

Summary
Of the new direct oral anticoagulants, direct factor Xa inhibitors are limited by the absence of a proven reversal agent. We assessed the safety, tolerability and impact on anticoagulation reversal of ciraparantag (PER977) alone and following a 60 mg dose of the FXa inhibitor edoxaban. Escalating, single IV doses of ciraparantag were administered alone and following a 60 mg oral dose of edoxaban in a double-blind, placebo-controlled fashion to healthy subjects. Serial assessments of the pharmacokinetics and pharmacodynamic effects of ciraparantag were performed. Eighty male subjects completed the study. Following edoxaban (60 mg), a single IV dose of ciraparantag (100 to 300 mg) demonstrated full reversal of anticoagulation within 10 minutes and sustained for 24 hours. Fibrin diameter within clots was restored to normal 30 minutes after a single dose of 100 to 300 mg ciraparantag as determined by scanning electron microscopy and change in fibrin diameter quantified by automated image analysis. Potentially related adverse events were periorbital and facial flushing and cool sensation following IV injection of ciraparantag. Renal excretion of ciraparantag metabolite was the main elimination route. There was no evidence of procoagulant activity following ciraparantag as assessed by D-dimer, prothrombin fragments 1.2, and tissue factor pathway inhibitor levels. In conclusion, ciraparantag in healthy subjects is safe and well tolerated with minor, non-dose limiting adverse events. Baseline haemostasis was restored from the anticoagulated state with doses of 100 to 300 mg ciraparantag within 10–30 minutes of administration and sustained for at least 24 hours.
Institution where the work was performed: Duke University Clinical Research Unit, Duke University Medical Center, Durham, NC USA
Keywords: Ciraparantag, edoxaban, anticoagulant, reversal, whole blood clotting time
Introduction
Target-specific, direct oral anticoagulants (DOACs) have been shown to be effective, safer and easier to use compared to the vitamin K antagonists (VKAs) for indications including stroke prevention in atrial fibrillation and the prevention and treatment of venous thromboembolism ( 1 ). A major barrier to their more widespread use is the absence of a reversal agent. This lack of an antidote to reverse anticoagulation or bleeding in patients with major haemorrhage or in need of emergent surgery is an important clinical need ( 2 ). Limiting the time a patient is off anticoagulant therapy for an invasive procedure could also be achieved with the availability of an effective reversal drug that has a rapid onset, completely reverses the anticoagulant, and allows anticoagulation to be reestablished.
Ciraparantag (originally PER977; Perosphere Inc., Danbury, CT, USA) is a small synthetic water-soluble new molecular entity that binds to heparin and the oral direct factor Xa (FXa) and factor IIa (FIIa) inhibitors by charge interaction removing these drugs from their intended target and allowing rapid re-establishment of normal blood coagulation ( 3 ). In pre-clinical models (rat tail transection bleeding model and ex vivo fresh human blood) ciraparantag consistently reverses the anticoagulant effects of unfractionated heparin, low-molecular-weight heparin (LMWH), fondaparinux, and oral FXa (apixaban, rivaroxaban, edoxaban) and FIIa (dabigatran) inhibitors ( 4 ).
This phase I study assessed the safety, tolerability, pharmacokinetic and pharmacodynamic effects of ciraparantag when administered alone and following a single therapeutic oral dose of edoxaban, an oral FXa inhibitor, in healthy volunteers.
Methods
This was a first in human phase I, randomised, double-blind, placebo-controlled evaluation of the safety, tolerability, pharmacokinetics and pharmacodynamic effects of escalating single doses of ciraparantag, administered alone, and after a minimum of four days administered to the same subject for a second time, following a single oral dose of edoxaban. Healthy non-smoking subjects age 18 to 65 were eligible. Subjects were excluded with any significant clinical history including family or personal history of clotting abnormality or excessive bleeding, thrombotic or vascular disease, personal history of major or minor bleeding episodes within the 3–6 months prior to screening, blood product or anticoagulant use within the prior three months, non-steroidal anti-inflammatory drug use within the prior two weeks, use of chronic medication or history of drug or alcohol dependence, human immunodeficiency virus or hepatitis B or C.
