiBu·dibeTTTT]: MALDI-TOF Mass (M+H)+ calcd for C90H118N21O60P8+ 2700.47; found 2702.94.
N3-(2-Cyanoethyl)-5´-O-(4, 4'-dimethoxytrityl)thymidine (22). Compound 21 (437 mg, 0.80 mmol) was rendered anhydrous by repeated coevaporation with dry pyridine (1 mL × 1), and dissolved in dry pyridine (8 mL). To the solution was added acetic anhydride (83 μL, 0.88 mmol), N,N-dimethtylaminopyridine (10 mg, 0.08 mmol), and the mixture was stirred at room temperature for 2 h. The reaction was quenched by addition of saturated aqueous NaHCO3. The mixture was partitioned between CHCl3 and aqueous NaHCO3. The organic phase was collected, dried over Na2SO4, filtered, and evaporated under reduced pressure. The residue was dissolved in DBU-CH3CN (8 mL, 1:9, v/v), and to the solution was added acrylonitrile (1.05 mL, 16.0 mmol). After being stirred at room temperature for 2 h, the mixture was partitioned between CHCl3 and H2O. The organic phase was collected, dried over Na2SO4, filtered, and evaporated under reduced pressure. To the residue was added 2.0 M ammonium in MeOH (10 mL), and the mixture was stirred at room temperature for 2 h. The mixture was evaporated under reduced pressure, chromatographed on a column of silica gel with CHCl3-MeOH (100:0–98:2, v/v) containing 1% Et3N to give the fractions containing 22. The fractions were collected and evaporated under reduced pressure. The residue was finally evaporated by repeated coevaporation three times each with toluene and CHCl3 to remove the last traces of Et3N to give 22 (289 mg, 60%). 1H NMR (CDCl3) δ 1.50 (s, 3H), 2.34–2.37 (m, 1H), 2.41–2.42 (m, 1H), 2.76 (t, 2H, J = 7.1 Hz), 3.39 (dd, 1H, J = 2.9 Hz, J = 10.3 Hz), 3.50 (dd, 1H, J = 3.2 Hz, J = 8.9 Hz), 3.80 (s, 6H), 4.05–4.06 (m, 1H), 4.26–4.30 (m, 2H), 4.59 (m, 1H), 6.42 (t, 1H, J = 6.7 Hz), 6.85 (d, 4H, J = 8.5 Hz), 7.25–7.32 (m, 7H), 7.40 (d, 2H, J = 7.3 Hz), 7.61 (s, 1H); 13C-NMR (CDCl3) δ 12.6, 16.2, 36.8, 41.2, 55.4, 63.5, 72.5, 85.5, 86.2, 87.2, 110.6, 113.5, 117.4, 127.3, 128.2, 128.3, 129.2, 130.2, 134.4, 135.41, 135.47, 144.4, 150.7, 158.9, 163.1. HRMS (ESI) m/z (M+Na)+: calcd for C34H35N3NaO7+ 620.2367; found, 620.2368.
