

Abstract
Background
Composition of the vaginal microbiota has significant influence on female urogenital health and control of infectious disease. Murine models are widely utilized to characterize host-pathogen interactions within the vaginal tract, however, the composition of endogenous vaginal flora remains largely undefined with modern microbiome analyses. Here, we employ 16S rRNA amplicon sequencing to establish the native microbial composition of the vaginal tract in adult C57Bl/6 J mice. We further interrogate the impact of estrous cycle and introduction of the human vaginal pathobiont, group B Streptococcus (GBS) on community state type and stability, and conversely, the impact of the vaginal microbiota on GBS persistence.
Results
Sequencing analysis revealed five distinctive community states of the vaginal microbiota dominated largely by Staphylococcus and/or Enterococcus, Lactobacillus, or a mixed population. Stage of estrus did not impact microbial composition. Introduction of GBS decreased community stability at early timepoints; and in some mice, GBS became the dominant bacterium by day 21. Endogenous Staphylococcus abundance correlated with GBS ascension into the uterus, and increased community stability in GBS-challenged mice.
Conclusions
The murine vaginal flora is diverse and fluctuates independently of the estrous cycle. Endogenous flora may impact pathogen colonization and dissemination and should be considered in urogenital infection models.
Electronic supplementary material
The online version of this article (10.1186/s12866-018-1341-2) contains supplementary material, which is available to authorized users.
Keywords: Vaginal microbiome, Murine model, Estrous cycle, Group B Streptococcus, 16S rRNA sequencing
Background
The vaginal microbiota is intimately linked to women’s health. In humans, the vaginal microbiota exists in 5 distinct community state types (CSTs) which are generally dominated by Lactobacillus spp. [1]. Composition of the microbiota varies in temporal stability, with greatest instability exhibited during menses [2]. Subcategory CST IV-A, a community dominated by facultative and strict anaerobes in place of Lactobacillus spp. [1], has been associated with increased incidence of vulvovaginal atrophy [3], and colonization by group B Streptococcus (GBS) [4]. Furthermore, vaginal dysbiosis, denoted clinically as bacterial vaginosis (BV), is characterized as a heterogeneous vaginal microbiota not dominated by a single taxon [5]. BV or high bacterial diversity has been associated with adverse health outcomes including preterm birth [6, 7], HIV acquisition [8, 9], and infection with other urogenital pathogens including Trichomonas vaginalis [10, 11], Chlamydia trachomatis [12], and Neisseria gonorrhoeae [13]. The vaginal microbiota represents a key constituent of host-pathogen interactions in the vaginal mucosa, with significant implications for understanding susceptibility to disease and optimizing prevention and treatment strategies.
Murine models of vaginal infection and colonization are commonly used to characterize microbial pathogenesis determinants, host immune responses, and therapeutic interventions for urogenital pathogens including HIV [14], group B Streptococcus [15], Candida albicans [16, 17], Trichomonas vaginalis [18], Gardnerella vaginalis [19], and Chlamydia trachomatis [20]. Despite the widespread use of this animal model, the commensal murine vaginal microbiota has yet to be longitudinally characterized with modern microbiome sequencing methods. Earlier culture-based studies identified Enterobacteriaceae, Streptococcus spp., Staphylococcus spp., Corynebacterium spp., and Lactobacillus spp. among the murine vaginal flora [21, 22], while 16S sequence-based studies have either focused on select microbes of interest [23] or have failed to account for estrus [24]. Variation in vaginal communities across time and in relation to estrus have not been described.
Integrating detailed knowledge of the mouse vaginal microbiota into host-pathogen infections can provide a more direct application for these models to human urogenital pathogens. In this study, we characterize the murine vaginal microbiota of the post-pubertal female C57Bl/6 J Jackson mouse over time to assess the composition and stability of the vaginal flora throughout the estrous cycle. We also introduce the vaginal commensal bacterium and opportunistic pathogen, GBS to evaluate microbiota changes in the context of a relevant human infection model.
