

Abstract
The patient-derived xenograft (PDX) model has been adopted as a major tool for studying tumorigenesis and differentiation in various carcinomas. In addition, it has been used in the development of anticancer agents. PDX models have been among the most meaningful tools used to understand the role of stromal cells and vascular cells in the body, which are major factors in cancer development and the application of therapeutic agents. Also, the establishment of PDX models from liposarcoma patients is considered to be important for understanding lipomagenesis and following drugs development. For these reasons, we developed patient-derived cell (PDC) and PDX models derived from 20 liposarcoma patients. The tissues of these patients were obtained in accordance with the principles of the Samsung Medical Center's ethics policy, and cell culture and xenografting onto the mice were performed under these principles. High-throughput drug screening (HTS) was carried out using established PDCs to select candidate drugs. Among the different candidate anticancer drugs, we tested the effect of bortezomib, which was expected to inhibit MDM2 amplification. First, we confirmed that the PDCs maintained the characteristics of liposarcoma cells by assessing MDM2 amplification and CDK4 overexpression using fluorescence in situ hybridization. Analysis of short tandem repeats and an array using comparative genomic hybridization confirmed that the PDX model exhibited the same genomic profile as that of the patient. Immunohistochemistry for MDM2 and CDK4 showed that the overexpression patterns of both proteins were similar in the PDX models and the PDCs. Specifically, MDM2 amplification was observed to be significantly correlated with the successful establishment of PDX mouse models. However, CDK4 expression did not show such a correlation. Of the anticancer drugs selected through HTS, bortezomib showed a strong anticancer effect against PDC. In addition, we observed that bortezomib suppressed MDM2 expression in a dose-dependent manner. In contrast, p21 tended to elicit an increase in PDC expression. Treatment of the PDX model with bortezomib resulted in an anticancer effect similar to that seen in the PDCs. These results support that PDCs and PDX models are among the most powerful tools for the development and clinical application of anticancer drugs for the treatment of liposarcoma patients.
Introduction
Patient-derived xenograft (PDX) models are becoming a common platform for research and clinical purposes [1]. The establishment of PDX models to study cancer biology and pharmacology is a common practice that has been successfully applied across many types of cancer [1], [2]. Engrafting human primary carcinomas is, in fact, the only method currently available that permits the propagation of a significant proportion of carcinomas [3], [4], [5], and has many advantages over tumor-derived cell lines maintained in vitro [6]. Both cell lines and PDX models permit the removal of non-neoplastic human cells from the human tumors. However, the surrounding tissue architecture is only partially maintained in PDXs [7], [8], with mouse stromal cells substituting for human stromal cells. In general, the results obtained using PDX models in mice show better preclinical and clinical concordance than those from cell lines [9].
Liposarcoma is a common type of soft tissue sarcoma. It accounts for 10% of pediatric sarcoma patients and 8% of adolescent and adult sarcoma patients [10]. Although the primary treatment mainly consists of a surgical resection, if a relapse or a metastatic event has occurred owing to an incomplete surgical resection, malignant tumors may occur. While chemotherapy is a useful therapy for malignant liposarcoma patients [11], no major agent specific to liposarcoma treatment has been discovered yet. Therefore, the development of new treatment strategies is necessary. Current first-line chemotherapy treatments include the single-agent use of anthracyclines (doxorubicin) or an anthracycline-based combination product (doxorubicin and ifosfamide or dacarbazine). Second-line treatment involves nonanthracycline combination treatment (gemcitabine and docetaxel) [12]. Recently, flavopiridol has been suggested as a potent alternative adjuvant agent for liposarcoma treatment [13].
Bortezomib (VELCADE; Millennium Pharmaceuticals Inc., Cambridge, MA, and Johnson & Johnson Pharmaceutical Research and Development, LLC, Raritan, NJ) is the first proteasome inhibitor to be approved and investigated in clinical trials. After phase II and III studies, it was approved for use in the United States and Europe for treating multiple myeloma patients who have received at least one prior therapy, and is currently being investigated in other hematological malignancies and solid tumors. Proteasome inhibition affects the ubiquitin-proteasome pathway, disrupting protein homeostasis and resulting in cell-cycle disruption, inhibition of transcription factors such as nuclear factor kappa-B, and antiangiogenic effects, which ultimately affect tumor growth and proliferation and result in apoptosis [14], [15].
In this study, we established PDC and PDX models from liposarcoma patients. Also, we attempted to evaluate whether these models could be useful tools for investigating anticancer drugs for treatment of liposarcoma patients.
Materials and Methods
Biospecimen Collection and Ethical Approval
Anonymized tumor tissues from patients 19 to 90 years old undergoing surgery or diagnostic core biopsy were collected with informed consent according to procedures approved by the Internal Review Board of the Samsung Medical Center (SMC). Patients at the SMC were recruited, and samples were collected as part of the Sarcoma study at SMC Hospital, Seoul, Korea (IRB no. 2013-07-122).
Tissue Processing
Primary surgical and core biopsy tissue samples were transported from the operating room on ice in cold Dulbecco's modified Eagle’s medium (DMEM) (Hyclone Technologies, South Logan, UT). A small piece of tumor tissue was obtained using a scalpel and fixed in 10% formalin-buffered saline (Fisher Scientific, Kalamazoo, MI) for histological analysis. Additional small fragments from different portions of the tissue were collected together, flash frozen in liquid nitrogen, and stored at −80°C for nucleic acid extraction. The remaining tissue was minced finely with scalpels and then mechanically disaggregated for 1 minute using a Stomacher 80 Biomaster (Seward Limited, Worthing, UK) in 1-2 ml cold DMEM. Aliquots from the resulting suspension of cells and organoids were used for xenotransplants (Figure 1).
Figure 1.

Schematic of the strategy for personalized treatment of liposarcoma patients.
Blood samples and tumor tissues were obtained from patients for primary cell culture, and a patient-derived tumor xenograft mice model was established. Isolated patient-derived tumor cells and tissues were analyzed using histology and genomic profiling, and used for drug screening.
Generation of the PDC Model
The tumor material was excised aseptically and then processed for primary tissue as described previously [16]. Briefly, one primary tissue sample (SA500) was enzymatically dissociated to single cells by sequential incubation in warm (37°C) collagenase (300 U/ml, Sigma-Aldrich) plus hyaluronidase (100 U/ml, Sigma) for 2.5 hours, 0.25% trypsin/EDTA (STEMCELL Technologies) for 4 minutes, and dispase (5 U/mL, STEMCELL Technologies) plus DNaseI (100 μg/ml, Sigma-Aldrich) for 4 minutes before being passed through a 40-μm filter.
Cell Culture
Dedifferentiated human liposarcoma cell lines LPS246 and LPS863 were provided by Dr. Dina Lev (MD Anderson Cancer Center). The patient-derived cells (PDCs) 11-013, 11-018, 11-079, 11-099, 11-106, 14-026, 14-032, 14-035, 14-076, 14-078, 15-023 were established by investigators at the Sarcoma Research Center at the Samsung Medical Center in Korea as described above. The isolation of MSCs from human tissues has been described previously [17]. The cells were maintained in DMEM (HyClone, South Logan, UT) supplemented with 10% fetal bovine serum.
PDC Identity Validation by Cytogenetics
PDCs from patients were subjected to chromosome analysis for validation by karyotyping and FISH. In brief, cells were incubated in 0.0075 M KCl hypotonic solution and fixed with a mixture of methanol and glacial acetic acid (1:3). G banding was performed 2× saline sodium citrate buffer for 2 minutes at 68°C, and Wright's solution staining was performed for 2 minutes. Metaphase images were captured by an Olympus BX61 microscope (Olympus Corporation, Tokyo, Japan) and analyzed using CytoVison software (Leica Biosystems Ltd., Newcastle, UK). Average banding was measured around 300 resolutions. For FISH analysis, a probe cocktail containing an MDM2-labeled, chromosome-specific painting probe was denatured and then hybridized to denatured tumor metaphase chromosomes using a Human Multicolor FISH kit (Meta Biosystem) according to the manufacturer's protocols. Briefly, slides were washed using posthybridization buffer, dehydrated through a series of ethanol incubations, and counterstained with 10 μl of DAPI. The signal detection and analysis were performed using a Metafer system and Metasystem's ISIS software (software for spectral karyotype).
