

Abstract
Psammaplin F, an unsymmetrical disulfide bromotyrosine, was isolated from the sponge Pseudoceratina purpurea in 2003. We reported here the first total synthesis of psammaplin F in 12% overall yield by employing Cleland’s reagent reduction as key step. The longest linear synthetic sequence starting from 3-bromo-4-hydroxybenzaldehyde and hydantoin was seven steps. In addition, a detailed X-ray crystal structure analysis of psammaplin A analogue 8b is given for the first time.
Keywords: psammaplin F, marine bromotyrosine, Cleland’s reagent, total synthesis, X-ray crystal structure
1. Introduction
The psammaplin disulfide bromotyrosine derivatives exhibit wide-ranging biological activities including anticancer activity [1,2], anti methicillin-resistant Staphylococcus aureus (MRSA) activity [3], antifungal activity [4] and antiangiogenic activity [5]. As a result, these natural products have become interesting targets for synthesis on account of their activities, but only a few successful synthetic routes have been reported [3,6,7] over the years. Psammaplin F (1) which has been isolated from the sponge Pseudoceratina purpurea, containing an oximic amide unit and an oxalamic amide moiety rarely found in marine organisms, is a potent histone deacetylase (HDAC) inhibitor (IC50 = 2.1 ± 0.4 nM) [1]. In addition, the combination of a medium chain fatty acid or salt having a carbon chain length of 6 to 20 carbon atoms and psammaplin F (1) in oral dosage form can promote absorption of psammaplin F (1) in the gastrointestinal tract cell lining [8]. Based on this consideration, and as a continuation of our chemical and biological investigations of marine bromotyrosine natural products, our interest was focused on the synthesis of psammaplin F (1), and in this paper the first total synthesis of compound 1 is presented.
2. Results and Discussion
2.1. Chemistry
Our retrosynthetic analysis of psammaplin F (1) is shown in Figure 1. It was envisioned that the natural product could be synthesized by concise unsymmetrical disulfide coupling [13] between the left hand oximic amide 2 and the right side oxalamic amide 3, followed by the hydrolysis of the ester.
Figure 1.

Retrosynthetic analysis of psammaplin F (1).
The synthetic route to oximic amide 2 is seen in Scheme 1. The synthesis started from commercially available 3-bromo-4-hydroxybenzaldehyde (4a) and hydantoin, whose Claisen-Schmidt reaction gave the corresponding benzalhydantoin 5a.
Scheme 1.

Synthetic route to the target compound 2.
After hydrolysis and oximation of key intermediate 5a, the resulting oximic acid 7a was coupled with cystamine dihydrochloride (9) in a one-pot reaction to give psammaplin A (8a) in 37% yield for the four steps.
The co-crystallization of psammaplin A (8a) with chitinase B could not fully confirm the skeletal configuration of psammaplin A due to its flexibility [4], therefore, psammaplin A analogues with different substituents in the phenyl ring were also synthesized by this procedure and single crystals of these compounds were grown in order to confirm the configuration of the skeleton. As a result, starting from 4-methylbenzaldehyde (4b) and hydantoin, psammaplin A analogue 8b was obtained in 44% yield for the four steps, a crystal of compound 8b was grown and the structure confirmed by X-ray crystal structure analysis, which showed that the oxime of these compounds has the (E)-configuration. In a word, the structure of synthetic psammaplin A (8a) was in accordance with natural psammaplin A reported by Piña et al. Psammaplin A (8a) was then reduced by Cleland’s reagent to afford oximic amide (2) in quantitative yield [14].
The synthesis of oxalamic amide 3 is outlined in Scheme 2. Cystamine dihydrochloride (9) was converted to bisamide 10 according to a Schotten-Baumann-like reaction followed by reduction to yield oxalamic acetate 11, which was reacted with aldrithiol (13) obtained from the oxidation of 2-pyridinethiol (12) to provide the desired oxalamic amide 3 [15].
Scheme 2.

Synthetic route to the target compound 3.
The convergent synthesis between oximic amide 2 and oxalamic amide 3 which is shown in Scheme 3 was achieved based on very simple and mild conditions. The obtained unsymmetrical disulfide 14 was then hydrolyzed by potassium hydroxide in THF-water mixture to give psammaplin F (1) [16]. By comparison of 1H-NMR and 13C-NMR data of synthetic and natural compounds as displayed in Table 1, we were able to prove that the synthetically material was identical to natural psammaplin F (1).
Scheme 3.

Synthetic route to psammaplin F (1).
Table 1.