Results
Murine vaginal microbiota can be categorized into distinct community state types
Little is known about the compositional stability of the murine vaginal flora. Mice have short estrous cycles that last 4–5 days and consist of four stages: proestrus, estrus, metestrus, and diestrus [25].To assess stability of the mouse vaginal microbiota in this context, we used one of the most commonly utilized mouse strains/ages and sources: post-pubertal 8-week-old female C57Bl/6 J mice, obtained from Jackson Laboratories. Mice were received at 7 weeks of age, randomized upon arrival into 5 mice per cage, and acclimated over a one-week period. We longitudinally sampled the vaginal microbiota every three days over a period of 15 days by lavaging with phosphate-buffered saline (PBS). We lavaged a second set of mice only twice, on days 0 and 15, to test whether frequent lavaging itself alters the microbiota. The corresponding estrous cycle stage of each lavage sample was determined by light microscopy. Samples were processed for 16S rRNA gene sequencing as detailed in the Methods section. Contaminants from sequencing reagents (primarily Pseudomonas, Geobacillus, and Sphingobium reads) or chloroplast and mitochondrial sequences were removed before assessing community composition.
For comparison to earlier human vaginal microbiome characterization studies [1, 2], samples were assigned murine community state types (mCST) by hierarchical clustering with Ward’s linkage of Euclidean distances (Fig. 1) (silhouette score of 0.732), with the rarefied OTU table at 1500 reads per sample. The most predominant community state type, mCST I, consisted of Staphylococcus-dominant flora. mCST II samples contained vaginal flora comprised primarily of both Staphylococcus and Enterococcus, while mCST III was predominantly Enterococcus. mCST IV samples were Lactobacillus-dominant and mCST V samples were not dominated by either Staphylococcus or Enterococcus and had higher alpha diversity (p < 0.001), Shannon diversity and observed operational taxonomic units (OTU). Additionally, there was one single sample dominated by Bifidobacterium, and this sample was excluded from subsequent analyses as it was thought to be contaminated.
Fig. 1.

Murine community state types and bacterial landscape of the murine vaginal microbiota. Samples are clustered by community state with Ward’s linkage of Euclidean distances (silhouette score of 0.732, sklearn). Estrous cycle stage is depicted by shades of purple and bacterial abundances are indicated by heatmap intensity corresponding to the colorbar ranging from purple to yellow
Murine vaginal community states are unstable
Interestingly, community states were relatively unstable in mice. Of mice that had at least two successfully sequenced samples (at least 1500 reads/sample), 70% had samples in at least two different community states (Fig. 2). mCST I appeared to be the most stable community state: for samples from consecutive timepoints that successfully sequenced, 12/14 mCST I samples were assigned mCST I at the next time point (Fig. 2). In comparison, only 1/5 samples in mCST II were also mCST II at the next consecutive time point.
Fig. 2.

Vaginal Microbiome Stability. Mice from the cohort sampled every three days are displayed ordered by mouse and time point (left). Missing samples indicate sequencing failure (< 1500 reads/sample) and point color depicts the community state type of the sample. Mice with fewer than two successfully sequenced samples were excluded. Bray-Curtis distances between all samples from each individual mouse (right)
Estrous cycle does not impact vaginal community state type
After filtering and rarefaction to 1500 reads per sample, few samples staged as diestrus and proestrus remained (19.5 and 31.1% respectively) compared to estrus and metestrus (64.1, and 66.6% respectively, Fig. 3). Though sequencing success may vary over estrous cycle stage, community state type was not significantly associated with different stages in the estrous cycle (X2 = 17.29, p = 0.138). A random forest classifier (scikit-learn) for estrous cycle stage performed on the rarefied OTU table achieved an accuracy of only 0.125, indicating that bacterial composition is a poor predictor of estrous cycle stage. Additionally, beta diversity clustering for estrous cycle stage was not significant and clustering by cage, stratified by day, was only significant on days 0 and 9. (Fig. 4).
Fig. 3.