Cell Viability
CCK8 (Dojindo, Kyoto, Japan) was used as a biomarker to measure the relative number of live cells in a culture population after experimental manipulation. All procedures were performed according to manufacturer's instructions. Briefly, cells seeded in wells of an opaque 96-well microtiter plate (5 × 103 cells/well) and grown in DMEM were incubated with PBS (as a control group) or each drug for the indicated time periods. Plates were incubated under standard cell culture conditions. Tests were performed in triplicate; three wells per plate with medium only served as blanks, and two wells with untreated cells served as controls. The plate was loaded into a dispensing fluorescence detector (Multiplate Reader, Mithras2, Berthold, Bad, Wildbad, Germany), and the relative light units at 340-380 nm/505 nm (CCK8: 450 nm) were measured. Statistical analysis of the results was performed using an unpaired Student's t test.
PDC-Based Chemical Screening and Analysis
PDCs grown in serum-free medium were seeded in 384-well plates at a density of 500 cells per well in duplicate for each treatment. The drug panel consisted of 60 anticancer agents (Selleckchem) targeting oncogenic signals. Two hours after plating, PDCs were treated with drugs in a four-fold and seven-point serial dilution series from 20 μM to 4.88 nM using a Janus Automated Workstation (PerkinElmer, Waltham, MA). After 6 days of incubation at 37°C in a 5% CO2 humidified incubator, cell viability was analyzed using an adenosine triphosphate monitoring system based on firefly luciferase (ATPLite 1step, PerkinElmer). Viable cells were estimated using an EnVision Multilabel Reader (PerkinElmer). During in vitro drug screening, control wells containing only cells and the dimethyl sulfoxide (DMSO) vehicle were included on each assay plate. These controls were used to calculate the relative cell viability for each plate and to normalize the data on a per-plate basis. Drug response curve (DRC) fitting was performed using GraphPad Prism 6 (GraphPad) and evaluated by measuring the half maximal inhibitory concentration (IC50) and area under the curve (AUC). In brief, each plate was normalized to the mean of the seven conditions on the plate with a DMSO control. After normalization, best-fit lines and the resulting IC50s were calculated using GraphPad: [log(inhibitor) vs. response – variable slope (four parameters)], Y = bottom + (top – bottom) / (1 + 10^ ((LogIC50-X)*HillSlope)). The AUC of each curve was determined using GraphPad Prism, ignoring regions defined by fewer than two peaks.
Western Blotting
Western blot analyses were performed as previously described [18], [19]. Briefly, cells were washed twice with PBS and then lysed via sonication in lysis buffer (Intron, Seoul, Korea). The samples were separated on 6%-15% SDS-PAGE gels and then transferred to nitrocellulose membranes (Protran BA83; Whatman). Immunoblotting was performed using the following primary antibodies and dilutions: anti–p-Akt (1:2000; Cell Signaling), anti–p-Akt (phospho Ser432) (1:1000; Cell Signaling), anti-mTor (1:2000; Cell Signaling), anti–p-mTor (1:1000; Cell Signaling), and anti–beta-actin (1:10000; Sigma, St. Louis, MO). Horseradish peroxidase–labeled rabbit anti-mouse (Abcam, diluted 1:5000) and goat anti-rabbit (Santa Cruz, diluted 1:2000) secondary antibodies were used. The proteins were visualized using an ECL detection system (Ab Frontier, Seoul, Korea).
Xenografting
Animals
NOD.Cg-PrkdcscidIl2rgtmlWjl/Sz (NOD-SCID IL2rγnull; NSG, Jackson Laboratories) mice were obtained from the DNA Link. The mice were maintained under specific pathogen-free conditions in accordance with the ethical guidelines for the care of these mice at the Laboratory Animal Research Center of the Samsung Biomedical Research Institute. The mice were 6-8 weeks of age at the time of transplant.
Subcutaneous transplants
Disaggregated cells and organoids were resuspended in 100-200 μl of a 1:1 v/v mixture of cold DMEM:Matrigel (BD Biosciences, San Jose, CA) and kept on ice until transplantation. For transplants of intact cell fragments, a small incision was made in the skin in the flank area, and then fragments were inserted using forceps. Xenograft-bearing mice were euthanized when the size of the tumors approached 1 ml in volume (adding together the sizes of individual growths when more than one was present). Serially transplanted aliquots represented ~0.1%-0.3% of the xenograft tumor volume.
Histology Experiment
The histological features of patients' tumor tissues stained with hematoxylin and eosin were analyzed for validation and comparison with tumor tissues from the PDX model. Immunohistochemistry (IHC) was performed using a previously published method (28). Slides were incubated with anti-CDK4 (Invitrogen, Waltham, MA) or anti-MDM2 (Thermo Fisher Scientific) overnight. After incubation, immunodetection was performed with the EnVision (DAKO, Glostrup, Denmark) visualization system according to manufacturer's instruction.
Short Tandem Repeat (STR) Analysis
To verify the PDX tumor tissues derived from each patients, an STR analysis were performed on different chromosomes at 16 loci [20]. Target DNA (10 ng) was amplified by multiplex polymerase chain reaction (PCR) using fluorescent dye-linked primers for the 16 loci: 13 autosomal STR loci (CSF1PO, D3S1358, D5S818, D7S820, D8S1179, D13S317, D16S539, D18S51, D21S11, FGA, TH01, TPOX, and vWA), 2 additional STR loci (D2S1338 and D19S433), and the amelogenin locus. Amplification was performed using an AmpFlSTR Identifiler PCR Amplification Kit (Applied Biosystems, Foster City, CA) according to the manufacturer's instructions. PCR products generated were mixed with an internal size standard (GS-500 LIZ, Applied Biosystems), electrophoresed on an ABI 3130xL Genetic Analyzer (Applied Biosystems), and analyzed with the GeneMapper 4.0 software using the supplied allelic ladders (Applied Biosystems).
Array-Based Comparative Genomic Hybridization (CGH)
Array-based CGH was performed using an Agilent Human Whole Genome CGH 8 × 60 K microarray (Agilent Technologies). Labeling and hybridization were performed using protocols provided by the manufacturer (Cancer Sci. 2013; 104; 631-638). Briefly, 0.5 μg of test or reference DNA was digested with Alu I and Rsa I (Promega, Madison, WI) and purified with the QIAprep Spin Miniprep kit (QIAGEN). Test and reference DNA samples were labeled by random priming with either Cy3-dUTP or Cy5-dUTP using the Agilent Genomic DNA Labeling Kit PLUS (Agilent Technologies). Following the labeling reaction, individually labeled test and reference samples were combined and concentrated using Amicon Ultra-0.5 centrifugal filters (Millipore, Billerica, MA). After denaturing the probe and preannealing it to human Cot-1 DNA, samples were hybridized at 65°C and 20 rpm rotation for 24 hours in a DNA Microarray Hybridization Oven (Agilent Technologies). Samples were washed in wash buffer 1 at room temperature for 5 minutes and wash buffer 2 at 37°C for 1 minute using Agilent Oligo CGH washes. All slides were scanned on an Agilent DNA microarray scanner. Data were obtained using Agilent Feature Extraction Software 9 and analyzed with Agilent CGH Analytics Version 6.5 software using the ADM-2 statistical algorithms with 6.0 sensitivity thresholds.