Comparison of 1H-NMR (600 MHz) and 13C-NMR (150 MHz) data in CD3OD of synthetic and natural psammalin F (1).
| Position | Synthetic psammaplin F | Natural psammaplin F[1] | ||
|---|---|---|---|---|
| H (δ) | C (δ, mult, J in Hz) | H (δ) | C (δ, mult, J in Hz) | |
| 2 | 2.85 (t, 6.0) | 38.7 | 2.84 (t, 6.0) | 38.6 |
| 2’ | 2.85 (t, 6.0) | 38.2 | 2.84 (t, 6.0) | 37.9 |
| 3 | 3.55 (t, 6.0) | 39.8 | 3.54 (t, 6.0) | 39.8 |
| 3’ | 3.55 (t, 6.0) | 40.0 | 3.54 (t, 6.0) | 40.1 |
| 5 | 166.0 | 166.0 | ||
| 5’ | 161.8 | 161.9 | ||
| 6 | 153.2 | 153.3 | ||
| 6’ | 159.0 | 159.2 | ||
| 7 | 3.80 (s) | 28.7 | 3.78 (s) | 28.8 |
| 8 | 130.6 | 130.7 | ||
| 9 | 7.34 (d, 1.8) | 134.5 | 7.35 (d, 2.0) | 134.6 |
| 10 | 110.5 | 110.6 | ||
| 11 | 153.7 | 153.9 | ||
| 12 | 6.77(d, 8.4) | 117.0 | 6.75 (d, 8.0) | 117.2 |
| 13 | 7.07 (dd, 1.8, 8.4) | 130.4 | 7.05 (dd, 2.0, 8.0) | 130.5 |
2.2. Crystal structure analysis of compound 8b
The molecular structure of compound 8b is shown in Figure 2. The detailed crystallographic data, selected bond lengths and angles for compound 8b are listed in Table 2 and Table 3, respectively.
Figure 2.

The molecular structure of the compound 8b with 30% probability of thermal ellipsoids.
Table 2.
Crystal data and structure refinement for compound 8b.
| Formula | C24 H30 N4 O4 S2 |
| Fw | 502.64 |
| Temp (K) | 295(2) |
| Cryst system | Triclinic |
| Space group | P-1 |
| a (Å) | 5.7159(9) |
| b (Å) | 9.6170(15) |
| c (Å) | 23.585(4) |
| α (°) | 94.030(2) |
| β (°) | 95.455(2) |
| γ (°) | 102.532(2) |
| V (Å3) | 1254.3(3) |
| Z | 2 |
| ρcalc (g cm-3) | 1.331 |
| Μ (mm-1) | 0.250 |
| F(000) | 532 |
| Limiting indices | -6<=h<=6, -11<=k<=5, -28<=l<=27 |
| Data / restraints / parameters | 4589 / 0 / 311 |
| θ range for data collection (°) | 1.74 to 25.66 |
| Completeness to theta = 25.66 | 97.2 % |
| Reflnscollected/unique | 6718 / 4589 |
| GOF | 1.049 |
| Final R indices [I>2σ(I)] | R1a = 0.0484, ωR2b = 0.1271 |
| R indices (all data) | R1 = 0.0595, ωR2 = 0.1364 |
Table 3.
Selected bond lengths (Å) and angles (°) for compound 8b.
| C(1)-C(2) | 1.516(4) | C(14)-N(4) | 1.446(3) |
| C(2)-C(3) | 1.361(4) | C(15)-O(4) | 1.222(3) |
| C(2)-C(7) | 1.380(4) | C(15)-N(4) | 1.337(3) |
| C(3)-C(4) | 1.393(4) | C(15)-C(16) | 1.500(3) |
| C(4)-C(5) | 1.375(3) | C(16)-N(3) | 1.271(3) |
| C(5)-C(6) | 1.379(3) | C(16)-C(17) | 1.501(3) |
| C(5)-C(8) | 1.518(3) | C(17)-C(18) | 1.510(3) |
| C(6)-C(7) | 1.382(4) | C(18)-C(23) | 1.372(3) |
| C(8)-C(9) | 1.507(3) | C(18)-C(19) | 1.376(4) |
| C(9)-N(1) | 1.274(3) | C(19)-C(20) | 1.387(4) |
| C(9)-C(10) | 1.502(3) | C(20)-C(21) | 1.370(5) |
| C(10)-O(2) | 1.223(3) | C(21)-C(22) | 1.360(5) |
| C(10)-N(2) | 1.327(3) | C(21)-C(24) | 1.518(4) |
| C(11)-N(2) | 1.445(3) | C(22)-C(23) | 1.395(4) |
| C(11)-C(12) | 1.509(3) | N(1)-O(1) | 1.384(2) |
| C(12)-S(1) | 1.817(2) | N(3)-O(3) | 1.388(2) |
| C(13)-C(14) | 1.518(3) | S(1)-S(2) | 2.0376(8) |
| C(13)-S(2) | 1.830(2) | ||