N3-(2-Cyanoethyl)-5´-O-(4, 4'-dimethoxytrityl)thymidine 3'-(2-cyanoethyl N, N-diisopropylphosphramidite) (23). Compound 22 (239 mg, 0.40 mmol) was rendered anhydrous by repeated coevaporation with dry pyridine (1 mL × 1), dry toluene (1 mL × 1), dry CH3CN (1 mL × 1), and dissolved in dry CH2Cl2 (4 mL). To the solution was added bis(diisopropylamino) (2-cyanoethoxy)phosphine (123 μL, 0.44 mmol), 1H-tetrazole (17 mg, 0.24 mmol), diisopropylamine (34 μL, 0.24 mmol), and the mixture was stirred at room temperature for 12 h. The reaction was quenched by addition of saturated aqueous NaHCO3. The mixture was partitioned between CHCl3 and aqueous NaHCO3. The organic phase was collected, dried over Na2SO4, filtered, and evaporated under reduced pressure. The residue was chromatographed on a column of silica gel with hexane-ethyl acetate (70:30–50:50, v/v) containing 1% Et3N to give the fractions containing 23. The fractions were collected and evaporated under reduced pressure. The residue was finally evaporated by repeated coevaporation three times each with toluene and CHCl3 to remove the last traces of Et3N to give 23 (179 mg, 58%). 1H NMR (CDCl3) δ 1.07 (d, 3H, J = 6.8 Hz), 1.17–1.19 (m, 9H), 1.44 (s, 3H), 2.35–2.37 (m, 1H), 2.43 (t, 1H, J = 6.2 Hz), 2.49–2.58 (m, 1H), 2.63 (t, 1H, J = 6.2 Hz), 2.76 (t, 2H, J = 7.1 Hz), 3.32–3.35 (m, 1H), 3.48–3.68 (m, 5H), 3.80, 3.81 (2s, 6H), 4.15–4.19 (m, 1H), 4.26–4.28 (m, 2H), 4.66–4.68 (m, 1H), 6.40–6.44 (m, 1H), 6.83–6.86 (m, 4H), 7.25–7.31 (m, 7H), 7.40-7.42 (m, 2H), 7.65, 7.69 (2s, 1H); 13C-NMR (CDCl3) δ 12.5, 16.2, 20.3, 20.4, 20.55, 20.61, 24.60, 24.66, 24.69, 24.7, 24.8, 36.8, 43.35, 43.43, 43.5, 55.41, 55.43, 58.2, 58.4, 63.1, 63.3, 73.5, 73.6, 73.8, 74.0, 85.6, 85.7, 85.9, 87.1, 110.6, 113.4, 117.4, 117.5, 117.7, 127.32, 127.35, 128.1, 128.3, 128.4, 130.29, 130.31, 130.3, 134.47, 134.51, 135.46, 135.51, 144.4, 150.67, 150.70, 158.9, 163.1; 31P NMR (CDCl3) 149.7, 150.1 (2s). HRMS (ESI) m/z (M+Na)+: calcd for C42H50N5NaO8P+ 820.3446; found, 820.3450.
Enzymes and ODNs. AmpliTaq Gold DNA polymerase was purchased from applied biosystemsTM, and dNTPs were purchased from Takara Bio, Inc. ODNs used in enzyme reactions and Tm experiments were purchased from Sigma-Aldrich Japan. ODNs 1 and 2 in Table 2 were synthesized by use of an ABI 392 DNA synthesizer.
Tm experiments. An appropriate ODN (2 μM) and its complementary 2 μM ssDNA 11-mer were dissolved in a buffer consisting of 1 M NaCl, 10 mM sodium phosphate, and 0.1 mM EDTA adjusted to pH 7.0. The solution was maintained at 80 °C for 10 min for complete dissociation of the duplex to single strands and cooled at the rate of UV-1700TM (Shimadzu) by increasing the temperature at the rate of 0.5 °C/min. During this process of annealing and melting, the absorption at 260 nm was recorded and used to draw UV melting curves. The Tm value was calculated as the temperature at which the first derivative of the UV melting curve had a maximum.
Single dNTP Insertion Reaction Using Taq DNA Polymerase. A mixture containing PCR Gold buffer, 1.8 mM MgCl2, and 0.25 unit AmpliTaq Gold DNA polymerase was incubated at 95 °C for 3 min and slowly cooled to room temperature. To the mixture was added 100 nM (final concentration) 5′-FAM-labeled primer/template, and 10 μM (final concentration) dNTP (N = A, C, G, or T). The mixture (10 μL) was incubated at 74 °C for 10 min, and the reactions were terminated by adding 30 μL of stop solution (95% formamide, 20 mM EDTA). After being gently vortexed, the samples were separated by electrophoresis using 20% denaturing polyacrylamide gel containing 7 M urea and visualized by Fujifilm FLA-7000.