Bortezomib Treatment
For bortezomib treatment, each tumor sample was engrafted subcutaneously into mice. When tumors reached 1000 mm3, they were harvested and cut into small pieces (~1×1×1-mm blocks) and implanted to other mice. To avoid biases at the start of treatment, tumor-bearing mice having similar characteristics (tumor volume, tumor growth rate, and mouse weight) were distributed evenly between the bortezomib-treated and vehicle-treated (NaCl) groups (Supplementary Figure 3). Once the tumor grew up to 100 mm3, mice were treated for 4 weeks (21 days). Tumor volumes were calculated according to the following formula: (l × w × h)/6π, where l is maximal length, w is maximal width perpendicular to l, and h is maximal height.
Supplementary Figure 3.

Tumor volume of liposarcoma patient-derived xenograft before drug treatments.
Mice were randomized and assigned to treatment groups. To avoid biases derived from excessively weighing any particular dimension, tumor volumes were calculated according to a set formula, (l × w × h)/6π, and mice were distributed evenly among groups according to tumor volume. Statistical analyses showed no biases at the start of drug trials.
Statistical Methods
Clinical variables were compared between successful and failed xenografts with χ2 and Fisher's exact tests. Clinical variables were compared among xenografts (P1 to P6) using the Kruskal-Wallis test. All statistical analyses were performed using GraphpadVer 5.0.
Results
Establishment of PDC and PDX Models
Between September 2011 and January 2015, 20 tumors [well-differentiated liposarcomas (WDLPSs) 5, dedifferentiated liposarcomas (DDLPSs) 15] were implanted in mice. Eligibility criteria were based on Fédération Nationale des Centres de Lutte Contre Le Cancer (FNCLCC) grading and included tumor size, tumor location, primary or recurrent tumors, and histological positivity for MDM2/CDK4 (Table 1). All tumors were liposarcomas, and 85% were DDLPSs. Histological examination showed that both CDK4 and MDM2 were detected in 65% of liposarcomas taken from patients (Table 1). Over 70% of LPSs exhibited MDM2 amplification (Table 1, Figure 2), and tumors were found in retroperitoneal area approximately 65% of the time (Table 1). Of these, we were able to culture primary cells from all patients. The expression of MDM2 in the primary cell line was also analyzed by FISH. As a result, it was confirmed that the expression of the MDM2 gene was maintained in all cells, as well as the tumor tissues obtained from the patients. To examine the efficacy of tumor xenograft in our mice, we examined the correlations between xenograft success and the factors listed in Table 1. The results of FISH showed that MDM2 expression was significantly positively correlated with tumor implantation success (Figure 3). In addition, we found that the tumor grade, classified by FNCLCC, was also correlated with successful tumor tissue grafting (Supplementary Table 1). On the other hand, there was no significant correlation between CDK4, a major liposarcoma marker, expression and tumor tissue xenograft success (Figure 3). These results show that amplification of MDM2 is not only an important factor affecting the malignancy and progression of liposarcoma [21], [22], [23] but also a crucial factor for xenograft efficiency in mice.
Table 1.
Clinical and Preclinical Information of Liposarcoma PDCs
| Clinical Information |
Experimental Information |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient ID | Sex | Age | SMC |
FNCLCC |
Primary |
Tumor |
Tumor |
MDM2 |
CDK4 |
Ki67 |
Cell |
MDM2 |
In vivo |
PDX |
|
| Dx | Grading | Recurrent | Size (cm) | Location | Histology | Histology | (%) | Passage | FISH | Tumor engraft | Passage | ||||
| 1 | 11GS-013 | F | 54 | DDLPS | 3/3 | 2nd recur | 49×30×11 | Retroperitoneum | Positive | Positive | 10 | p.36 | Positive (100) | 3/3 | G1 |
| 2 | 11GS-018 | M | 75 | DDLPS | 2/3 | primary | 39×34×15 | Retroperitoneum | - | Positive | 30 | p.19 | Positive (100) | 3/3 | G1 |
| 3 | 11GS-079 | M | 71 | DDLPS | 3/3 | 1st recur | 14×13×13 | Kidney and colon | - | Positive | 20 | p.102 | Positive (100) | 5/5 | G1 |
| 4 | 11GS-099 | M | 66 | DDLPS | - | 1st recur | 7×6×5.5 | Kidney Retroperitoneum | Positive | Positive | - | P.59 | Positive (100) | 5/5 | G1 |
| 5 | 11GS-106 | F | 77 | DDLPS | 3/3 | Primary | 18×14×11 9×9×3 |
Kidney | Positive | Positive | 15 | p.43 | Positive (100) | - | - |
| 6 | 14GS-022 | M | 66 | DDLPS | 2/3 | 2nd recur | 8×7 | Retroperitoneum | Positive | Positive | - | p.6 | N.D | 2/8 | G2 |
| 7 | 14GS-026 | F | 77 | DDLPS | 3/3 | 1st recur | 5.7×3×2.5 | back | Positive | Positive | - | p.47 | Positive (100) | 3/3 | G1 |
| 8 | 14GS-032 | F | 49 | DDLPS | 2/3 | Primary | 18×15×10 | Retroperitoneum | Positive | Positive | - | p.27 | Positive(100) | 21/22 | G3 |
| 9 | 14GS-034 | M | 35 | WDLPS | 2/3 | Primary | 42×33×16 | Retroperitoneum | Positive | Positive | - | p.13 | N.D | - | - |
| 10 | 14GS-035 | M | 46 | DDLPS | 2/3 | Primary | 34×24×6 | Intraabdominal and kidney | Positive | Positive | - | p.15 | Positive (100) | - | - |
| 11 | 14GS-054 | F | 52 | DDLPS | 2/3 | 4th recur | 9×7×5 | Retroperitoneum | Positive | Positive | - | p.11 | Positive (37.5) | - | - |
| 12 | 14GS-057 | F | 65 | WDLPS | 1/3 | 3rd recur | 5×4×4 | Retroperitoneum pancreas | Positive | Positive | - | p.9 | Positive (19) | - | - |
| 13 | 14GS-076 | F | 52 | DDLPS | 2/3 | 3rd recur | 16×14×11 11×7×6 |
Small bowel | Positive | Positive | 60 | P27 | Positive (100) | 33/34 | G3 |
| 14 | 14GS-078 | M | 76 | WDLPS | 1/3 | Primary | 18×17×5 | Retroperitoneum | Positive | Positive | - | P9 | Positive (100) | - | - |
| 15 | 14GS-079 | M | 77 | WDLPS | 1/3 | Primary | 30×26×8 5.5×4×2.5 |
Perirenal mass, Retroperitoneum | Positive | Positive | - | P12 | Positive (6) | - | - |
| 16 | 14GS-084 | F | 59 | DDLPS | 2/3 | Primary | 20×19×8.5 | Retroperitoneum | - | - | P17 | N.D | 23/23 | G5 | |
| 17 | 15GS-002 | F | 61 | DDLPS | 3/3 | Primary | 30×16 | Retroperitoneum | Positive | - | 35 | P11 | N.D | 19/19 | G3 |
| 18 | 15GS-006 | M | 67 | DDLPS | 1/3 | 4th recur | 6×6 5×4.2 | Peristomach Kidney | - | - | - | P7 | N.D | - | - |
| 19 | 15GS-022 | M | 56 | WDLPS | 1/3 | 1st recur | 9.8×5×3.5 | Retroperitoneum | - | - | - | P6 | N.D | - | - |
| 20 | 15GS-023 | F | 61 | DDLPS | 3/3 | Primary | 15×8×6 | Retroperitoneum | Postivie | - | 10 | P7 | Positive (76.5) | 15/15 | G2 |
Figure 2.