| C(3)-C(2)-C(7) | 117.0(3) | O(4)-C(15)-C(16) | 120.5(2) |
| C(3)-C(2)-C(1) | 121.2(3) | N(4)-C(15)-C(16) | 117.6(2) |
| C(7)-C(2)-C(1) | 121.9(3) | N(3)-C(16)-C(15) | 114.33(19) |
| C(2)-C(3)-C(4) | 121.9(3) | N(3)-C(16)-C(17) | 127.5(2) |
| C(5)-C(4)-C(3) | 120.7(3) | C(15)-C(16)-C(17) | 118.11(19) |
| C(4)-C(5)-C(6) | 117.8(2) | C(16)-C(17)-C(18) | 112.58(18) |
| C(4)-C(5)-C(8) | 120.6(2) | C(23)-C(18)-C(19) | 117.3(2) |
| C(6)-C(5)-C(8) | 121.6(2) | C(23)-C(18)-C(17) | 122.0(2) |
| C(5)-C(6)-C(7) | 120.6(3) | C(19)-C(18)-C(17) | 120.7(2) |
| C(2)-C(7)-C(6) | 122.0(3) | C(18)-C(19)-C(20) | 121.2(3) |
| C(9)-C(8)-C(5) | 112.76(18) | C(21)-C(20)-C(19) | 121.4(3) |
| N(1)-C(9)-C(10) | 113.78(18) | C(22)-C(21)-C(20) | 117.5(3) |
| N(1)-C(9)-C(8) | 126.7(2) | C(22)-C(21)-C(24) | 120.8(3) |
| C(10)-C(9)-C(8) | 119.52(18) | C(20)-C(21)-C(24) | 121.7(4) |
| O(2)-C(10)-N(2) | 122.1(2) | C(21)-C(22)-C(23) | 121.6(3) |
| O(2)-C(10)-C(9) | 121.86(19) | C(18)-C(23)-C(22) | 121.0(3) |
| N(2)-C(10)-C(9) | 116.09(18) | C(9)-N(1)-O(1) | 113.18(18) |
| N(2)-C(11)-C(12) | 110.54(18) | C(10)-N(2)-C(11) | 124.39(19) |
| C(11)-C(12)-S(1) | 114.69(15) | C(16)-N(3)-O(3) | 112.38(18) |
| C(14)-C(13)-S(2) | 110.52(16) | C(15)-N(4)-C(14) | 122.4(2) |
| N(4)-C(14)-C(13) | 113.57(19) | C(12)-S(1)-S(2) | 105.05(8) |
| O(4)-C(15)-N(4) | 121.9(2) | C(13)-S(2)-S(1) | 104.27(8) |
The single crystal X-ray diffraction analysis of compound 8b reveals that it crystallizes in the triclinic crystal P-1 space group and the 2,2'-disulfandiyldiethanamino chain links two 3-(4-methylphenyl)-2-oximidopropionyl groups via the amide group C-N bond. In this structure, all bond lengths and bond angles are comparable to those previously reported [17,18]. The bond angles 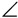 C(9)-C(8)-C(5) and C(16)-C(17)-C(18) are 112.76(18) and 112.58(18)°, respectively. This suggests that the dihedral angles between the amide and oxime group plane and phenyl group are 75.02 and 71.79°, respectively. The two benzene rings are close to parallel, with a dihedral angle of 3.92°. The torsion angles of C(12)-S(1)-S(2)-C(13) and C(11)-C(12)-S(1)-S(2) are -88.50(11) and 77.45(16)°, respectively, which leads to the conclusion that the 2,2'-disulfandiyldiethanamino chain is seriously twisted. It is interesting that the overall shape of the compound 8b takes on zigzag-like chain.
C(9)-C(8)-C(5) and C(16)-C(17)-C(18) are 112.76(18) and 112.58(18)°, respectively. This suggests that the dihedral angles between the amide and oxime group plane and phenyl group are 75.02 and 71.79°, respectively. The two benzene rings are close to parallel, with a dihedral angle of 3.92°. The torsion angles of C(12)-S(1)-S(2)-C(13) and C(11)-C(12)-S(1)-S(2) are -88.50(11) and 77.45(16)°, respectively, which leads to the conclusion that the 2,2'-disulfandiyldiethanamino chain is seriously twisted. It is interesting that the overall shape of the compound 8b takes on zigzag-like chain.