4. Conclusions
In this study, we have synthesized the fully protected monomers (TBz, dCphth, dAphth, and dGiBu·dibe), and an ODN using these monomers. The TBz monomer can be obtained efficiently from T in three steps. The dCphth and dAphth monomers were obtained from the corresponding N-unprotected phosphoroamidite units by facile acylation. The full protection of dG was achieved and used in an addition reaction with glyoxal at the N1- and 2-N-amino groups. To examine the stability of the fully protected monomers toward addition of acrylonitirile, we carried out the reaction of these monomers with excess acrylonitirile in DBU/CH3CN. As a result, TBz suppressed the addition of acrylonitrile. Full deprotection of the protecting groups used in the oligonucleotide synthesis revealed the presence of side reactions associated with dGiBu·dibe. Further improvement was necessary to avoid such side reactions. In contrast, it was found that an ODN having TCE formed an unstable DNA duplex in comparison with the unmodified DNA duplex. In addition, TCE on the template exhibited complete inhibitory effect on chain elongation of a primer in DNA polymerase reactions.
Acknowledgements
This work was supported by Grant-in-Aids for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan and in part by Health Sciences Research Grants for Research on Psychiatric and Neurological Diseases and Mental Health from the Ministry of Health, Labor and Welfare of Japan, and by the global COE project.
Footnotes
Sample Availability: Samples of the compounds are available from the authors.
References
- 1.Ozsolak F., Platt A.R., Jones D.R., Reifenberger J.G. Sass, L.E., McInerney P., Thompson J.F., Bowers J., Jarosz M., Milos P.M. Direct RNA sequencing. Nature. 2009;461:814–819. doi: 10.1038/nature08390. [DOI] [PubMed] [Google Scholar]
- 2.Kurreck J. RNA interference: From basic research to therapeutic applications. Angew. Chem. Int. Ed. 2009;48:1378–1398. doi: 10.1002/anie.200802092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosi N.L., Giljohann D.A., Thaxton C.S., Lytton-Jean A.K.L., Han M.S., Mirkin C.A. Oligonucleotide-modified gold nanoparticles for intracellular gene reguration. Science. 2006;312:1027–1030. doi: 10.1126/science.1125559. [DOI] [PubMed] [Google Scholar]
- 4.Gibson D.G., Glass J.I., Lartigue C., Noskov V.N., Chuang R.Y., Algire M.A., Benders G.A., Montague M.G., Ma L., Moodie M.M., Merryman C., Vashee S., Krishnakumar R., Assad-Garcia N., Andrews-Pfannkoch C., Denisova E.A., Young L., Qi Z.Q., Segall-Shapiro T.H., Calvey C.H., Parmar P.P., Clyde A. Hutchison C.A., III, Smith H.O., Venter J.C. Creation of a Bacterial Cell Controlled by a Chemically Synthesized Genome. Science. 2010;329:52–56. doi: 10.1126/science.1190719. [DOI] [PubMed] [Google Scholar]
- 5.Gibson D.G., Benders G.A., Andrews-Pfannkoch C., Denisova E.A., Baden-Tillson H., Zaveri J., Stockwell T.B., Brownley A., Thomas D.W., Algire M.A., Merryman C., Young L., Noskov V.N., Glass J.I., Venter J.C., Hutchison C.A., III, Smith H.O. Complete Chemical Synthesis, Assembly, and Cloning of a Mycoplasma genitalium Genome. Science. 2008;319:1215–1220. doi: 10.1126/science.1151721. [DOI] [PubMed] [Google Scholar]