Characterization of liposarcoma PDCs
Phenotype characterization of liposarcoma primary cells derived from patients. Phase contrast micrographs of the PDC model. FISH imaging showing MDM2 amplification. Representative Q-banded metaphase. Chromosome showing typical karyotypes of liposarcoma cells.
Figure 3.

Effects of CDK4 overexpression and MDM2 amplification on tumor xenograft.
MDM2 and CDK4 levels were examined for correlation of expression between patient tumors and tumor xenograft models. We analyzed the results statistically using an ANOVA. MDM2 (FISH) high: MDM2 positivity 76.5%-100%; MDM2 (FISH) low: MDM2 (FISH) positivity <37.5%; MDM2 (FISH) negative: N.D. CDK4 (+): histology (IHC +); CDK4 (−): histology (IHC −). Data shown represent the mean ± SEM (**P = .0097).
We then maintained the established PDCs and animals under the following conditions for further analysis. To establish the PDC model, we enzymatically dissociated primary tumor tissues to single cells and sequentially seeded them in tissue culture dishes. For maintenance of the primary cells, we changed with fresh media once every 3 days. Tumor fragments (2-3 mm in diameter) were implanted orthotopically, under the subcutaneous tissue, into two to five NSG mice (Supplementary Figure 1A). To preserve tumor architecture and minimize confounding factors, samples were implanted without disaggregation or additives. The PDX tumors could be observed by palpation (Supplementary Figure 1).
Supplementary Figure 1.

Establishment of a PDX model using liposarcoma PDCs and histology of xenograft tumors.
(A) In vivo tumorigenesis from patient-derived tumor cells (11GS-013, 11GS-079, 11GS-099). A total of 2 × 107 cells were subcutaneously injected into immunodeficient mice, NOD/Shi-scid IL-2Rγnull. (B) Hematoxylin and eosin sections comparing primary tumors, orthotropic xenografts, subcutaneously injected xenografts, and xenografts implanted into the kidneys. Immunohistochemistry of tumor xenografts from mice using anti-CDK4 and anti-MDM2. Tumor features were preserved in the xenografts over the course of serial passages.
When tumors reached ~1500 mm3 in diameter, mice were humanely euthanized, and tumor tissues were prepared for serial xenografting. Euthanization was performed earlier if the mouse became sick or was getting old. Upon sacrifice of the mice, tumor diameters ranged from approximately 1000 to 1500 mm3 (Supplementary Figure 1A). Most tumors xenografts were DDLPSs. The efficiency of the tumor growth rate reached 94.2% for DDLPSs and 0% for WDLPSs at the initial passage. As determined by histologically examining recipient mice (PDX mouse generation 0, G0), 75% of grafts formed viable tumors in these mice (Table 3). The average latency period from the day of implantation in mice until passage into the first generation (G1) was highly variable, ranging from 3 to 8 months. The time was generally shorter for DDLPS tumors and became shorter with each sequential passage. At each passage, samples were fixed for histological analyses, and when sufficient material was available, additional samples were frozen separately for molecular studies (Figure 5, Supplementary Figure 1B).
Table 3.
In Vivo Tumorigenic Potential
| PDX Class | Tumor Growth Rate (%) |
|---|---|
| DD/WDLPS | |
| DDLPS PDX | 131/139 (94.2) |
| WDLPS PDX | 0/9 (0) |
| PDX Generation | |
| Primary PDX (G1) | 36/48 (75.0) |
| Secondary PDX (G2) | 55/59 (93.2) |
| Tertiary PDX (G3) | 34/35 (97.1) |
| Quaternary PDX (G4) | 3/3 (100) |
| Quinary PDX (G5) | 3/3 (100) |
| Total PDX | 131/148 (88.5) |
Figure 5.

Characterization of liposarcoma patient-derived xenograft.
(A) Representation of STRs in patient tumors and corresponding tumor xenografts. Each locus was selected from locations of human chromosomes. (B) Paired copy number analyses of patient tumors (parental) and patient-derived tumor xenografts (Xeno-G1, G2, G3). (C) IHCof patient and tumor xenografts was compared and evaluated for the expression of CDK4 and MDM2. Primary tumor (Xeno-G0), xenografted F1 tumor (Xeo-G1), xenografted F2 (Xeno-G2), and xenografted F3 (Xeno-G3).
High-Throughput Drug Screening (HTS) for Predicting drug response to PDC
Using a high-throughput culture system, we searched the antitumor activity of a 28-drug panel (Table 2, Figure 4A). Among them, bortezomib, panobinostat, AYU922, and ceritinib showed inhibitory effects on liposarcoma cell growth. First, we selected five kinds of drugs known to affect cell growth and the cell cycle to assess their inhibitory effects on liposarcoma proliferation: PKI-587 (a PI3K inhibitor), foretinib (a c-Met inhibitor), and bortezomib (a proteasome inhibitor). Also, since liposarcoma is characterized by overexpression of CDK4, we examined the inhibitory effects of the CDK4 inhibitor LY2835219 on liposarcoma growth [24]. Unexpectedly, our models showed resistance to the kinase inhibitor, but all cells were sensitive to bortezomib treatment, which significantly inhibited cell growth (Figure 4, B and C). Based on these initial results, we validated the antitumor effect of bortezomib, which showed an IC50 of less than 3.0 nM, in an in vitro cell viability assay (Figure 4C). In accordance with the preliminary results, we confirmed that most liposarcoma cells were highly sensitive to bortezomib (mean growth = 100.0% vs. 16.04% for control and bortezomib-treated cells, respectively; 95% CI = 70.0%-97.92%; P = .0435). However, despite the presence of CDK4 amplification in the patient-derived tumor cells, cells treated with LY2835219, the CDK4 inhibitor, did not significantly suppress cell proliferation when compared with control cells (mean growth = 100.0% vs. 80.23% for control and LY2835219-treated cells, respectively; 95% CI = 1.828%-37.72%; P = .377) (Figure 4B).
Table 2.
High-Throughput Drug Screening of LPS PDCs.