Furthermore, intermolecular hydrogen bond interactions [O-H…O] are formed by hydrogen atoms attached to oxygen atoms in the oxime groups and the oxygen atoms associated with carboxyl groups of a neighboring molecule, leading to the formation of ladder-link chains which run along the crystallographic an axis. In addition, intermolecular hydrogen bond interactions [Csp2-H…O] between the benzene ring hydrogen atom and the oxygen atom of the oxime group are further favorable to stabilize the ladder-link chains, as depicted in Figure 3. All intermolecular hydrogen bond interaction information is shown in Table 4.
Figure 3.

The 1D chain structure of compound 8b constructed via intermolecular hydrogen bond interactions (green dash line).
Table 4.
Intermolecular hydrogen bond lengths (Å) and bond angles (°).
| D-H...A | d(D-H) | d(H-A) | d(D-A) |
D-H...A |
|---|---|---|---|---|
| O(1)-H(1)...O(4)#1 | 0.82 | 1.87 | 2.671(2) | 167.0 |
| O(3)-H(3A)...O(2)#2 | 0.82 | 1.90 | 2.711(2) | 171.5 |
| C(4)-H(4)...O(3)#2 | 0.93 | 2.53 | 3.229(3) | 133 |
Symmetry transformations used to generate equivalent atoms: #1 -x+1, -y+1, -z+1; #2 -x+3, -y+2, -z+1.
3. Experimental
3.1. General
All melting points (mp) were determined on an electrically heated X4 digital visual melting point apparatus and were uncorrected. IR spectra were recorded on a Bruker IR-27G spectrometer using KBr pellets. NMR spectra were recorded on a Bruker ARX 300 or AV 600 MHz spectrometer at room temperature in DMSO-d6 or CD3OD with tetramethylsilane as an internal standard. Chemical shifts were reported in ppm (δ). Mass spectra (MS) were determined on Finnigan MAT/USA spectrometer (LC-MS). High-resolution mass spectra were obtained on Bruker micrOTOF-Q in ESI mode (HR-ESI-MS). Column chromatography was performed with silica gel 60 (200-300 mesh). TLC board (Alugram silica gel G/UV254) was purchased from Macherey-Nagel GmbH & Co. All evaporations were carried out under reduced pressure. All reagents and solvents are commercially available and were used as received.
General procedure for the synthesis of (E,E)-N,N’-bis(3-(substituted phenyl)-2-oximidopropionyl)cyst- amines 8. To a solution of oximic acid 7 (3.66 mmol) in THF (25 mL) was added HOBt (3.66 mmol) at room temperature. After stirring for 40 min, a mixture of cystamine dihydrochloride (9) (1.83 mmol), DMAP (1.10 mmol) and TEA (1 mL) in CH3OH (6 mL) were added dropwise at 0 °C, followed by slow dropwise addition of a solution of DCC (4.03 mmol) in THF (25 mL). The resulting mixture was stirred for 1 h and the temperature was then allowed to warm to room temperature overnight. After the reaction was over, the reaction mixture was left standing in a refrigerator overnight and then filtered, the obtained filtrate was washed with brine, dried over MgSO4, filtered and concentrated to give 8.
Psammaplin A (8a). The residue was purified by column chromatography (silica gel, petroleum ether-AcOEt 1:1) to give 8a (0.84 g, 69%) as a white solid, m.p. 170-172 °C (lit. [19] 172-174 °C). IR (KBr): 3384, 2927, 1654, 1626, 1579, 1535, 1493, 1421, 1359, 1284, 1209, 1044, 1015, 984, 801 cm-1; 1H-NMR (300 MHz, CD3OD) δ: 2.81 (4H, t, J = 6.8 Hz), 3.52 (4H, t, J = 6.8 Hz), 3.79 (4H, s), 6.76 (2H, d, J = 8.4 Hz), 7.07 (2H, dd, J = 8.4 Hz, J = 2.1 Hz), 7.36 (2H, d, J = 2.1 Hz); 13C-NMR (75 MHz, CD3OD) δ: 28.7, 38.5, 39.5, 110.5, 117.0, 130.4, 130.6, 134.4, 153.1, 153.7, 165.8. LC-MS m/z: 660.9 [M-H]-.