- 6.Beaucage S.L., Iyer R.P. Advances in the synthesis of oligonucleotides by the phosphoramidite approach. Tetrahedron. 1992;48:2223–2311. doi: 10.1016/S0040-4020(01)88752-4. [DOI] [Google Scholar]
- 7.McBride L.J., Iyer R.P. An investigation of several deoxynucleoside phosphoramidites useful for synthesizing deoxyoligonucleotides. Tetrahedron Lett. 1983;24:245–248. doi: 10.1016/S0040-4039(00)81376-3. [DOI] [Google Scholar]
- 8.Matteucci M.D., Iyer R.P. Synthesis of deoxyoligonucleotides on a polymer support. J. Am. Chem. Soc. 1981;103:3185–3191. doi: 10.1021/ja00401a041. [DOI] [Google Scholar]
- 9.Beaucage S.L., Caruthers M.H. Deoxynucleoside phosphoramidites—A new class of key intermediates for deoxypolynucleotide synthesis. Tetrahedron Lett. 1981;22:1859–1862. doi: 10.1016/S0040-4039(01)90461-7. [DOI] [Google Scholar]
- 10.Sinha N.D., Biernat J., McManus J., Köster H. Polymer support oligonucleotide synthesis XVIII1.2): use of β-cyanoethyl-N,N-dialkylamino-/N-morpholino phosphoramidite of deoxynucleosides for the synthesis of DNA fragments simplifying deprotection and isolation of the final product. Nucleic Acids Res. 1984;12:4539–4557. doi: 10.1093/nar/12.11.4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sinha N.D., Biernat J., Köster H. β-Cyanoethyl N,N-dialkylamino/N-morpholinomonochloro phosphoamidites, new phosphitylating agents facilitating ease of deprotection and work-up of synthesized oligonucleotides. Tetrahedron Lett. 1983;24:5843–5846. doi: 10.1016/S0040-4039(00)94216-3. [DOI] [Google Scholar]
- 12.Boal J.H., Wilk A., Harindranath N., Max E.E., Kempe T., Beaucage S.L. Cleavage of oligodeoxyribonucleotides from controlled-pore glass supports and their rapid deprotection by gaseous amines. Nucleic Acids Res. 1996;24:3115–3117. doi: 10.1093/nar/24.15.3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tener G.M. 2-Cyanoethyl Phosphate and its Use in the Synthesis of Phosphate Esters. J. Am. Chem. Soc. 1961;83:159–168. doi: 10.1021/ja01462a032. [DOI] [Google Scholar]
- 14.Solomon J.J., Cote I.L., Wortman M., Decker K., Segal A. In vitro alkylation of calf thymus DNA by acrylonitrile. Isolation of cyanoethyl-adducts of guanine and thymine and carboxyethyl-adducts of adenine and cytosinestar, open. Chem. Biol. Interact. 1984;51:167–190. doi: 10.1016/0009-2797(84)90028-0. [DOI] [PubMed] [Google Scholar]
- 15.Prokopczyk B., Bertinato P., Hoffmann D. Synthesis and kinetics of decomposition of 7-(2-cyanoethyl)guanine and O6-(2-cyanoethy)guanine, markers for reaction of acrylonitrile and 3-(methylnitrosamino)propionitrile with DNA. Carcinogenesis. 1988;9:2125–2128. doi: 10.1093/carcin/9.11.2125. [DOI] [PubMed] [Google Scholar]
- 16.Mag M, Engels J.W. Synthesis and structure assignments of amide protected nucleosides and their use as phosphoramidites in deoxyoligonucleotide synthesis. Nucleic Acids Res. 1988;16:3525–3543. doi: 10.1093/nar/16.8.3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ogilvie K.K., Beaucage S.L. Fluoride ion promoted deprotection and transesterification in nucleotide triesters. Nucleic Acids Res. 1979;7:805–823. doi: 10.1093/nar/7.3.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chambers R.W. The chemistry of pseudouridine. IV. Cyanoethylation. Biochemistry. 1965;4:219–226. doi: 10.1021/bi00878a007. [DOI] [Google Scholar]