The starting and maximum doses of ciraparantag tested were selected based on toxicology studies in animals. There were a total of eight dosing cohorts (► Figure 1 ) each enrolling ten subjects randomised in an 8:2 ratio of ciraparantag:placebo (saline injection). There were two dosing periods in each cohort; injection of ciraparantag alone (Period 1) and injection of ciraparantag approximately 3 hours (h) following a single 60 mg oral dose of edoxaban (Period 2). Within each cohort, there was a minimum four-day wash out interval between day 1 dosing in Period 1 and in Period 2. Co-administration of edoxaban and ciraparantag was allowed in Period 2 only if the single dose of ciraparantag was well tolerated in Period 1. Ciraparantag or placebo was administered as a sterile aqueous solution injected by slow intravenous (i.v.) injection into an indwelling catheter in a double-blind manner in both dosing periods.
Figure 1: Study design.
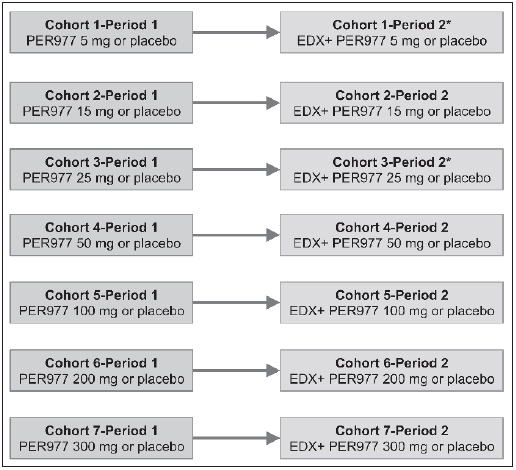
. All cohorts: n=10 (8 active + 2 placebo); 7 days between patient dosing within a cohort and 3 days between cohorts; in Period 2, ciraparantag or placebo administered approximately 3 h post edoxaban (EDX). The 25 mg dosing cohort was repeated using the WBCT to achieve a total of eight dosing cohorts.
Serial assessments of the pharmacodynamic effects of ciraparantag were to be accomplished by assessing commonly used whole blood laboratory tests, namely whole blood point-of-care (PoC) prothrombin time (PT) (ITC Hemochron Signature Elite ® , International Technidyne Corp., Edison, NJ, USA) and thromboelastography reaction time (TEG-R) (TEG ® 5000 Thromboelastograph ® Hemostasis Analyzer System, Haemonetics, Braintree, MA, USA). It was found during the course of this study that ciraparantag cannot be studied in blood collected with sodium citrate, oxalate, EDTA or heparin because of the molar excess of these anions that overwhelm and disrupt the ciraparantag anticoagulant complex, thereby freeing the anticoagulant in the plasma and making plasma-based assays non-representative of physiological conditions. It was found that activators such as kaolin and celite also adsorb ciraparantag, significantly reducing the active concentration of soluble ciraparantag in a blood sample and making kaolin and celite-based assays insensitive to quantitating reversal by ciraparantag. Additionally, due to the observation that blood collected in glass tubes for biomarker testing was clotting more rapidly with increasing doses of ciraparantag even while PoC-PT and TEG-R reflected little or no change (see Suppl. Table 1, available online at www.thrombosis-online.com ), it was decided that a whole blood clotting time (WBCT) ( 11 ) might provide the best measure of anticoagulation and its reversal by ciraparantag. The WBCT was performed as traditionally described ( 11 ) in glass tubes without activator or anticoagulant and with clotting time assessed by the tilt-tube visual inspection method using three tubes with 0.5 ml of whole blood in each. Because enhanced clotting of anticoagulated blood was visually apparent beginning in Cohort 4 (ciraparantag 50 mg), whole blood clotting time was introduced in Period 2 of Cohort 4 and all remaining cohorts. In addition, clot fibrin integrity (CFI) using scanning electron microscopy (SEM) and quantified by automated image analysis of fibrin diameter was added by protocol amendment in the higher dose cohorts. Assessment of CFI was performed on clots formed during measurement of the WBCT in order to validate the restoration of fibrin’s ability to form normal clots. Clots from the WBCT determination were glutaraldehyde in sodium cacodylate buffer and stored under refrigeration for critical point drying, freeze fracture and CFI imaging by SEM. Automated image analysis of the CFI SEMs (MATLAB, MathWorks) was used to quantify mean fibrin diameter. The potential of inducing a procoagulant effect by ciraparantag when administered alone in Period 1 was assessed by measurement of D-dimer (Imuclone ® D-dimer ELISA kit, Sekisui Diagnostics, LLC, Stamford, CT, USA) and prothrombin fragments 1.2 (Enzygnost ® F1+2; Siemens Healthcare Diagnostic Products GmbH, Marburg, Germany). Tissue factor pathway inhibitor (TFPI) was also measured (ab108904 TFPI Human ELISA Kits, Abcam, Cambridge, MA, USA). ciraparantag and its major metabolite, (1,2-bis(3-aminopropyl)piperazine (BAP), as well as edoxaban and its major metabolite were measured in plasma and urine. Safety and tolerability, including adverse events, vital signs, pulse oximetry, electrocardiograms, clinical laboratory testing, fecal occult blood testing, and concomitant medications were assessed in all subjects who received at least one dose of study medication.
