

Brown-Vialetto-Van Laere syndrome (BVVL) is a rare, progressive neurodegenerative disease with fewer than 100 cases reported in the literature. It is characterized by pontobulbar palsy and sensorineural hearing loss.1 The age at onset varies from infancy to early adulthood, commonly presenting with cranial nerve VII–XII palsies and deafness. Other findings include gait ataxia, limb weakness, optic atrophy, epilepsy, and respiratory compromise.1 The genetic etiology of BVVL has recently been linked to mutations affecting the riboflavin transporter genes SLC52A2 and SLC52A3, which code for human riboflavin transporters RFVT2 and RFVT3, respectively.2 Consequentially, several studies have reported improved clinical outcomes with riboflavin supplementation in patients confirmed to have mutations in the SLC52A2 and SLC52A3 genes.3–7 In this report, we describe a case of BVVL in a 6-year-old girl with dramatic visual recovery and neurologic improvement after delayed initiation of riboflavin supplementation. Consent was obtained from the child's parents.
Case report
A 6-year-old girl was seen in the pediatric ophthalmology clinic after failing a vision screening test. Her family reported increasing clumsiness over the past year, resulting in frequent falls. On examination, her best-corrected visual acuity was 20/150 in each eye monocularly and 20/100 binocularly. She also was noted to have a small-angle exophoria with decreased stereopsis. The remainder of her ophthalmologic examination was unremarkable.
Visual evoked potential (VEP) testing demonstrated reduced amplitudes (∼60% of normal) and prolonged latencies (∼40 ms increased) on flash VEP and reduced acuity on spatial-sweep VEP testing. MRI of the patient’s brain and orbits revealed no intracranial abnormalities, but the optic nerves and tracts appeared mildly decreased in size. Based on these findings, the presumed diagnosis was bilateral optic nerve hypoplasia.
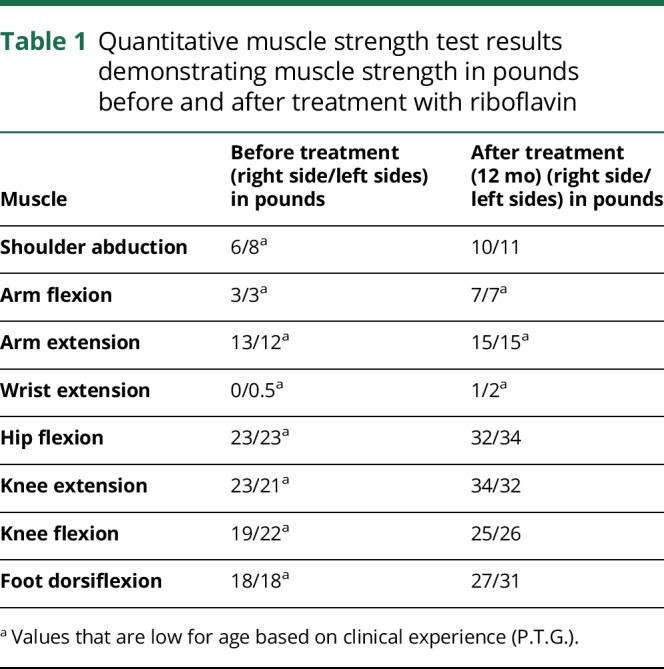
Approximately 13 months after initial presentation, the patient presented to the emergency department with progressive worsening in fine and gross motor skills. Physical examination demonstrated decreased visual acuity of 20/400 bilaterally and horizontal nystagmus in primary gaze that dampened with directional gaze. Sensory testing was normal and strength testing (table 1) was decreased for age with +1 tendon reflexes. She had bilateral dysmetria and prominent ataxia. She was born to healthy, nonconsanguineous parents without relevant family history, and had one healthy 4-year-old brother.
Table 1.
Quantitative muscle strength test results demonstrating muscle strength in pounds before and after treatment with riboflavin
Laboratory testing included normal thyroid function testing, serum lactate and pyruvate ratio, and serum ammonia levels. Total plasma carnitine was slightly low for age at 34 μmol/L. Quantitative serum acylcarnitine profile was normal. Serum amino acid profile exhibited nonspecific abnormalities including mildly low glutamine and arginine levels and slightly elevated proline and α-aminobutyric acid levels. CSF studies were normal with the exception of low homovanillic acid and 5-hydroxyindoleacetic acid. Chromosomal microarray was normal and genetic testing for Friedreich ataxia was negative.