- 19.Solomon J.J., Segal A. Direct alkylation of calf thymus DNA by acrylonitrile. Isolation of cyanoethyl adducts of guanine and thymine and carboxyethyl adducts of adenine and cytosine. Environ. Health Perspect. 1985;62:227–230. doi: 10.1289/ehp.8562227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Capaldi D.C., Gaus H., Krotz A.H., Arnold J., Carty R.L., Moore M.N., Scozzari A.N., Lowery K., Cole D.L., Ravikumar V.T. Synthesis of high quality antisense drug. Addition of acrylonitrile to phosphorothioate oligonucleotides: Adduct characterization and avoidance. Org. Process Res. Dev. 2003;7:832–838. doi: 10.1021/op020090n. [DOI] [Google Scholar]
- 21.Wilk A., Grajkowski A., Philips L.R., Beaucage S.L. The 4-[N-(2,2,2-trifluoroacetyl)amino]butyl group as an alternative to the 2-cyanoethyl group for phosphate protection in the synthesis of oligodeoxyribonucleotides. J. Org. Chem. 1999;64:7515–7522. doi: 10.1021/jo990835w. [DOI] [Google Scholar]
- 22.Umemoto T., Wada T. Nitromethane as a scavenger of acrylonitrile in the deprotection of synthetic oligonucleotides. Tetrahedron Lett. 2005;46:4251–4253. doi: 10.1016/j.tetlet.2005.04.066. [DOI] [Google Scholar]
- 23.Chang C.A., Horn T. An improved deprotection procedure of amine-containing oligonucleotides from acrylonitrile modification. Nucleos. Nucleot. Nucleic Acids. 1999;18:1205–1206. doi: 10.1080/07328319908044663. [DOI] [Google Scholar]
- 24.de Koning M.C., Ghisaidoobe A.B.T., Duynstee H.I., Ten Kortenaar P.B.W., Filippov D.V., van der Marel G.A. Simple and efficient solution-phase synthesis of oligonucleotides using extractive work-up. Org. Process Res. Dev. 2006;10:1238–1245. [Google Scholar]
- 25.Sekine M., Fujii M., Nagai H., Hata T. An improved method for the synthesis of N3-benzoylthymidine. Synthesis. 1987;12:1119–1121. [Google Scholar]
- 26.Tsunoda H., Ohkubo A., Taguchi H., Seio K., Sekine M. Synthesis and properties of DNA oligomers containing 2´-deoxynucleoside N-oxide derivatives. J. Org. Chem. 2008;73:1217–1224. doi: 10.1021/jo7021845. [DOI] [PubMed] [Google Scholar]
- 27.Beier M., Pfleiderer W. Nucleotides, Part LXI, Phthaloyl strategy: A new concept of oligonucleotide syntheis. Helv. Chim. Acta. 1999;82:633–644. doi: 10.1002/(SICI)1522-2675(19990407)82:4<633::AID-HLCA633>3.0.CO;2-U. [DOI] [Google Scholar]
- 28.Kume A., Iwase R., Sekine M., Hata T. Cylic diacyl groups for protection of the N6-amino group of deoxyadenosine in oligodeoxynucleotide synthesis. Synthesis. 1984;12:8525–8538. doi: 10.1093/nar/12.22.8525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kume A., Sekine M., Hata T. Further improvements of oligodeoxyribonucleotides synthesis: synthesis of tetradeoxyadenylate on a new silica gel support using N6-phthaloyldeoxyadenosine. Chem. Lett. 1983;12:1597–1600. [Google Scholar]
- 30.Kume A., Sekine M., Hata T. Phthaloyl group: A new amino protecting group of deoxyadenosine in oligonucleotide synthesis. Tetrahedron Lett. 1982;23:4365–4368. doi: 10.1016/S0040-4039(00)85601-4. [DOI] [Google Scholar]