Study oversight
The study was sponsored by Perosphere, co-sponsored by Daiichi-Sankyo and performed by Duke Clinical Research Institute (DCRI). It was conducted according to the protocol and statistical analysis plan (Clinicaltrials.gov # NCT01826266). All authors and the sponsors were responsible for designing the study protocol and analysis plan. Data collection and entry was performed by DCRI. All authors had full access to the data and analyses for compilation of this report. Manuscript drafts were prepared by the authors with editorial assistance from a professional medical writer (K. Martin-Glenmere Research, Inc) paid for by Perosphere. All authors vouched for the accuracy and completeness of the data reported and the fidelity of the study to the protocol, and all authors made the decision to submit the manuscript for publication.
The protocol was approved by the Copernicus Group Independent IRB, Durham, NC, USA ( http://www.cgirb.com/ ) and was conducted under the guidelines of the Declaration of Helsinki and Good Clinical Practice. All subjects provided written informed consent.
Statistical evaluation
The safety population consisted of all subjects who received study medication in either period. The pharmacokinetic (PK) population consisted of all subjects who received ciraparantag in either period and had sufficient PK samples in either or both periods. The pharmacodynamic (PD) population consisted of all subjects who received both edoxaban and ciraparantag in Period 2 and had sufficient PD samples assessed. Whole blood clotting time and clot fibrin diameter measurements were analysed by one-way analysis of variance (ANOVA) Tukey’s post-hoc Honestly Significant Difference in means (StatPlus, AnalystSoft) with statistical significance established at p<0.05.
Results
The study was conducted at the Duke Clinical Research Unit in Durham, NC, USA. Of the 83 subjects who were randomised into the study, 80 (96.4 %) completed the study. Sixty-seven subjects were randomised to receive ciraparantag, and 16 were randomised to placebo. Three (4.5 %) of the subjects who received ciraparantag discontinued prematurely; two withdrew consent, and one was discharged before dosing and was not included in the Safety Population. None of the placebo subjects discontinued prematurely. Mean age was 31 and 32, respectively, in each treatment group, 100 % were male, 30 % were Caucasian and 70 % were African American.
Safety assessment
Adverse events, whether considered related or not to ciraparantag, are presented in ► Table 1 . There were no serious adverse events and only one subject discontinued due to an adverse event. This subject, in, Cohort 5, presented with an elevated creatinine phosphokinase at check-in prior to Period 2 which was attributed to a period of extreme exercise and muscle cramps not considered related to ciraparantag. All adverse events were mild in severity with the exception of a report of moderate muscle cramp in the right anterior thigh in one subject in Cohort 5 (same subject as above) during the no treatment phase and not considered related to ciraparantag, and one report of moderate headache in one subject in Cohort 7 during Period 1 considered related to ciraparantag. There were no significant changes in blood counts, blood chemistries (electrolytes, bilirubin, hepatic enzymes) or electrocardiograms. The majority of adverse events that were considered related to ciraparantag included flushing or a cool temperature sensation around the face and dysgeusia after injection.