| Drug | Target | IC50 (nmol/l) |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 11GS_013 | 11GS_018 | 11GS_079 | 11GS_099 | 11GS_106 | 14GS_026 | 14GS_032 | 14GS_035 | 14GS_076 | 14GS_078 | LPS224 | LPS246 | FDA | ||
| Imatinib (Gleevec) | BCR/ABL | >20,000 | 7246 | 10,160 | 6693 | 7741 | 7247 | - | 9347 | 12,840 | 14,270 | 8141 | 11,310 | o |
| Dasatinib (BMS-354825) | BCR/ABL | 35 | - | - | - | - | - | - | 19 | 47 | 25 | 2798 | 5749 | o |
| Nilotinib (AMN-107) | BCR/ABL | 3089 | 2559 | 979 | 1977 | 2709 | 3018 | 2196 | 2506 | 1764 | 2250 | 2018 | 2300 | o |
| Sunitinib Malate (Sutent) | BCR/ABL | 2079 | 1277 | 1241 | 1017 | 1037 | 468 | 767 | 1564 | 2737 | 2356 | 1851 | 1230 | o |
| PD 0332991 (Palbociclib) | CDK4/6 | 6812 | 2866 | 598 | - | 2885 | 308 | 6939 | - | 8389 | - | 5146 | 4395 | o |
| LY2835219 | CDK4/6 | 1566 | 470 | 2720 | 2551 | 131 | 231 | 552 | 621 | 2148 | - | 444 | 171 | o |
| LEE011 | CDK4/6 | - | >20,000 | 119 | 170 | 11,250 | - | - | 438 | - | - | 25 | 1078 | x |
| BKM120 (NVP-BKM120) | PI3 | 1404 | 458 | 294 | 363 | 451 | 241 | 420 | 318 | 235 | 747 | 413 | 559 | x |
| AZD2014 | mTOR | 169 | 158 | 27 | 135 | 117 | 66 | 49 | 105 | 106 | 193 | 130 | 63 | x |
| Everolimus (RAD001) | mTOR | - | 7964 | 354 | 8513 | 1414 | 11,310 | 49 | - | 10,010 | - | - | 2258 | o |
| PF-05212384 (PKI-587) | PI3K & mTOR | 11 | 93 | 196 | 790 | 0 | - | - | 2 | - | 10 | 27 | 24 | x |
| BEZ235 | PI3K & mTOR | - | 5 | 12 | 46 | - | 11 | - | 21 | 1 | 14 | 48 | 32 | x |
| Bortezomib (Velcade) | Proteasome | - | - | - | 2 | 11 | - | 11 | - | - | 9 | - | 7 | o |
| Panobinostat (LBH589) | HDAC | 36 | 18 | 17 | 10 | 22 | 31 | 19 | 15 | 27 | 23 | 14 | 2 | o |
| Cabozantinib (XL184) | VEGFR2 | 2141 | 1429 | 2450 | 1131 | 5838 | 4310 | 2424 | 8892 | 1861 | 7233 | 1426 | 2190 | o |
| Crizotinib (PF-02341066) | c-Met,ALK | 3223 | 2538 | 761 | 766 | 1576 | 1539 | 641 | 3122 | 4897 | 2307 | 918 | 1042 | o |
| vandetanib | VEGFR | 7030 | 3966 | 3616 | 2233 | 2385 | 1756 | 2315 | 3207 | 6449 | 5667 | 2205 | 2430 | o |
| Axitinib | VEGFR1,FLT1 | 1178 | 935 | 462 | 433 | 888 | 1296 | - | 1372 | 1235 | 871 | 809 | 483 | o |
| Foretinib (XL880) | c-MET,KDR | 249 | 256 | 341 | 198 | 368 | 556 | 258 | 775 | 1220 | 357 | 137 | 162 | x |
| regorafenib | VEGFR,c-KIT | 3114 | 7626 | 3185 | - | 9472 | - | - | 6828 | 8788 | 5952 | 5790 | 4536 | o |
| Trametinib | MEK1 | 19 | 155 | 3 | 98 | 5 | 18 | 98 | - | 16,370 | 49 | 99 | 64 | o |
| Pazopanib HCl | VEGFR, PDGFR, FGFR, c-Kit | 4769 | 1061 | 2194 | 4126 | 2762 | 6415 | 1947 | 6142 | 3982 | 1976 | 2234 | 1438 | o |
| Dovitinib (TKI-258) | c-Kit,VEGFR, FGFR | 214 | 232 | 1103 | 431 | 298 | 668 | 282 | 822 | 1356 | 177 | 303 | 196 | x |
| Erlotinib HCl | EGFR | >20,000 | 2721 | 955 | 2908 | 2341 | 4795 | - | 6296 | - | - | 9910 | 16,080 | o |
| Afatinib (BIBW2992) | EGFR,HER2 | - | 717 | 784 | 891 | 1170 | 1120 | 580 | 1212 | 2006 | 1765 | 1501 | 1283 | o |
| Gefitinib (Iressa) | EGFR | 42,640 | - | 6271 | 6827 | 8518 | 6963 | - | - | 12,730 | - | 8899 | 11,050 | o |
| Neratinib (HKI-272) | HER2 & EGFR | 1848 | 441 | 100 | 448 | 776 | 200 | 181 | 780 | 1573 | 193 | 548 | 524 | x |
| vemurafenib | BRAF | >20,000 | 10,160 | 5945 | 19,130 | 12,250 | 11,130 | 9782 | 17,880 | 13,430 | 12,550 | >20,000 | 16,310 | o |
Figure 4.

Drug responses of liposarcoma PDCs.
(A) Each response hit is represented as a box graph. Dose-response analyses were carried out using GraphPad Prism 6 software program (detailed in Materials and Methods). Each column represents drugs, which are clustered by color according to targeting pathway or molecules. Bar graphs represent the hits for the AUC (y-axis). (B). Comparison of the IC50 of drugs screened in PDCs. On each graph, the y-axis represents the cell survival (%) of the indicated drugs. (C) Proteasome inhibitor exhibited cytotoxicity in PDCs. A liposarcoma cell line and PDCs were treated with vehicle or bortezomib (0-80 nM) for 48 hours. The effects of these drugs on proliferation were assayed by a CCK8 experiment. The IC50 values of both drugs were calculated. The y-axis represents the percentage of cells under each condition. Each point was analyzed in triplicate. (D) Western blot analysis of GS11-079 PDC and LPS246 cell line. Cells were treated with bortezomib (0-10 μM) for 48 hours. Lysates were subjected to an analysis of the efficacy of bortezomib in inhibiting the proteasomal degradation pathway.
Next, we tested if certain downstream targets were downregulated upon exposure to bortezomib. As shown in Figure 4, C and D, we observed that bortezomib dose-dependently downregulated MDM2 in tumor cells. Liposarcoma is characterized by amplification of MDM2 on chromosome 12. As an E3 ligase, MDM2 regulates p53 in the cytoplasm through proteasomal lysis [25], [26], [27]. Moreover, it has been hypothesized that inhibition of MDM2 induces the activity of p53, thereby acting to treat liposarcoma [18], [19], [22]. We therefore observed the therapeutic effect of bortezomib, a proteasome inhibitor, based on these hypotheses. Indeed, the expression level of MDM2 decreased in a dose-dependent manner with bortezomib treatment in the PDC model. Conversely, p21, a downstream molecule of p53, was found to increase in the 11GS-079 PDC model. However, we did not observe any changes in the levels of activated or nonactivated p53. Indeed, we did not find any difference in expression levels of proteins except those MDM2 in the LPS246 cell line, which was in agreement with the in vitro results from the cell viability assay (Figure 4, C and D). Also, activated AKT, ERK, and mTOR, all molecules related to cell growth, did not show variations in either PDC line after bortezomib treatment. These results indicate that the inhibitory effects of bortezomib treatment in liposarcoma may be due to an activation of p53 followed by activated p21, as well as inactivation of MDM2 [24], [28], [29]. Taken together, the upregulation of the MDM2 pathway seen in liposarcoma patients can be considerably modulated by bortezomib treatment, which results in activation of p21 and downregulation of MDM2 (Figure 4D) [18], [30].
In LPS246, the liposarcoma cell line, we did not observe change of cell viability according to treatment of bortezomib between concentrations from 0 nM to 20 nM, whereas in 11GS079 PDC, the viability was decreased significantly among these doses. Indeed, at around 1.0 nM, 11GS079 was already diminishing its survival curve. From these results, we suspected that there might be differences which discriminated the response and/or sensitivity toward bortezomib treatment. Next, when we expanded the dose of bortezomib to 40 nM on LPS246 cell line, it was observed that the activity of p53 (acetylated p53) was sustained in that dose.
From these results, we found that bortezomib suppressed liposarcoma cell growth effectively in response to the activation of p53. It can also suggest that this drug, as a proteasome inhibitor, may have anticancer effect mainly by p53-p21 pathways (Supplementary Figure 4).
Supplementary Figure 4.
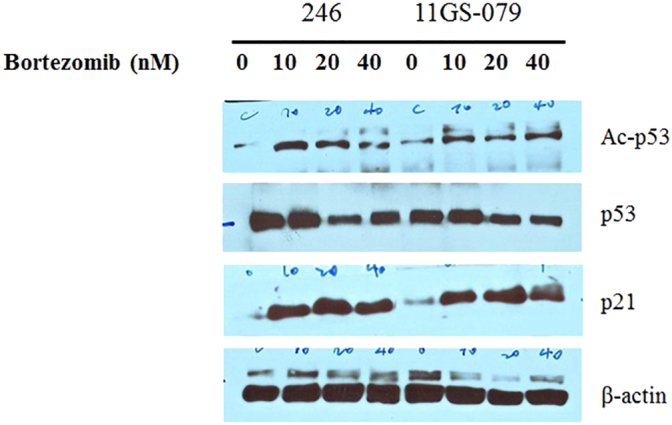
Drug response to activation of p53, the acetylated p53.