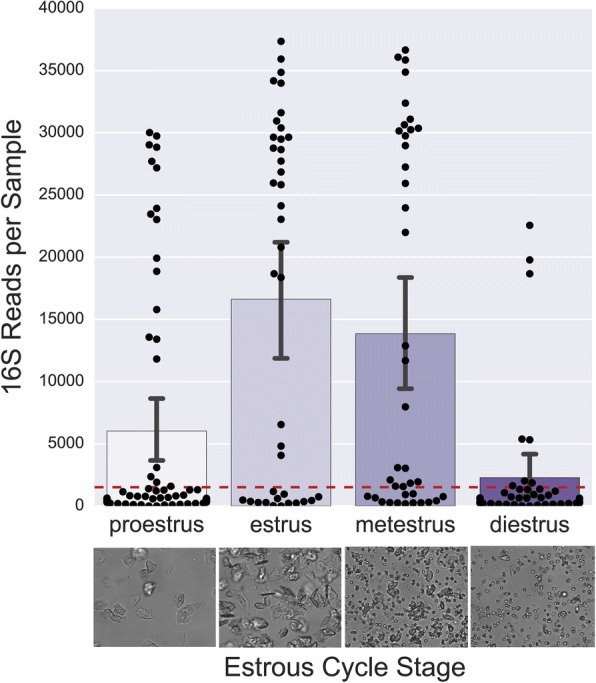
Estrous cycle stage influences 16S rRNA sequencing success. 16S rRNA sequences after removing contaminants, mitochondria, and chloroplast sequences (top). Images (bottom) depict representative microscopy images from vaginal lavages in the different estrous cycle stages, Magnification: 200X
Fig. 4.

Significant clustering by mCST, but not estrous stage. PCoA Plots of the Bray-Curtis distance matrix of mCST (top left), estrous stage (top right), and cage (bottom). Clustering by mCST was significant (p = 0.001, pseudo-F-statistic = 52.13, by PERMANOVA with 999 permutations), while clustering by estrous stage was not (p = 0.089, pseudo-F statistic = 1.69, by PERMANOVA 999 permutations). Clustering by cage was only significant on days 0 (p = 0.002, pseudo-F-statistic = 3.00) and 9 (p = 0.001, pseudo-F-statistic = 35.5)
Group B Streptococcus challenge destabilizes vaginal community states
To investigate how urogenital pathogens may perturb the murine vaginal microbiota, we longitudinally sampled a second cohort of mice experimentally challenged with GBS and compared them to an uninfected control group. These mice received intraperitoneal injection of 0.5 mg β-estradiol 24 h before infection to synchronize estrus and promote colonization [26]. Mice were lavaged and vaginally inoculated with 1 × 107 colony forming units (CFU) of a well characterized human serotype III GBS isolate COH1 in 10 μl PBS or 10 μl PBS alone (control), then lavaged 3, 7, 14, and 21 days after infection for microbiome sampling. GBS CFUs were also monitored by plating. On days 3, 14, and 21, twelve mice per group were sacrificed to determine bacterial tissue burdens. Lavage samples were processed for 16S rRNA sequencing following the same protocol as the staging cohort.
Although these mice were ordered in a separate shipment, they exhibited similar vaginal microbiota and community-state type clustering (Additional file 1). Community state type mCST I consisted of Staphylococcus-dominant flora and mCST II samples contained vaginal flora primarily comprised of Staphylococcus and Enterococcus, and in some cases, Streptococcus reads from GBS colonization. mCST VI samples were dominated by GBS (Streptococcus), while mCST III was predominantly Enterococcus. Lactobacillus comprised most of the flora in mCST IV samples and mCST V samples consisted of a mix of bacteria. Streptococcus 16S reads were only abundant in GBS-colonized mice and deblurred 16S sequences mapped to Streptococcus agalactiae (GBS) in Genbank. To preserve more samples for longitudinal analysis, samples were rarefied to 500 reads per sample (rarefaction plots in Additional file 2).
Though mCST VI represented the GBS-dominant state, GBS CFU determined by plating of vaginal lavage samples was not significantly different at day 3 or 7 across all mCSTs (ANOVA p = 0.68, p = 0.75, respectively)(Fig. 5a). However, by day 21 GBS CFU were only detected in mice with mCST VI where GBS had completely overtaken the vaginal flora (Fig. 5a). Initially, GBS challenge significantly destabilized the vaginal microbiota; pairwise Bray-Curtis distances between consecutive days for individual mice revealed significantly increased distances in the GBS-challenged mice for days 3 (distance from day 0 to day 3) and 7 (distance between days 3 and 7) (Fig. 5c). By day 21, the majority of the GBS mice had GBS-dominant flora (mCST VI), significantly reducing the pairwise distance between days 14 and 21 compared to PBS mice. To look specifically at how changes in prominent taxa were contributing to the initial increased pairwise sample distance with GBS colonization while minimizing the effects of compositionality, we took the log ratio of Staphylococcus relative abundance over Streptococcus relative abundance or GBS CFU and Enterococcus relative abundance over Streptococcus relative abundance or GBS CFU. Correlating these log ratios to the Bray-Curtis distances between the day 0 and day 3 samples within mice revealed that a decrease in the ratio of Staphylococcus to Streptococcus or GBS CFU was significantly correlated with increased distance between samples. Conversely, the ratio of Enterococcus to Streptococcus had no significant correlation with Bray-Curtis distance and the log ratio of Enterococcus relative abundance to GBS CFU was correlated with reduced distance between samples (Fig. 6). As the ratio of Staphylococcus to GBS is associated with community instability (distance), this suggests that turnover of Staphylococcus for GBS is contributing to the increase in pairwise distances.