Table 1.
Summary of adverse events (safety population).
| Variable | Placebo (N=16) | Cira-parantag (N=66) | Total (N=82) |
|---|---|---|---|
| Number of AEs | 18 | 204 | 222 |
| Number of subjects with AE | 10 (62.9 %) | 49 (74.2 %) | 59 (72 %) |
| Number of subjects with TEAE | 10 (62.5 %) | 49 (74.2 %) | 59 (72 %) |
| Number of subjects with SAE | 0 | 0 | 0 |
| Number of subjects who died | 0 | 0 | 0 |
| Number of subjects with AE leading to study drug discontinuation | 0 | 0 | 0 |
| Number of subjects with AE related to study drug | 8 (50 %) | 38 (57.6 %) | 46 (56.1 %) |
| Related to Ciraparantag/Placebo | 7 (43.8 %) | 36 (54.5 %) | 43 (52.4 %) |
| Related to edoxaban | 2 (12.5 %) | 5 (7.6 %) | 7 (8.5 %) |
| Number of AEs per subject | |||
| 0 | 6 (37.5%) | 17 (25.8 %) | 23 (28.0 %) |
| 1 | 5 (31.3%) | 10 (15.2 %) | 15 (18.3 %) |
| 2–3 | 4 (25.0 %) | 16 (24.2 %) | 20 (24.4%) |
| 4–5 | 1 (6.3 %) | 9 (13.6 %) | 10 (12.2 %) |
| >5 | 0 | 14 (21.2 %) | 14 (17.1 %) |
AE = adverse event; TEAE = treatment emergent adverse event(defined as an AE that started on or after study drug administration); SAE = serious adverse event.
Pharmacokinetic assessment
Ciraparantag and its primary metabolite 1,4-bis(3-aminopropyl)-piperazine (BAP), demonstrated dose-proportional pharmacokinetics based on maximal concentration (C max ) and half life (t ½ ). At the lowest dose of ciraparantag administered, 5 mg, the average C max was approximately 170 ng/ml in serum with a t ½ of approximately 12 minutes (min) and at the highest ciraparantag dose, 300 mg, the average C max was approximately 10,600 ng/ml in serum and the average t ½ was approximately 19 min. BAP reached time to maximal concentration (T max ) 20–30 min after ciraparantag administration, and C max ranged from ∼90 ng BAP/ml serum at a 5 mg ciraparantag dose to 1,100 ng BAP/ml serum at a 300 mg ciraparantag dose. Ciraparantag was metabolised and approximately 30 % was recovered as BAP in pooled urine samples in the first 24 hours (h).
Pharmacodynamic assessment
Findings during the initial 4 cohorts of this study indicated that PoC-PT (which contains celite activator) and TEG-R (which uses kaolin activator) were not acceptable biomarkers due to their in-sensitivity and high variability (see Suppl. Table 1, available online at www.thrombosis-online.com ). Samples for the TEG-R test were observed to be clotting faster as ciraparantag dose increased, and yet the PoC-PT values showed no change. The TEG-R showed sufficiently high variability between time points and between subjects to render it clinically useless. Mean PoC-PT and TEG-R showed no change over time within cohorts and across escalating dose cohorts. Thus, we employed glass-activated whole blood clotting time (WBCT) ( 5 ), absent any chemical or biologic activating agents, performed manually and reproducibly by trained technicians starting in Period 2 of Cohort 4 and remaining cohorts. Edoxaban, 60 mg, resulted in a significant elevation of the WBCT (► Figure 2A ) which correlated well with edoxaban pharmacokinetics whereas administration of ciraparantag in the absence of edoxaban had no effect on the WBCT (► Figure 2 B). Additional studies correlating the WBCT with PT and increasing concentrations of edoxaban are included in the Suppl. Material (Suppl. Figures 1 and 2, available online at www.thrombosis-online.com ).
Figure 2: Correlation of WBCT after a 60 mg dose of edoxaban (A) and lack of effect of ciraparantag administration on WBCT (B).