(E,E)-N,N’-bis(3-(4-methylphenyl)-2-oximidopropionyl)cystamine (8b). The residue was purified by column chromatography (silica gel, petroleum ether-AcOEt 2:1) to give 8b (0.70 g, 76%) as a white solid, m.p. 162-163 °C; IR (KBr): 3392, 3327, 2928, 2852, 1657, 1627, 1576, 1531, 1426, 1204, 1123, 1022, 783 cm-1; 1H-NMR (300 MHz, DMSO-d6) δ: 2.20 (6H, s), 2.78 (4H, t, J = 6.8 Hz), 3.38 (4H, m), 3.73 (4H, s), 7.01 (4H, d, J = 8.1 Hz), 7.06 (4H, d, J = 8.1 Hz), 8.02 (2H, t, J = 5.7 Hz), 11.7 (2H, s); 13C-NMR (75 MHz, DMSO-d6) δ: 20.9, 28.8, 37.3, 38.5, 128.9, 129.1, 134.0, 135.3, 152.3, 163.6; LC-MS m/z: 503.3 [M+H]+.
(E)-3-(3-bromo-4-hydroxyphenyl)-2-(hydroxyimino)-N-(2-mercaptoethyl)propanamide (2). To a solution of psammaplin A (8a, 0.60 g, 0.90 mmol) in CH3OH (30 mL) were added 1M KOH (90 µL) and Cleland’s reagent (0.42 g, 2.70 mmol) at room temperature. The reaction was quenched with 0.5 M HCl at 0 °C after 1 h and extracted with CH2Cl2. The organic layer was washed with water, brine, dried over MgSO4, filtered and concentrated. The desired compound 2, obtained as a yellow oil (0.60 g, quant.) was used directly in the next reaction. IR (KBr): 3384, 2930, 1655, 1627, 1535, 1493, 1421, 1363, 1285, 1214, 1043, 1009, 801 cm-1; 1H-NMR (600 MHz, DMSO-d6) δ: 2.24 (1H, t, J = 7.5 Hz), 2.54 (2H, q, J = 7.5 Hz ), 3.28 (2H, m), 3.69 (2H, s), 6.83 (1H, d, J = 8.4 Hz), 7.00 (1H, dd, J = 8.4 Hz, J = 2.1 Hz), 7.29 (1H, d, J = 2.1 Hz), 8.05 (1H, t, J = 6.0 Hz), 9.99 (1H, s), 11.8 (1H, s). 13C-NMR (150 MHz, DMSO-d6) δ: 27.3, 28.0, 42.4, 109.2, 116.5, 129.1, 129.5, 133.1, 152.2, 152.7, 163.5. LC-MS m/z: 333.0 [M+H]+.
N,N’-bis(ethoxyoxalyl)cystamine (10). To a solution of cystamine dihydrochloride (9, 1.00 g, 4.44 mmol) in 1 M K2CO3 (14 mL) was added dropwise ethyl oxalyl monochloride (1.46 g, 10.6 mmol) in CH2Cl2 at 0 °C. After stirring for 1 h, the mixture was extracted with CH2Cl2. The organic layer was washed with water, brine, dried over MgSO4, filtered and concentrated. The residue was recrystallized by petroleum ether-acetone 2:1 to give 10 (1.45 g, 93%) as a white solid, m.p. 102-103 °C; IR (KBr): 3337, 2970, 2932, 1733, 1681, 1535, 1436, 1368, 1309, 1289, 1220, 1058, 1024, 865 cm-1; 1H-NMR (300 MHz, DMSO-d6) δ: 1.25 (6H, t, J = 7.2 Hz), 2.83 (4H, t, J = 6.6 Hz), 3.42 (4H, m), 4.22 (4H, q, J = 7.2 Hz), 9.01 (2H, t, J = 5.7 Hz); 13C-NMR (75 MHz, DMSO-d6) δ: 14.1, 36.5, 38.7, 62.3, 157.3, 160.8; LC-MS m/z: 353.1 [M+H]+.
N-ethoxyoxalyl-2-mercaptoethanamine (11). To a solution of bisamide 10 (0.40 g, 1.14 mmol) in C2H5OH (30 mL) were added 1M KOH (90 µL) and Cleland’s reagent (0.51 g, 3.42 mmol) at room temperature. The reaction was quenched with 0.5 M HCl at 0 °C after 40 min and extracted with CH2Cl2. The organic layer was washed with water, brine, dried over MgSO4, filtered and concentrated. The desired compound 11, obtained as a yellow oil (0.40 g, quant.), was used directly in the next reaction. IR (KBr): 3421, 2966, 2927, 1734, 1682, 1528, 1468, 1386, 1292, 1210, 1111, 1014, 858 cm‑1. 1H-NMR (600 MHz, DMSO-d6) δ: 1.25 (3H, t, J = 7.2 Hz), 2.40 (1H, t, J = 7.8 Hz), 2.56 (2H, q, J = 7.8 Hz), 3.27 (2H, m), 4.21 (2H, q, J = 7.2 Hz), 8.99 (1H, t, J = 6.0 Hz); 13C-NMR (150 MHz, DMSO-d6) δ: 14.3, 23.2, 42.8, 62.5, 157.5, 161.0. LC-MS m/z: 178.0 [M+H]+.