EEG lacked a well-defined posterior dominant rhythm suggestive of mild generalized cerebral dysfunction. Nerve conduction and EMG showed small median compound muscle action potentials (CMAPs) but normal peroneal CMAPs. Sensory testing revealed absent median nerve action potentials from digit 2 and small for age sural nerve action potentials. These findings suggested a non-length-dependent axonal peripheral neuropathy or brachial plexopathy (table 2). The clinical presentation of optic atrophy, disproportionate upper extremity weakness, and ataxia, combined with the nerve conduction study findings suggesting an axonal peripheral neuropathy, raised suspicion for BVVL syndrome. Repeat MRI brain imaging did not demonstrate any change. Based on successful therapeutic interventions from previous reports, empiric treatment of 300 mg riboflavin (30 mg/kg) 3 times a day and coenzyme Q10 supplementation was initiated at that time, 16 months after the patient’s first presentation to the ophthalmologist with vision loss.1 When the patient demonstrated clinical improvement, riboflavin was increased to 500 mg (50 mg/kg/d) 3 times a day.
Table 2.
Nerve conduction and EMG study results showing evidence of non-length-dependent axonal neuropathy

The clinical diagnosis of BVVL was subsequently confirmed by whole-exome sequencing, which identified compound heterozygous variants in SLC52A2. The first variant is paternally inherited and designated as c.245G>C:p.Arg82Pro. The second is a maternally inherited variant designated as c.1140delG:p.Leu381CysfsX9, which creates a frameshift starting with codon leucine 481 to cysteine, and results in a premature stop codon at position 9 of the new reading frame.
Ocular examination 18 months after initiation of riboflavin showed improvement in visual acuity to 20/20 bilaterally and complete resolution of nystagmus. Flash VEP testing demonstrated improved amplitudes (∼82%, right eye; ∼74%, left eye) and latencies in both eyes. Neurologic examination demonstrated improved coordination, gait, and muscle strength. Muscle strength measurements performed pretreatment and after 12 months of riboflavin treatment are shown in table 1. A formal hearing study showed no impairment.
Of note, subsequent targeted genetic testing also confirmed the same compound heterozygous variants in the patient's 4-year-old brother who exhibited mild symptoms including tremor when holding heavy objects and occasional choking episodes. At 2 years of age, he was found to have a significant hyperopic refractive error (∼+7.0 diopters) causing a 30-prism-diopter refractive esotropia and strabismic amblyopia in the left eye. Prescription glasses improved the esotropia, and he began atropine penalization treatment for amblyopia. A preferential look test performed at age 3 showed mild residual amblyopia of the left eye, and he was switched to patching therapy for treatment of amblyopia. After a thorough discussion with the family regarding the progressive nature of BVVL, the younger sibling was initiated on 200 mg (30 mg/kg/d) of riboflavin taken 3 times daily and 100 mg of CoQ10 taken twice daily. He has remained asymptomatic. At his last assessment at age 6, his minor neurologic abnormalities had resolved and no additional findings had appeared. His hearing remained normal and his ophthalmologic findings were stable, with flash VEP testing showing mild reduction of amplitude suggestive of early, asymptomatic BVVL syndrome.
Discussion
BVVL syndrome is a treatable genetic condition caused by mutations affecting the genes that code for riboflavin transporters. Riboflavin is absorbed in the small intestine by human riboflavin transporters RFVT1 and RFVT3, and is exchanged in the brain via a third transporter, RFVT2. Riboflavin is the precursor for essential flavo-coenzymes (flavin mononucleotide [FMN] and flavin adenine dinucleotide [FAD]). FMN and FAD are important coenzymes that catalyze the various electron transfer reactions involved in fatty acid oxidation. Interestingly, recent work has identified mutations in the genes SLC52A2 (coding for RFVT2) and SLC52A3 (coding for RFVT3) as the cause of many cases of BVVL syndrome.2,3 As a result of this discovery, treatment with high-dose riboflavin supplementation has significantly improved morbidity and mortality rates in a condition that was previously often fatal due to respiratory failure. Furthermore, Foley et al. demonstrated an increase in riboflavin levels in 9 out of the 10 patients assessed before and after initiation of riboflavin therapy.4
A review of the literature revealed 47 cases where riboflavin treatment was initiated; however, only 38 reports documented the clinical treatment response (24 RFVT2 cases; 14 RFVT3 cases).5–9 Clinical improvement was reported in 28 patients (74%) and stabilization without progression was documented in the remaining 10 patients (26%).
In regards to the effect of riboflavin treatment on optic atrophy, only 3 of the 17 cases with optic atrophy provided documentation on specific visual function change after treatment.6,7,9 A previously reported case of a 2-year-old girl with the SLC52A2 mutation showed visual acuity improvement to 20/300 after 6 months of riboflavin treatment.9 Mild visual improvement after initiating treatment was also seen in 2 patients with SLC52A2 mutations (11-year-old girl; 2-year-old boy).4 In this report, we describe a 6-year-old girl who exhibited remarkable visual recovery from 20/400 to 20/20, as well as improvement on VEP studies. To our knowledge, this could be one of the most dramatic recoveries in visual function reported after treatment with riboflavin.