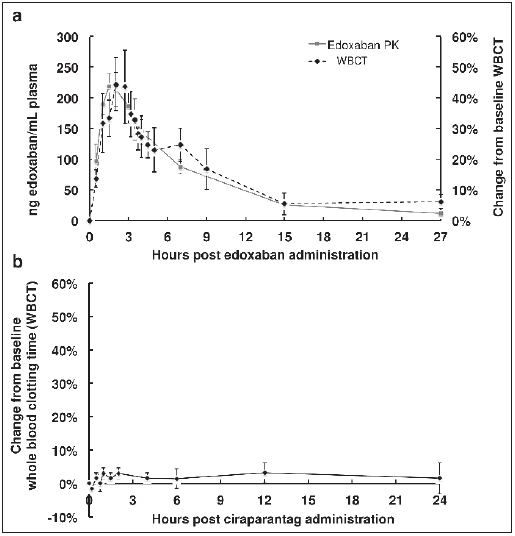
Effect of ciraparantag assessed by whole blood clotting time and clot structure
A single i.v. dose of increasing concentrations of ciraparantag (25, 50, 100, 200 and 300 mg), showed a progressive reversal of edoxaban-induced anticoagulation as measured using the WBCT versus placebo beginning with the 50 mg dose (► Figure 3 ). Reversal of anticoagulation persisted for the full testing period of 24 h following ciraparantag administration. Significant reversal of anticoagulation was noted within 10 min of ciraparantag administration in subjects administered 50 mg and essentially full reversal was noted with increased doses of 100–300 mg in Cohorts 5 to 7 (see Suppl. Table 2 for statistical differences, available online at www.thrombosis-online.com )
Figure 3: Mean WBCT following a single i.v. dose of 25, 100, and 300 mg ciraparantag versus pooled saline placebo.
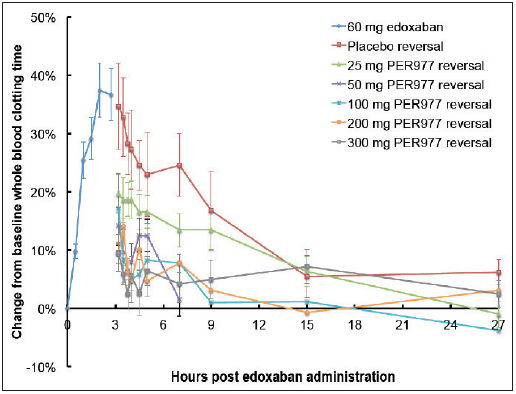
. Data in subjects administered a single oral dose of edoxaban 60 mg (see Suppl. Table 2 for statistical assessment of differences, available online at www.thrombosis-online.com ).
Visual examination of clots demonstrated the anticoagulant effect of edoxaban on clot formation and restoration of clot formation following a single IV dose of 100 mg ciraparantag (► Figure 4A ). Additionally, fibrin structure was examined via scanning electron microscopy. Fibrin strands were poorly developed following edoxaban and were restored to baseline levels within 30 min following administration of a single i.v. dose of 100 mg ciraparantag (► Figure 4B ) and was indistinguishable from placebo subjects at baseline. Automated image analysis of the SEMs determined that edoxaban anticoagulation reduced mean fibrin diameter from ∼250 nm in the native state to ∼125 nm. Twenty-five mg of ciraparantag produced a partial reversal effect increasing fibrin diameter to ∼200 nm, while 100 and 300 mg of ciraparantag yielded full reversal restoring fibrin diameter to ∼250 nm, statically indistinguishable (p>0.05) from native clots.
Figure 4: Visual examination of blood clots from WBCT assay pre-and post- ciraparantag versus saline.