N-ethoxyoxalyl-2-(2-pyridyldithio)ethanamine (3). To a solution of aldrithiol (13) (3.01 g, 13.7 mmol) in C2H5OH (45 mL) were added dropwise glacial AcOH (1.6 mL) and oxalamic acetate 11 (1.60 g, 4.55 mmol) in C2H5OH (20 mL) under N2 atmosphere. The mixture was stirred for 24 h at room temperature. After evaporation of the solvent, the residue was dissolved in CH2Cl2 and washed with water, brine, and dried over MgSO4. The organic layer was concentrated to afford the crude residue, which was purified by column chromatography (silica gel, petroleum ether-acetone 5:1) to give 3 (1.04 g, 80%) as a white solid, mp 65-66 °C. IR (KBr): 3412, 3214, 2971, 2915, 1729, 1706, 1572, 1538, 1453, 1411, 1369, 1309, 1237, 1182, 1112, 1023, 992, 762 cm-1; 1H-NMR (600 MHz, DMSO-d6) δ: 1.25 (3H, t, J = 7.2 Hz), 2.96 (2H, t, J = 6.6 Hz), 3.44 (2H, m), 4.22 (2H, q, J = 7.2 Hz), 7.23 (1H, m), 7.73 (1H, m), 7.80 (1H, m), 8.46 (1H, m), 9.10 (1H, t, J = 6.0 Hz); 13C-NMR (150 MHz, DMSO-d6) δ: 14.1, 36.9, 38.4, 62.3, 119.8, 121.5, 138.0, 149.9, 157.2, 159.2, 160.7; LC-MS m/z: 287.3 [M+H]+.
((E)-N-3-(3-bromo-4-hydroxyphenyl)-2-oximidopropionyl-N’-ethoxyoxalyl)cystamine (14). To a solution of oxalamic amide 3 (0.26 g, 0.90 mmol) in C2H5OH (20 mL) was added dropwise a solution of oximic amide 2 (0.10 g, 0.30 mmol) in C2H5OH (10 mL) under a N2 atmosphere. The whole mixture was stirred for 12 h at room temperature. After removal of the solvent, the residue was dissolved in CH2Cl2 and washed with water, brine and dried over MgSO4. The organic layer was concentrated to afford the crude product, which was purified by column chromatography (silica gel, petroleum ether-acetone 3:1) to give 14 (0.10 g, 66%) as a white solid, m.p. 57-58 °C; IR (KBr): 3378, 2961, 2933, 2873, 1742, 1687, 1658, 1620, 1534, 1416, 1291, 1213, 1115, 1017, 984, 802 cm-1; 1H-NMR (300 MHz, DMSO-d6) δ: 1.24 (3H, t, J = 7.2 Hz), 2.81 (4H, t, J = 6.2 Hz), 3.42 (4H, m), 4.20 (2H, q, J = 7.2 Hz), 6.81 (1H, d, J = 8.1 Hz), 7.00 (1H, dd, J = 8.1 Hz, J = 1.8 Hz), 7.27 (1H, d, J = 1.8 Hz), 8.06 (1H, t, J = 6.0 Hz), 8.10 (1H, t, J = 6.0 Hz, 10.0 (1H, s), 11.8 (1H, s); 13C-NMR (75 MHz, DMSO-d6) δ: 14.2, 28.0, 36.7, 37.3, 38.5, 38.7, 62.4, 109.2, 116.5, 129.2, 129.5, 133.1, 152.2, 152.7, 157.4, 160.8, 163.6; LC-MS m/z: 508.3 [M+H]+; HR-ESI-MS: calcd for C17H22BrN3O6S2Na+ [M+Na]+ 530.0026, found 530.0021.
Psammaplin F (1). To a solution of unsymmetrical disulfide 14 (0.2 g, 0.39 mmol) in THF (8 mL), 3.3 M KOH (1.65 mL) was added dropwise, the mixture was stirred for 1.5 h at 0 °C. The resulting mixture was adjusted to pH 1 with 0.5 M HCl at 0 °C and extracted with AcOEt. The organic layer was washed with water, brine, dried over MgSO4, filtered and concentrated. The residue was triturated in Et2O, filtered out and dried in vacuo to give 1 (0.09 g, 50%) as a white solid, m.p. 122-124 °C; IR (KBr): 3371, 1652, 1535, 1420, 1290, 1209, 1044, 985 cm-1; 1H-NMR (600 MHz, CD3OD) δ: 2.85 (4H, t, J = 6.0 Hz), 3.55 (4H, t, J = 6.0 Hz), 3.80 (2H, s), 6.77 (1H, d, J = 8.4 Hz), 7.07 (1H, dd, J = 8.4 Hz, J = 1.8 Hz), 7.34 (1H, d, J = 1.8 Hz); 13C-NMR (150 MHz, CD3OD) δ: 38.2, 38.7, 39.8, 40.0, 110.5, 117.1, 130.4, 130.6, 134.5, 153.2, 153.7, 161.8, 159.0, 166.0; LC-MS m/z: 477.9 [M-H]-; HR-ESI-MS: calcd for C15H18BrN3O6S2Na+ 501.9713, found 501.9719.