Fig. 5.

GBS colonization across community states. a GBS CFU recovered from vaginal lavage fluid and grouped according to mCST classification on day 0. b GBS CFU recovered from tissues on day 3 and grouped according to mCST classification on day 0 c Bray-Curtis distance of vaginal lavage sequences of GBS and PBS groups over time. Statistical significance indicated with asterisk (p < 0.05, Mann-Whitney U Test)
Fig. 6.
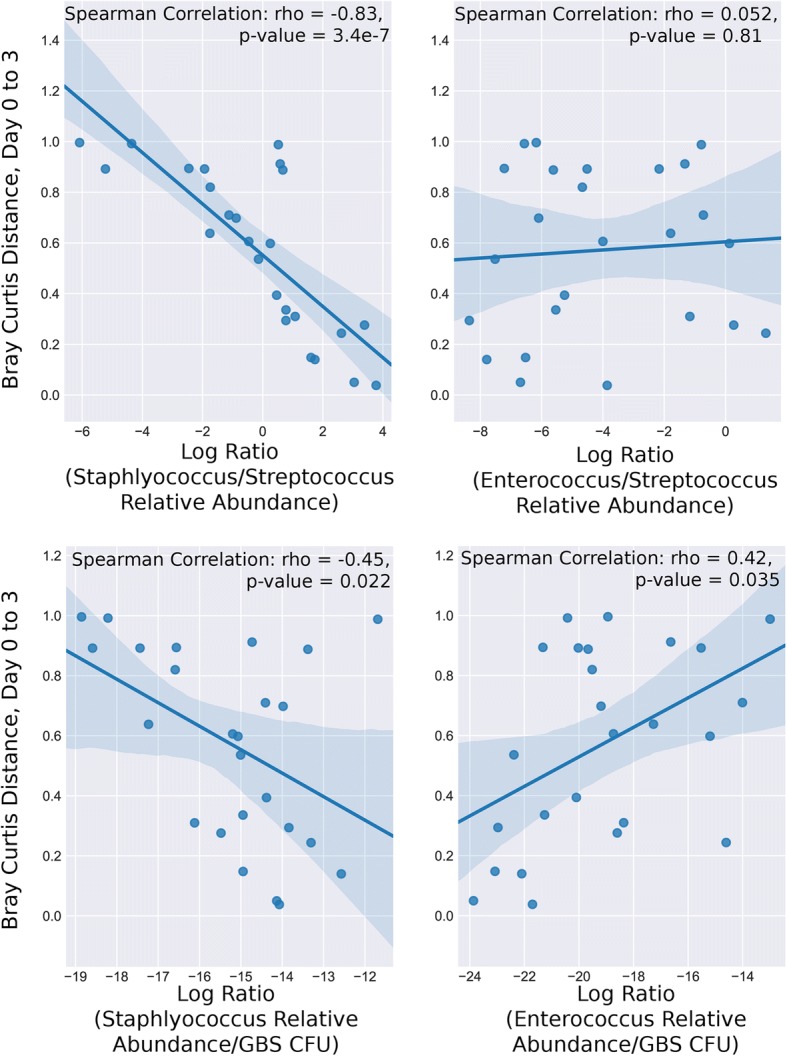
Community instability induced by GBS colonization correlates with Staphylococcus GBS turnover. Spearman correlations of pairwise Bray-Curtis distance between day 0 and day 3 samples with the log ratio of Staphylococcus relative abundance over Streptococcus relative abundance (top left) or GBS CFU (bottom left) and Enterococcus relative abundance over Streptococcus relative abundance (top right) or GBS CFU (bottom right)
For GBS-challenged mice, only 8/27 samples assigned to mCST I remained mCST I at the next consecutive time point. In comparison, control PBS mice had 19/27 mCST I samples assigned mCST I at the next consecutive time point. Additionally, mCST I mice exhibited significantly higher CFU/g in the uterus on day 3 (Fig. 5b) and uterus CFU was significantly correlated with Staphylococcus relative abundance (spearman, p = 0.0027), but this difference was nullified at later time points. By day 21, only mCST VI mice had GBS CFU remaining in vaginal, cervical, and uterine tissue (Additional file 3).