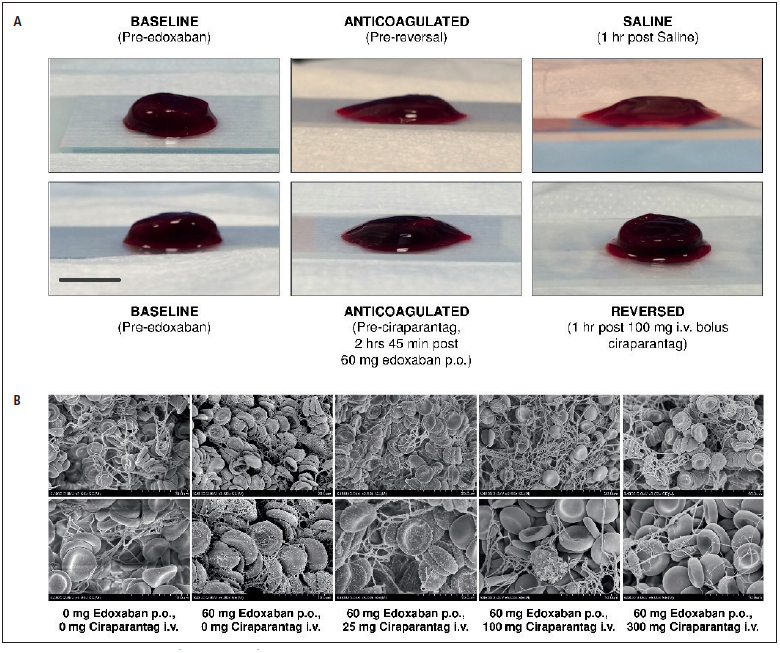
. Scale bar: 1 inch (A) and clot fibrin structure pre- and post- ciraparantag (B) (upper and lower photos represent lower and higher magnification. Computer algorithm-based quantification of clot fibrin diameter showed a significant difference between saline vs native fibrin (p<0.001); between ciraparantag 25 mg vs native fibrin (p<0.01); and between ciraparantag 25 mg vs ciraparantag 300 mg (p<0.05).
Assessment of pro-coagulant effects of ciraparantag
To assess whether administration of ciraparantag induces a prothrombotic state, D-dimer, prothrombin fragment 1.2, and tissue factor pathway inhibitor were measured at 12 and 24 h post administration of ciraparantag and compared to baseline levels (► Figure 5 ). Changes in the levels of these indicators variably fluctuated over time and in no consistent manner.
Figure 5: Ciraparantag effect on D-dimer, prothrombin fragment F1.2 and tissue factor pathway inhibitor (TFPI).
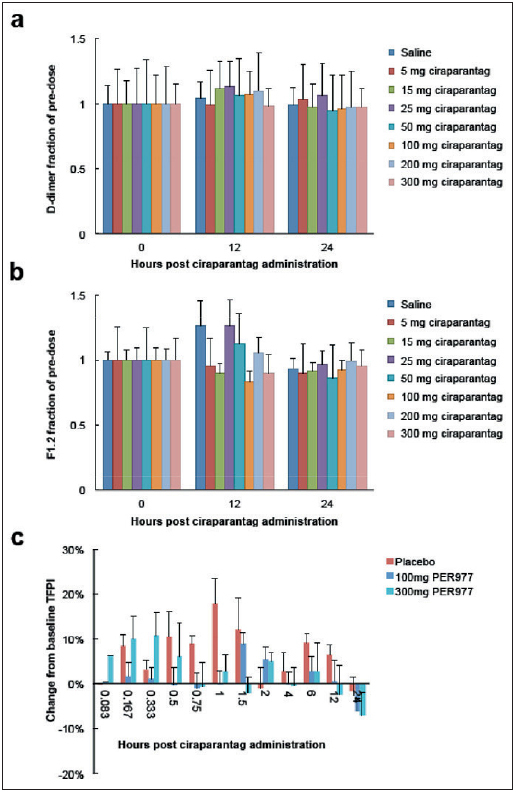
. Lower doses of ciraparantag (PER977) are deleted from panel C to make it visually more clear.