3.2. Single crystal X-ray determination of compound 8b
A colorless platelet crystal of the title compound with approximate dimensions 0.36 × 0.25 × 0.08 mm was used for data collection on a Bruker APEX CCD area diffractometer using a graphite-monochromated Mo-Kα radiation (0.71073 Å) at 295 (2) K in the ω-2θ scan mode. An empirical absorption correction was applied to the data using the SADABS program [20]. A total of 6718 reflections were collected in the range of 1.74º < θ < 25.66º, of which 4589 reflections were independent with Rint = 0.0158 and 3743 observed reflections with I > 2σ(I) were used in the succeeding refinements. The structures were solved by direct methods and refined by full-matrix least-squares methods on F2 using the SHELXTL crystallographic software package [21,22,23]. All non-hydrogen atoms were refined anisotropically. The hydrogen atoms were placed in calculated positions and refined by using a riding mode. Supplementary crystallographic data have been deposited with the Cambridge Crystallographic Data Centre as CCDC No. 786045 (compound 8b) which contains the supplementary crystallographic data for this paper. The data can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html or from the Cambridge Crystallographic Data centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax (+44) 1223-336-033; E-mail: deposit@ccdc.cam.ac.uk.
4. Conclusions
In conclusion, we have reported here the first total synthesis of psammaplin F (1) from simple starting materials and under mild reaction conditions. The overall yield of psammaplin F (1) from 3-bromo-4-hydroxybenzaldehyde (4) and hydantoin in seven steps was 12%. In addition, the absolute configuration of (E,E)-N,N’-bis(3-(4-methylphenyl)-2-oximidopropionyl)cystamine (8b) was confirmed for the first time by X-ray crystal structure analysis.
Acknowledgements
This work was supported by China International Science and Technology Cooperation Plan (2009DEA31200) and National Science and Technology Key Specific Project for Significant Creation of New Drugs (2009ZX09301-012).
Footnotes
Sample Availability: Samples are available from the authors.
References and Notes
- 1.Piña I.C., Gautschi J.T., Wang G.Y.S., Miranda L.S., Francis J.S., Dennis F., Susan C.K., Lidia C.S., Stacy W.R., Larry B.P., Kenneth W.B., Phillip C. Psammaplins from the sponge Pseudoceratina purpurea: inhibition of both histone deacetylase and DNA methyltransferase. J. Org. Chem. 2003;68:3866–3873. doi: 10.1021/jo034248t. [DOI] [PubMed] [Google Scholar]
- 2.Kim D., Lee I.S., Jung J.H., Lee C.O., Choi S.U. Psammaplin A, a natural phenolic compound, has inhibitory effect on human topoisomerase II and is cytotoxic to cancer cells. Anticancer Res. 1999;19:4085–4090. [PubMed] [Google Scholar]
- 3.Nicolaou K.C., Hughes R., Pfefferkorn J.A., Barluenga S., Roecker A.J. Combinatorial synthesis through disulfide exchange: discovery of potent psammaplin A type antibacterial agents active against methicillin-resistant staphylococcus aureus (MRSA) Chem. Eur. J. 2001;7:4280–4295. doi: 10.1002/1521-3765(20011001)7:19<4280::AID-CHEM4280>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 4.Tabudravu J.N., Eijsink V.G.H., Gooday G.W., Jaspars M., Komander D., Legg M., Synstad B., van Aalten D.M.F. Psammaplin A, a chitinase inhibitor isolated from the Fijian marine sponge Aplysinella rhax. Bioorg. Med. Chem. 2002;10:1123–1128. doi: 10.1016/S0968-0896(01)00372-8. [DOI] [PubMed] [Google Scholar]
- 5.Shim J.S., Lee H.S., Shin J., Kwon H.J. Psammaplin A, a marine natural product, inhibits aminopeptidase N and suppresses angiogenesis in vitro. Cancer Lett. 2004;203:163–169. doi: 10.1016/j.canlet.2003.08.036. [DOI] [PubMed] [Google Scholar]