- 31.Ohkubo A., Sakamoto K., Miyata K., Taguchi H., Seio K., Sekine M. Convenient synthesis of N-unprotected deoxynucleoside 3'-phosphoramidite building blocks by selective deacylation of N-acylated species and their facile conversion to other N-functionalized derivatives. Org. Lett. 2005;7:5389–5392. doi: 10.1021/ol051949z. [DOI] [PubMed] [Google Scholar]
- 32.Matsuzaki J., Kohno K., Tahara S., Sekine M., Hata T. Solid phase synthesis of oligodeoxyribonucleotides utilizing the phenylthio group as a phosphate protecting group. Bull. Chem. Soc. Jpn. 1987;60:1407–1413. doi: 10.1246/bcsj.60.1407. [DOI] [Google Scholar]
- 33.Matsuzaki J., Hotoda H., Sekine M., Hata T., Higuchi S., Nishimura Y., Tsuboi M. Self-complementary tetradeoxyribonucleoside triphosphates convenient chemical preparation and spectroscopic studies in solution. Tetrahedron. 1986;42:501–513. doi: 10.1016/S0040-4020(01)87448-2. [DOI] [Google Scholar]
- 34.Sekine M., Matsuzaki J., Hata T. A new type of protection mode for the guanine residue by using 1,2-diisobutyryloxyethylene group. Tetrahedron Lett. 1982;23:5287–5290. doi: 10.1016/S0040-4039(00)85820-7. [DOI] [Google Scholar]
- 35.Shimizu M., Saigo K., Wada T. Solid-phase synthesis of oligodeoxyribonucleoside boranophosphates by the boranophosphotriester method. J. Org. Chem. 2006;71:4262–4269. doi: 10.1021/jo0603779. [DOI] [PubMed] [Google Scholar]
- 36.Adamiak R.W., Barciszewska M.Z., Biala E., Grzéskowiak K., Kierzek R., Kraszewski A., Markiewicz W.T., Wiewiórowski M. Nucleoside-3'-phosphotriesters as key intermediates for the oligoribonucleotide synthesis. III. An improved preparation of nucleoside 3'-phosphotriesters, their 1H NMR characterization and new conditions for removal of 2-cyanoethyl group. Nucleic Acids Res. 1976;3:3397–3408. doi: 10.1093/nar/3.12.3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peng C.G., Damha M.J. Polymerase-directed synthesis of 2´-deoxy-2´-fluoro-β-D-arabinonucleic acids. J. Am. Chem. Soc. 2009;131:3958–3964. doi: 10.1021/ja8065896. [DOI] [PubMed] [Google Scholar]
- 38.Kutyavin I.V. Use of base-modified duplex-stabilizing deoxynucleoside 5´-triphosphates to enhance the hybridization properties of primers and probes in detection polymerase chain reaction. Biochemistry. 2008;47:13666–13673. doi: 10.1021/bi8017784. [DOI] [PubMed] [Google Scholar]
- 39.El-Sagheer A.H., Brown T. Synthesis and polymerase chain reaction amplification of DNA strands containing an unnatural triazole linkage. J. Am. Chem. Soc. 2007;129:5310–5311. doi: 10.1021/ja069100g. [DOI] [PubMed] [Google Scholar]
- 40.Gaffney B.L, Marky L.A., Jones R.A. The influence of the purine 2-amino group on DNA conformation and stability-II. Synthesis and physical characterization of d[CGT(2-NH2)ACG], d[CGU(2-NH2)ACG], and d[CGT(2-NH2)AT(2-NH2)ACG] Tetrahedron. 1984;40:3–13. doi: 10.1016/0040-4020(84)85098-X. [DOI] [Google Scholar]
- 41.Seio K., Negishi T., Negishi K., Sekine M. Mild and facile deprotection for the synthesis of oligodeoxynucleotide incorporating a 6-O-ethyldeoxyguanosine. Lett. Org. Chem. 2005;2:179–183. doi: 10.2174/1570178053202982. [DOI] [Google Scholar]