Discussion
Ciraparantag is a small synthetic water-soluble cationic molecule designed to bind directly to specific anionic molecules such as heparin and low-molecular-weight heparin via non-covalent hydrogen bonding and charge-charge interactions ( 3 , 4 ). It binds similarly to the new oral Xa inhibitors, edoxaban, rivaroxaban and apixaban, and to the oral thrombin inhibitor, dabigatran, but not to argatroban or warfarin ( 3 ). Pre-clinical animal studies have shown that it reverses the coagulopathy induced by these anticoagulants and reduces blood loss to baseline levels in animal models of anticoagulant-induced bleeding ( 3 ). Ciraparantag is cleaved by circulating peptidases to its major metabolite, 1,4-bis(3-aminopro-pyl)piperazine, which is renally eliminated. Ciraparantag has no significant binding to plasma proteins, does not interact substantially with the hepatic cytochrome enzymes or membrane transporters and has no binding to a selected spectrum of cardiovascular and antiepileptic drugs ( 3 ).
In this phase I study, ciraparantag was shown to be safe and well tolerated at doses three-fold higher than are required to achieve its intended pharmacodynamic effects. The most common side effects observed were periorbital and facial warmth, flushing and dysgeusia associated with an i.v. injection which was not a limiting factor. No consistent trends were observed in D-dimer, prothrombin fragment 1.2 or TFPI levels, all of which remained substantially unchanged over a 24-h period following injection of ciraparantag. Most importantly, ciraparantag significantly reversed anticoagulation induced by a therapeutic dose of edoxaban within 10 min following injection (first time point measured) which was further reduced to less than 10 % above baseline within 30 min. This return to normal haemostasis persisted over 24 h following a single i.v. dose of study drug.
Because plasma-based assays are unreliable for measuring the reversal effect of ciraparantag we resorted to a whole blood clotting assay without activators (the WBCT). This assay showed excellent correlation with edoxaban pharmacokinetics (► Figure 2 ) and excellent reproducibility in the hands of investigators (see Suppl. Material, available online at www.thrombosis-online.com ).
What is known about this topic?
Direct oral factor Xa (FXa) inhibitors do not currently have a reversal agent.
Having a reversal agent may lead to greater use of the direct oral FXa inhibitors.
Different approaches to developing a reversal agent are in progress, with one agent currently undergoing phase 3 evaluation and a second in an earlier stage of development.
What does this paper add?
Ciraparantag is a small molecule that binds to a FXa inhibitor (edoxaban) through charge-charge interaction and removes edoxaban from its target (i.e. FXa).
Ciraparantag shows progressively greater reversal of edoxaban anticoagulated healthy controls in a dose response manner.
Ciraparantag appears to be safe with no significant or dose-limiting side effects.
Ciraparantag shows no evidence of creating a prothrombotic state.
To further illustrate reversal of the anticoagulant activity of edoxaban, clot photographs and scanning electron micrographs (SEMs) of clot structure were obtained from the clots formed at the end of the WBCT assay. These SEMs clearly show the reduced and disordered fibrin structure formed by clots in the presence of edoxaban (without reversal), and those formed after administration of ciraparantag restoring clot and fibrin structure to baseline appearance.
The lack of a reversal agent for the new oral anticoagulants is a barrier to their more widespread use, although some argue that such an agent is not necessary ( 6 ). Based on very limited data, many experts suggest the use of prothrombin complex concentrates for the treatment of major bleeding ( 2 ), although these come with associated risks, especially that of inducing thrombosis ( 7 ). Recently, an anti-dabigatran Fab fragment ( 8 , 9 ) was approved for clinical use to reverse dabigatran and a modified, inactive FXa protein ( 10 ) for reversal of the Xa inhibitor is in clinical trials. The dabigatran reversal agent was shown to be effective in an ongoing phase 3 trial ( 11 ) and the Xa inhibitor reversal agent reverses anticoagulation in Xa inhibitor treated healthy controls ( 12 ), although questions have been raised about the duration of effect, immunogenicity and prothrombotic potential of the latter agent ( 13 ).
In summary, ciraparantag is given as a single i.v. injection, has a prolonged reversal effect lasting up to 24 h, has no evidence of a prothrombotic effect and is well tolerated. Ciraparantag does not bind to a selected group of cardiovascular and antiepileptic drugs, but more testing will be completed with other drugs to confirm the absence of a specific off-target drug interaction. Additional human studies are ongoing assessing the ability of ciraparantag to neutralize other anticoagulants.