Western blot analysis of GS11-079 PDC and LPS246, liposarcoma cell line. Cells were treated with bortezomib (0-80 μM) for 48 hours. Lysates were subjected to an analysis of the anticancer effect of bortezomib in response to p53, p21 pathway.
Orthotopic PDX Model Resembles Patient Tumors Histologically and Retains Liposarcoma Gene Expression
In the case of liposarcoma, which originates from the soft tissue of muscle, most of orthotopic tumor grafted site would be the subcutaneous tissue. For these reasons, we xenografted patients' liposarcoma tissues onto mice subcutaneously and evaluated the growth of these tumors in the PDX mouse model. Over ~90% of the xenograft lines growing orthotopically grew subcutaneously within a manageable time frame (Supplementary Figure 1A, Table 3). Detailed analyses done by a clinical pathologist specializing in soft tissue sarcoma showed that the PDX model retained not only the histology but also the expression of CDK4 and MDM2, corresponding to the patients' tumors (Supplementary Figure 1B). Even within tumors xenografted in the kidneys of mice, IHC analysis showed that expression of CDK4 and MDM2, and cytological characteristics were preserved in the respective tumor xenografts (Supplementary Figure 1B). Next, we performed microsatellite STR analyses and CGH array to determine whether the PDX model retained the gene expression pattern of the original tumors. We performed PCR using STR markers for following the chromosomal exchange between tumor tissues from patients and from mice that occurred in established PDX models. As a result, the expression levels of STR markers were observed at the same chromosome loci in parental tissues and successive PDX models (e.g., between parental tissue, first-generation PDX model and second-generation PDX model, Figure 5A). In addition, CGH array results showed that both gain and loss loci occurred in the same region on each chromosome (Figure 5B). Furthermore, no changes were observed in the histological findings of the tumor tissues between parental tissues and each generation of PDX model, and all tissues maintained characteristic CDK4 and MDM2 levels specific to liposarcoma (Figure 5C, Supplementary Figure 5). These results demonstrate that the genomes of the patient and tumor xenografted mouse models exhibited the same profiles and chromosome alterations, suggesting that this PDX model may serve as a useful tool for pre-clinical trials.
Supplementary Figure 5.





IHC results of tumor xenografts.
The patient-derived tumor tissues xenografted on mice were stained with IHC of MDM2. IHC was carried out serially about tumor tissues engrafted on mice (from G0 to G3) in order to determine whether it maintained the inherent character of liposarcoma. (A) Primary tumor (14GS-032: Xeno-G0), (B) xenografted F1 tumor (14GS-032: Xeo-G1), (C) xenografted F2 (14GS-032: Xeno-G2), (D) xenografted F3 (14GS-032: Xeno-G3), and (E) xenografted F1 tumor (15GS-002-2: Xeo-G1).
PDX Model Reproduces the Drug Responsiveness of DDLPS
Among the PDX models, we selected three, involving the patient-derived primary cells 14GS-076, 14GS-084, and 15GS-023, for evaluating the anticancer effect of bortezomib (Supplementary Figure 2). For the drug trial, 32 mice were implanted with ~10 mm3 tumor fragments, and 8-10 weeks after implantation, tumor volume measurements began. Tumor growth rates varied considerably among the three PDX models, and drug administration started when the average tumor size reached approximately 100 mm3. Bortezomib trials were carried out for ~21 days. During the trials, tumor measurements were taken twice weekly. Mice were weighed weekly, and drug administration was adjusted accordingly. Bortezomib profoundly suppressed growth of patient-derived xenograft liposarcoma tumors (Figure 6, A-C). As indicated by the lack of difference in the average tumor volume before drug treatment between groups, tumor volumes showed no bias from the start point of bortezomib treatment (Supplementary Figure 3). Overall, these results show that bortezomib was effective against liposarcoma in our PDX models.
Supplementary Figure 2.

Three PDCs used in PDX models were selected for drug treatment.
Three patient-derived primary cells (14GS-076, 14GS-084, and 15GS-023) that were maintained in mice were selected for evaluating the anticancer effect of certain drugs. These PDXs maintained expression of CDK4 and MDM2 in their tumor tissues.
Figure 6.

Bortezomib treatment inhibited patient-derived xenografts in NSG mice.
Relative tumor growth of PDX models treated with vehicle (control) (A) 14-GS076, n = 5; (B) 14GS-084, n = 4; (C) 15GS-023, n = 5, or bortezomib (0.3 mg/kg) (A) 14-GS076, n = 5; (B) 14GS-084, n = 6; (C) 15GS-023, n = 7. Mice were treated with either bortezomib (0.3 mg/kg body weight, in DMSO/PBS) or DMSO/PBS. The mice were monitored and weighed three times a week. Data shown represent the mean ± SEM (*P < .05; **P < .01).
Discussion
Our liposarcoma PDX mouse model reproduced the histological features and molecular genetics of human liposarcoma. Additionally, our results with FDA-approved drugs establish a proof of principle for the evaluation of molecularly targeted therapies for liposarcoma in mice. Using HTS screening data, we evaluated an agent in clinical development, bortezomib, which showed remarkable activity against DDLPS [29], [31].
Concerning on the 11GS-079 primary and LPS246 cell line, acetylated form of p53 was sustained through treatment of bortezomib. Many studies showed the results about another pathway for regulating cell fate by inhibition of protesome activity. [32], [33], [34] Strauss et al. said that bortezomib treatment resulted in cells moving into an aberrant mitosis, indicative of mitotic catastrophe that may contribute to increased sensitivity to bortezomib. Also, they suggested that different cell cycle effects were observed by abrogating the G2-M cell cycle checkpoint. Mainly p53 activation and suppression of NF-kB activity affects to cell death in multiple myeloma and non-Hodgkins lymphoma, but also identifying the cell death derived from mitotic catastrophe may help understaning drug efficacy of bortezomib treatment [35]. Indeed, many groups suggested the effect of bortezomib on in vitro models, independent of p53. However, contrary to other reports, our results well presented the correlation between activation of p53 and viability of cell lines according to treatment with bortezomib. Meanwhile whether another pathway might be included in anticancer effect in LPS cell lines, it may be beyond the scope of this study.
Our PDX model reproduced not only the histology of the patient tumors but also finer characteristics, such as the tumor architecture as well as cytological and nuclear features. In STR and DNA copy number alterations, the PDX models showed similarities to their corresponding tumors derived from patients. In addition, these studies failed to reveal any confident point mutations or indels in the xenograft that could not be found in the original patient tumor. Overall, these results show that PDX mouse models are faithful models of the corresponding tumors in humans.
Moreover, the PDX models retained MDM2 and CDK4 expression seen in patients' tumor tissues. MDM2 is a key repressor of p53. Degradation of p53 is mediated by the E3 ligase function of MDM2 [36]. In a previous study, we suggested that protein level of MDM2 increased according to the dedifferentiation of liposarcoma [37]. In this study, we found that bortezomib treatment interfered with MDM2 expression, and it correlated with p21 expression in PDC model 11GS-079. Also, our results showed that bortezomib treatment potently inhibited tumor growth in a liposarcoma xenograft mouse model. These results in a validated tumor xenograft model support the fact that bortezomib is a worthy potential anticancer agent in clinical development and led us to predict that bortezomib will be similarly effective against liposarcoma in humans.