- 6.Hoshino O., Murakata M., Yamada K. A convenient synthesis of a bromotyrosine derived metabolite, psammaplin A, from psammaplysilla SP. Bioorg. Med. Chem. Lett. 1992;2:1561–1562. doi: 10.1016/S0960-894X(00)80429-1. [DOI] [Google Scholar]
- 7.Godert A.M., Angelino N., Woloszynska-Read A., Morey S.R., James S.R., Karpf A.R., Sufrin J.R. An improved synthesis of psammaplin A. Bioorg. Med. Chem. Lett. 2006;16:3330–3333. doi: 10.1016/j.bmcl.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 8.Thomas W.L., Edel O’T., Orlagh F. Solid oral dosage form containing an enhancer. 20070292512 (A1) U.S. Patent. 2007 Dec 20;
- 9.Thenmozhiyal J.C., Wong P.T.H., Chui W.K. Anticonvulsant activity of phenylmethylenehydantoins: a structure-activity relationship study. J. Med. Chem. 2004;47:1527–1535. doi: 10.1021/jm030450c. [DOI] [PubMed] [Google Scholar]
- 10.Naoyuki T., Hiroyuki M. Method for producing 4-hydroxyphenylpyruvic acid. 2003104932 (A) Japan Patent. 2003 Apr 9;
- 11.Knapp S., Amorelli B., Darout E., Ventocilla C.C., Goldman L.M., Huhn R.A., Minnihan E.C. A family of mycothiol analogues. J. Carbohydr.Chem. 2005;24:103–130. [Google Scholar]
- 12.Leino R., Lönnqvist J.E. A very simple method for the preparation of symmetrical disulfides. Tetrahedron Lett. 2004;45:8489–8491. doi: 10.1016/j.tetlet.2004.09.100. [DOI] [Google Scholar]
- 13.Fournie-Zaluski M.C., Coric P., Turcaud S., Lucas E., Noble F., Maldonado R., Roques B.P. Mixed-inhibitor-prodrug as a new approach toward systemically active inhibitors of enkephalin-degrading enzymes. J. Med. Chem. 1992;35:2473–2481. doi: 10.1021/jm00091a016. [DOI] [PubMed] [Google Scholar]
- 14.Kemp D.S., Galakatos N.G., Dranginis S., Ashton C., Fotouhi N., Curran T.P. Peptide synthesis by prior thiol capture. 4. Amide bond formation: the effect of a side-chain substituent on the rates of intramolecular O, N-acyl transfer. J. Org. Chem. 1986;51:3320–3324. doi: 10.1021/jo00367a013. [DOI] [Google Scholar]
- 15.Zugates G.T., Anderson D.G., Little S.R., Lawhorn I.E.B., Langer R. Synthesis of poly(β-amino esters) with thiol-reactive side chains for DNA delivery. J. Am. Chem. Soc. 2006;128:12726–12734. doi: 10.1021/ja061570n. [DOI] [PubMed] [Google Scholar]
- 16.Kotoku N., Tsujita H., Hiramatsu A., Mori C., Koizumi N., Kobayashi M. Efficient total synthesis of bastadin 6, an anti-angiogenic brominated tyrosine-derived metabolite from marine sponge. Tetrahedron. 2005;61:7211–7218. [Google Scholar]
- 17.Dhar S., Nethajiand M., Chakravarty A.R. Designing molecules for PDT: red light-induced DNA cleavage on disulfide bond activation in a dicopper (II) complex. Dalton Trans. 2005:344–348. doi: 10.1039/b413410a. [DOI] [PubMed] [Google Scholar]
- 18.Katti K.V., Singh P.R., Barnes C.L. Transition-metal chemistry of main-group hydrazides. Part 2. A new oxime thiosemicarbazide framework as a novel SN multifunctional tripodal ligand for palladium (II): synthetic and X-ray crystal structural investigations. J. Chem. Soc. Dalton Trans. 1993:2153–2156. [Google Scholar]
- 19.Arabshahi L., Schmitz F.J. Brominated tyrosine metabolites from an unidentified sponge. J. Org. Chem. 1987;52:3584–3586. doi: 10.1021/jo00392a016. [DOI] [Google Scholar]
- 20.Sheldrick G. Program SADABS: Area-Detector Absorption Correction. University of Göttingen; Göttingen, Germany: 1996. [Google Scholar]
- 21.Sheldrick G.M. SHELXL-97. University of Göttingen; Göttingen, Germany: 1997. [Google Scholar]
- 22.Sheldrick G.M. SHELXL-97. University of Göttingen; Göttingen, Germany: 1997. [Google Scholar]
- 23.Sheldrick G.M. A short history of SHELXL. Acta Cryst. 2008;A64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]