Acknowledgements
Medical writing and editorial support was provided by Kathryn Martin, Pharm.D., Glenmere Research, Montebello NY. Selected coagulation studies performed by the Duke Hemostasis & Thrombosis Core Laboratory, Director Thomas Ortel, MD.
Funding Statement
Financial support: This study was funded by Peropshere, Inc. Danbury CT; Clinicaltrials.gov number NCT01826266.
Footnotes
Conflicts of interest Dr. Ansell is a consultant to Perosphere, Inc. Drs. Bakhru, Costin, Laulicht, and Steiner are employees of Perosphere, Inc. Drs. Lanz, Mercuri, Grosso and Dishy are employees of Daiichi-Sankyo Pharma Development, the co-sponsor of this study. Dr. Brown was an employee of Daiichi Sankyo Pharma Development at the time this work was performed. Dr. Noveck is an employee of Duke Clinical Research Unit, the clinical research organization which conducted the study.
Online Supplementary Material (PDF)
Online Zusatzmaterial (PDF)
References
- 1.Weitz JI, Eikelboom JW, Samama MM. New Antithrombotic Drugs: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guideline. Chest. 2012;141 02:e120S–e151S.. doi: 10.1378/chest.11-2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaatz S, Kouides PA, Garcia DA et al. Guidance on the emergent reversal of oral thrombin and factor Xa inhibitors. Am J Hematol. 2012;87 02:S141–S145.. doi: 10.1002/ajh.23202. [DOI] [PubMed] [Google Scholar]
- 3.Laulicht B, Bakhru S, Jiang Xet al. Antidote for new oral anticoagulants: Mechanism of action and binding specificity of PER977Internat’l Soc Thromb & Haemost2013. Abstracthttp://www.eventure-online.com/eventure/publicAbstractView.do?id=226718&congressId=6839andhttp://www.perosphere.com/content/presentations/documents/Perosphere_ISTH_Talk.pdf
- 4.Laulicht B, Bakhru S, Lee Cet al. Small molecule antidote for anticoagulants Circulation 2012126Abstract#11395. [Google Scholar]
- 5.Didisheim P. Tests of blood coagulation and hemostasis: The coagulation (clotting) tim. J Am Med Assoc. 1966;198:1299. [PubMed] [Google Scholar]
- 6.Eerenberg ES, Levi M, Buller HR. Contra: Antidotes for novel anticoagulants? Do we really need the. Thromb Haemost. 2012;108:623–624. doi: 10.1160/TH12-05-0298. [DOI] [PubMed] [Google Scholar]
- 7.Dickneite G, Hoffman M. Reversing the new oral anticoagulants with prothrombin complex concentrates (PCCs): what is the evidence. Thromb Haemost. 2014;111:189–198. doi: 10.1160/TH13-05-0431. [DOI] [PubMed] [Google Scholar]
- 8.Schiele F, van Ryn J, Canada K et al. A specific antidote for dabigatran: functional and structural characterization. Blood. 2013;121:3554–3562. doi: 10.1182/blood-2012-11-468207. [DOI] [PubMed] [Google Scholar]
- 9.http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm467300.htmAccessed March 9, 2016.
- 10.Lu G, DeGuzman FR, Hollenbach SJ et al. A novel antidote for reversal of anticoagulation by direct and indirect inhibitors of coagulation factor Xa. Nat Med. 2013;19:446–451. doi: 10.1038/nm.3102. [DOI] [PubMed] [Google Scholar]
- 11.Pollack CV, Reilly PA, Eikelboom J et al. Idarucizumab for dabigatran reversal. N Engl J Med. 2015;373:511–520. doi: 10.1056/NEJMoa1502000. [DOI] [PubMed] [Google Scholar]
- 12.Siegal DM, Curnutte JT, Connolly SJ et al. Andexanet alfa for the reversal of factor Xa inhibitor activity. N Engl J Med. 2015;373:2413–2424. doi: 10.1056/NEJMoa1510991. [DOI] [PubMed] [Google Scholar]
- 13.Ansell J. Blocking Bleeding: Reversing Anticoagulant Therap. Nat Med. 2013;19:402–404. doi: 10.1038/nm.3157. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Online Supplementary Material (PDF)
Online Zusatzmaterial (PDF)