The magnitude of visual recovery raises many questions regarding the downstream cellular effect of the SLC52A2 and SLC52A3 mutations. Variable neurohistopathologic findings of neuronal loss, myelinated fiber loss, and gliosis in the brainstem cranial nerve nuclei and anterior horns of the spinal cord with accompanying nerve root atrophy were reported in 2 patients with genetically confirmed SLC52A3 gene mutations who were not treated with supplemental riboflavin.10 Given the magnitude of visual improvement exhibited in the present case, the mutation in BVVL syndrome likely does not result in an isolated axonal loss. We hypothesize a combined axonal neuropathy with coinciding reactive demyelination as a result of microglia and macrophage infiltration. However, initiation of riboflavin supplementation may halt further axonal death and reduce additional microglia- and macrophage-induced inflammation. Consequently, areas with myelin loss may undergo remyelination, producing a recovery in neuronal function as exhibited in our patient.
It is critical to consider BVVL syndrome in the differential diagnosis in patients presenting with cranial neuropathies, balance difficulties, and sensorineural hearing loss. Treatment with riboflavin supplementation should be initiated immediately while awaiting genetic confirmation. In addition, extensive genetic counseling should be provided to affected families, and genetic screening for at-risk family members should be recommended to identify any asymptomatic siblings with positive molecular results who may benefit from prophylactic riboflavin treatment.
Glossary
- BVVL
Brown-Vialetto-Van Laere syndrome
- CMAP
compound muscle action potential
- FAD
flavin adenine dinucleotide
- FMN
flavin mononucleotide
- VEP
visual evoked potential
Author contributions
A.K. Bamaga: data collection, literature review, writing the manuscript. R.N. Maamari: data collection, literature review, writing the manuscript. S.M. Culican: data interpretation, critical revision of the manuscript. M. Shinawi: data interpretation, critical revision of the manuscript. P.T. Golumbek, data interpretation, critical revision of the manuscript.
Study funding
No targeted funding reported.
Disclosure
The authors report no disclosures relevant to the manuscript. Go to Neurology.org/N for full disclosures.
References
- 1.Jaeger B, Bosch AM. Clinical presentation and outcome of riboflavin transporter deficiency: mini review after five years of experience. J Inherit Metab Dis 2016;39:559–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Green P, Wiseman M, Crow YJ, et al. Brown-Vialetto-Van Laere syndrome, a ponto-bulbar palsy with deafness, is caused by mutations in C20orf54. Am J Hum Genet 2010;86:485–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnson JO, Gibbs JR, Megarbane A, et al. Exome sequencing reveals riboflavin transporter mutations as a cause of motor neuron disease. Brain 2012;135:2875–2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Foley AR, Menezes MP, Pandraud A, et al. Treatable childhood neuronopathy caused by mutations in riboflavin transporter RFVT2. Brain 2014;137:44–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bosch AM, Abeling NGG, Ijlst L, et al. Brown-Vialetto-Van Laere and Fazio Londe syndrome is associated with a ribo-flavin transporter defect mimicking mild MADD: a new in-born error of metabolism with potential treatment. J Inherit Metab Dis 2011;34:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haack TB, Makowski C, Yao Y, et al. Impaired riboflavin transport due to missense mutations in SLC52A2 causes Brown-Vialetto-VanLaere syndrome. J Inherit Metab Dis 2012;35:943–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cosgrove J, Datta S, Busby M. Adult onset Brown-Vialetto-VanLaere syndrome with opsoclonus and a novel heterozygous mutation: a case report. Clin Neurol Neurosurg 2015;128:1–3. [DOI] [PubMed] [Google Scholar]
- 8.Davis A, Josifova D, Lloyd-Owen S, et al. Brown-Vialetto-VanLaere syndrome: a 28-year follow-up. J Neurol Neurosurg Psychiatry 2016;87:681–682. [DOI] [PubMed] [Google Scholar]
- 9.Shashi V, Petrovski S, Schoch K, et al. Sustained therapeutic response to riboflavin in a child with a progressive neurological condition, diagnosed by whole-exome sequencing. Cold Spring Harb Mol Case Stud 2015;1:a000265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Manole A, Jaunmuktane Z, Hargreaves I, et al. Clinical, pathological and functional characterization of riboflavin-responsive neuropathy. Brain 2017;140:2820–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]