The PDX may represent nearly pure populations of human tumor cells because the only cellular compartment that can regenerate itself in PDX mouse models is the neoplastic compartments [38], [39], [40], [41]. Thus, PDX models are instrumental for exploring new cellular and molecular targeting agents and whether the target of the drug was successfully inhibited in the tumor, which is a challenge in patients. PDX models can also be used to determine optimal drug combinations and to develop for variable patients' tailored drug treatment. The exploitation of differences between humans and rodents may render xenografted tumor-bearing mice useful for the identification of the effects of drugs and for developing novel drugs. In addition, PDX models can be used to study cell therapies against liposarcoma [42]. Finally, PDX models may be useful for the evaluation and applications the novel tumor markers in liposarcoma patients [43].
In summary, we established a liposarcoma PDX mouse model for the validation of drug regimens for application as human chemotherapies. We suggest that the PDX mouse models could advance preclinical drug evaluation of drug regimens and improve oncology drug development for liposarcoma patients.
The following are the supplementary data related to this article.
Supplementary material
Acknowledgement
The authors thank SMC LARC facility staff, and we thank Jae Ro Park, who provided assistance in writing. This work was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (NRF-2015R1D1A1A01059879).
References
- 1.Hidalgo M, Amant F, Biankin AV, Budinska E, Byrne AT, Caldas C, Clarke RB, de Jong S, Jonkers J, Maelandsmo GM. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov. 2014;4:998–1013. doi: 10.1158/2159-8290.CD-14-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Malaney P, Nicosia SV, Dave V. One mouse, one patient paradigm: new avatars of personalized cancer therapy. Cancer Lett. 2014;344:1–12. doi: 10.1016/j.canlet.2013.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jung J, Lee CH, Seol HS, Choi YS, Kim E, Lee EJ, Rhee JK, Singh SR, Jun ES, Han B. Generation and molecular characterization of pancreatic cancer patient-derived xenografts reveals their heterologous nature. Oncotarget. 2016;7:62533–62546. doi: 10.18632/oncotarget.11530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krempley BD, Yu KH. Preclinical models of pancreatic ductal adenocarcinoma. Chin Clin Oncol. 2017;6:25. doi: 10.21037/cco.2017.06.15. [DOI] [PubMed] [Google Scholar]
- 5.Taromi S, Kayser G, von Elverfeldt D, Reichardt W, Braun F, Weber WA, Zeiser R, Burger M. An orthotopic mouse model of small cell lung cancer reflects the clinical course in patients. Clin Exp Metastasis. 2016;33:651–660. doi: 10.1007/s10585-016-9808-8. [DOI] [PubMed] [Google Scholar]
- 6.Goodspeed A, Heiser LM, Gray JW, Costello JC. Tumor-derived cell lines as molecular models of cancer pharmacogenomics. Mol Cancer Res. 2016;14:3–13. doi: 10.1158/1541-7786.MCR-15-0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burgenske DM, Monsma DJ, MacKeigan JP. Patient-derived xenograft models of colorectal cancer: procedures for engraftment and propagation. Methods Mol Biol. 2018;1765:307–314. doi: 10.1007/978-1-4939-7765-9_20. [DOI] [PubMed] [Google Scholar]
- 8.Okada S, Vaeteewoottacharn K, Kariya R. Establishment of a patient-derived tumor xenograft model and application for precision cancer medicine. Chem Pharm Bull (Tokyo) 2018;66:225–230. doi: 10.1248/cpb.c17-00789. [DOI] [PubMed] [Google Scholar]
- 9.Tentler JJ, Tan AC, Weekes CD, Jimeno A, Leong S, Pitts TM, Arcaroli JJ, Messersmith WA, Eckhardt SG. Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol. 2012;9:338–350. doi: 10.1038/nrclinonc.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beird HC, Wu CC, Ingram DR, Wang WL, Alimohamed A, Gumbs C, Little L, Song X, Feig BW, Roland CL. Genomic profiling of dedifferentiated liposarcoma compared to matched well-differentiated liposarcoma reveals higher genomic complexity and a common origin. Cold Spring Harb Mol Case Stud. 2018;4 doi: 10.1101/mcs.a002386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garcia-Ortega DY, Villa-Zepeda O, Martinez-Said H, Cuellar-Hubbe M, Luna-Ortiz K. Oncology outcomes in retroperitoneal sarcomas: prognostic factors in a retrospective cohort study. Int J Surg. 2016;32:45–49. doi: 10.1016/j.ijsu.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 12.Choi Y, Yun MS, Lim SH, Lee J, Ahn JH, Kim YJ, Park KH, Park YS, Lim HY, An H. Gemcitabine and docetaxel combination for advanced soft tissue sarcoma: a nationwide retrospective study. Cancer Res Treat. 2018;50:175–182. doi: 10.4143/crt.2016.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luke JJ, D'Adamo DR, Dickson MA, Keohan ML, Carvajal RD, Maki RG, de Stanchina E, Musi E, Singer S, Schwartz GK. The cyclin-dependent kinase inhibitor flavopiridol potentiates doxorubicin efficacy in advanced sarcomas: preclinical investigations and results of a phase I dose-escalation clinical trial. Clin Cancer Res. 2012;18:2638–2647. doi: 10.1158/1078-0432.CCR-11-3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen D, Frezza M, Schmitt S, Kanwar J, Dou QP. Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Curr Cancer Drug Targets. 2011;11:239–253. doi: 10.2174/156800911794519752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shahshahan MA, Beckley MN, Jazirehi AR. Potential usage of proteasome inhibitor bortezomib (Velcade, PS-341) in the treatment of metastatic melanoma: basic and clinical aspects. Am J Cancer Res. 2011;1:913–924. [PMC free article] [PubMed] [Google Scholar]
- 16.Daigeler A, Klein-Hitpass L, Chromik MA, Muller O, Hauser J, Homann HH, Steinau HU, Lehnhardt M. Heterogeneous in vitro effects of doxorubicin on gene expression in primary human liposarcoma cultures. BMC Cancer. 2008;8:313. doi: 10.1186/1471-2407-8-313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang HM, Sung JH, Choi YS, Lee HJ, Roh CR, Kim J, Shin M, Song S, Kwon CH, Joh JW. Enhancement of the immunosuppressive effect of human adipose tissue-derived mesenchymal stromal cells through HLA-G1 expression. Cytotherapy. 2012;14:70–79. doi: 10.3109/14653249.2011.613926. [DOI] [PubMed] [Google Scholar]
- 18.Ravandi F, Gojo I, Patnaik MM, Minden MD, Kantarjian H, Johnson-Levonas AO, Fancourt C, Lam R, Jones MB, Knox CD. A phase I trial of the human double minute 2 inhibitor (MK-8242) in patients with refractory/recurrent acute myelogenous leukemia (AML) Leuk Res. 2016;48:92–100. doi: 10.1016/j.leukres.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saha MN, Jiang H, Jayakar J, Reece D, Branch DR, Chang H. MDM2 antagonist nutlin plus proteasome inhibitor velcade combination displays a synergistic anti-myeloma activity. Cancer Biol Ther. 2010;9:936–944. doi: 10.4161/cbt.9.11.11882. [DOI] [PubMed] [Google Scholar]
- 20.Heo EJ, Cho YJ, Cho WC, Hong JE, Jeon HK, Oh DY, Choi YL, Song SY, Choi JJ, Bae DS. Patient-derived xenograft models of epithelial ovarian cancer for preclinical studies. Cancer Res Treat. 2017;49:915–926. doi: 10.4143/crt.2016.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kammerer-Jacquet SF, Thierry S, Cabillic F, Lannes M, Burtin F, Henno S, Dugay F, Bouzille G, Rioux-Leclercq N, Belaud-Rotureau MA. Differential diagnosis of atypical lipomatous tumor/well-differentiated liposarcoma and dedifferentiated liposarcoma: utility of p16 in combination with MDM2 and CDK4 immunohistochemistry. Hum Pathol. 2017;59:34–40. doi: 10.1016/j.humpath.2016.08.009. [DOI] [PubMed] [Google Scholar]
- 22.Rayburn E, Zhang R, He J, Wang H. MDM2 and human malignancies: expression, clinical pathology, prognostic markers, and implications for chemotherapy. Curr Cancer Drug Targets. 2005;5:27–41. doi: 10.2174/1568009053332636. [DOI] [PubMed] [Google Scholar]
- 23.Ware PL, Snow AN, Gvalani M, Pettenati MJ, Qasem SA. MDM2 copy numbers in well-differentiated and dedifferentiated liposarcoma: characterizing progression to high-grade tumors. Am J Clin Pathol. 2014;141:334–341. doi: 10.1309/AJCPLYU89XHSNHQO. [DOI] [PubMed] [Google Scholar]
- 24.Castro ME, Ferrer I, Cascon A, Guijarro MV, Lleonart M, Ramon y Cajal S, Leal JF, Robledo M, Carnero A. PPP1CA contributes to the senescence program induced by oncogenic Ras. Carcinogenesis. 2008;29:491–499. doi: 10.1093/carcin/bgm246. [DOI] [PubMed] [Google Scholar]
- 25.Li M, Brooks CL, Wu-Baer F, Chen D, Baer R, Gu W. Mono- versus polyubiquitination: differential control of p53 fate by Mdm2. Science. 2003;302:1972–1975. doi: 10.1126/science.1091362. [DOI] [PubMed] [Google Scholar]
- 26.Michael D, Oren M. The p53-Mdm2 module and the ubiquitin system. Semin Cancer Biol. 2003;13:49–58. doi: 10.1016/s1044-579x(02)00099-8. [DOI] [PubMed] [Google Scholar]
- 27.Ofir-Rosenfeld Y, Boggs K, Michael D, Kastan MB, Oren M. Mdm2 regulates p53 mRNA translation through inhibitory interactions with ribosomal protein L26. Mol Cell. 2008;32:180–189. doi: 10.1016/j.molcel.2008.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carnero A, Beach DH. Absence of p21WAF1 cooperates with c-myc in bypassing Ras-induced senescence and enhances oncogenic cooperation. Oncogene. 2004;23:6006–6011. doi: 10.1038/sj.onc.1207839. [DOI] [PubMed] [Google Scholar]
- 29.Perez M, Peinado-Serrano J, Garcia-Heredia JM, Felipe-Abrio I, Tous C, Ferrer I, Martin-Broto J, Saez C, Carnero A. Efficacy of bortezomib in sarcomas with high levels of MAP17 (PDZK1IP1) Oncotarget. 2016;7:67033–67046. doi: 10.18632/oncotarget.11475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wagner AJ, Banerji U, Mahipal A, Somaiah N, Hirsch H, Fancourt C, Johnson-Levonas AO, Lam R, Meister AK, Russo G. Phase I trial of the human double minute 2 inhibitor MK-8242 in patients with advanced solid tumors. J Clin Oncol. 2017;35:1304–1311. doi: 10.1200/JCO.2016.70.7117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu Y, Wang L, Wang L, Wu X, Wu X, Gu Y, Shu Y, Sun Y, Shen Y, Xu Q. Preferential cytotoxicity of bortezomib toward highly malignant human liposarcoma cells via suppression of MDR1 expression and function. Toxicol Appl Pharmacol. 2015;283:1–8. doi: 10.1016/j.taap.2014.12.015. [DOI] [PubMed] [Google Scholar]
- 32.Dabiri Y, Kalman S, GS GX, Wu X, Gu Y, Shu Y, Sun Y, Shen Y, Xu Q. Preferential cytotoxicity of bortezomib toward highly malignant human liposarcoma r cells. Sci Rep. 2015;7:5423. doi: 10.1016/j.taap.2014.12.015. [DOI] [PubMed] [Google Scholar]
- 33.Yerlikaya A, Okur E, Baykal AT, Ac Ac, Shu Y, Sun Y, Shen Y, Xu Q. Preferential cytotoxicity of bortezomib toward highly malignant human liposarcoma r cellsia suppressio. J Proteomics. 2015;113:315–325. [Google Scholar]
- 34.Yerlikaya A, Okur E, Tar Tar AT, Ac Ac, Shu Y, Sun Y, Shen Y, Xu Q. Preferential cytotoxicity of bortezomib toward highly malignant human liposarcoma r c. Data Brief. 2015;1:56–59. doi: 10.1016/j.taap.2014.12.015. [DOI] [PubMed] [Google Scholar]
- 35.Strauss SJ, Higginbottom K, JK Jc, Ac Shu Y, Sun llen P, Schenkein D, Lister TA, Joel SP. The Proteasome inhibitor bortezomib acts independently of p53 and induces cell death via apoptosis and mitotic catastrophe in B-cell lymphoma cell lines. Cancer Res. 2007;67:2783. doi: 10.1158/0008-5472.CAN-06-3254. [DOI] [PubMed] [Google Scholar]
- 36.Moll UM, Petrenko O. The MDM2-p53 interaction. Mol Cancer Res. 2003;1:1001–1008. [PubMed] [Google Scholar]
- 37.Hwang JA, Yang HM, Hong DP, Joo SY, Choi YL, Park JH, Lazar AJ, Pollock RE, Lev D, Kim SJ. Gankyrin is a predictive and oncogenic factor in well-differentiated and dedifferentiated liposarcoma. Oncotarget. 2014;5:9065–9078. doi: 10.18632/oncotarget.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Allaway RJ, Fischer DA, de Abreu FB, Gardner TB, Gordon SR, Barth RJ, Colacchio TA, Wood M, Kacsoh BZ, Bouley SJ. Genomic characterization of patient-derived xenograft models established from fine needle aspirate biopsies of a primary pancreatic ductal adenocarcinoma and from patient-matched metastatic sites. Oncotarget. 2016;7:17087–17102. doi: 10.18632/oncotarget.7718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dobrolecki LE, Airhart SD, Alferez DG, Aparicio S, Behbod F, Bentires-Alj M, Brisken C, Bult CJ, Cai S, Clarke RB. Patient-derived xenograft (PDX) models in basic and translational breast cancer research. Cancer Metastasis Rev. 2016;35:547–573. doi: 10.1007/s10555-016-9653-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hasmim M, Bruno S, Azzi S, Gallerne C, Michel JG, Chiabotto G, Lecoz V, Romei C, Spaggiari GM, Pezzolo A. Isolation and characterization of renal cancer stem cells from patient-derived xenografts. Oncotarget. 2016;7:15507–15524. doi: 10.18632/oncotarget.6266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lai Y, Wei X, Lin S, Qin L, Cheng L, Li P. Current status and perspectives of patient-derived xenograft models in cancer research. J Hematol Oncol. 2017;10:106. doi: 10.1186/s13045-017-0470-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee H, Jo EB, Kim SJ, Yang HM, Kim YM, Sung YC, Park JB, Hong D, Park H, Choi YL. Therapeutic strategies for locally recurrent and metastatic de-differentiated liposarcoma with herpes simplex virus-thymidine kinase-expressing mesenchymal stromal cells. Cytotherapy. 2017;19:1035–1047. doi: 10.1016/j.jcyt.2017.05.008. [DOI] [PubMed] [Google Scholar]
- 43.Jung J, Seol HS, Chang S. The generation and application of patient-derived xenograft model for cancer research. Cancer Res Treat. 2018;50:1–10. doi: 10.4143/crt.2017.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material