Discussion
Like humans, laboratory mouse strain C57Bl/6 J exhibit distinct vaginal microbiota community states (here named mCSTs) generally dominated by single bacterial taxa (Fig. 1). Three of the murine vaginal microbiota mCSTs were dominated by bacteria from different genera of Gram-positive facultative anaerobes: Staphylococcus, Enterococcus, and Lactobacillus. This dramatic variation in vaginal flora composition within an in-bred strain of mice raises the question of whether the commensal microbiota should be monitored in murine vaginal colonization and infection models. It is also likely that vaginal microbiota varies across vivaria, vendor, and mouse strain. Previous studies have examined the vaginal microbiota of BALB/c [24] and ICR mice [21]. In the ICR study, culture-based techniques revealed frequent presence of Streptococcus/Enterococcus, Staphylococcus, Lactobacillus, and Gram-negative rods [21]. In the BALB/c study, 16S amplicon sequencing described the most abundant phyla were Firmicutes and Proteobacteria, with distinctive separation of vaginal communities into two subclusters, one of which was dominated by Streptococcus [24]. Although the consistent presence of organisms such as Staphylococcus, Enterococcus, and Lactobacillus suggest there may be a core set of vaginal organisms across mouse strains, direct comparison of our results with these studies is difficult due to differences in sampling and analyses and smaller sample size (10 BALB/c mice and 27 ICR mice). Our findings indicate that the urogenital pathogens under study may encounter and interact with completely different commensal vaginal flora, even within the same cage of mice, potentially introducing variation and influencing experimental outcomes.
Numerous studies have examined constituents controlling vaginal persistence in mice including innate and adaptive immune responses [27–29] and GBS regulatory and virulence factors [30–32]. Additionally, several studies have noted GBS in the vaginal lumen in close proximity to native vaginal flora [29, 33]; however, none to date have examined the role of native vaginal microbiota in this model. Our study did not establish a definitive role for the microbiota in experimental GBS challenge, and this may be due to the high inoculum dose utilized in this model, which typically achieves > 90% colonization within the first week post-infection [26]. Future studies should examine a titration of inoculum to reveal more subtle contributions of the endogenous flora on GBS persistence. Nonetheless, we did note the otherwise relatively stable Staphylococcus-dominated CST I was perturbed by GBS challenge. Mice with mCST I also exhibited increased GBS ascension into uterine tissue by day 3. In multiple human studies, GBS has been co-isolated with Staphylococcus spp. in both pregnant and non-pregnant women [4, 34, 35]. Additionally, in invasive GBS polymicrobial infections, S. aureus is the most frequently co-isolated organism [36, 37]. One study demonstrated induction of S. aureus toxic shock syndrome toxin-1 by GBS culture supernatants in vitro [38], but whether this phenomenon occurs in the vaginal mucosa, or whether GBS virulence is also impacted, remains to be described.
In humans, vaginal CST does not generally correlate with GBS colonization status, except for the sub-group CST IV-A, a non-Lactobacillus dominant state [4]. Antagonism between Lactobacillus and GBS has been reported in both in vitro [39–41] and in vivo [42] model systems. Oral probiotics containing Lactobacillus have demonstrated efficacy in controlling GBS colonization in pilot human trials [43, 44]. Likewise, in mice dominated by Lactobacillus (CST IV), we were unable to detect any GBS beyond day 7, however, our sample size was too small (n = 2) to observed statistical differences. Furthermore, we observed that GBS was able to completely overtake the vaginal microbiota in some mice (> 80% of 16S reads), even 21 days post-infection. These mice had significantly higher GBS CFU over the course of the study than mice with flora that was not taken over by GBS on day 21 (student’s t-test, t-stat = 2.45, p = 0.018).
To our knowledge, the acquisition and maturation of the murine vaginal microbiota has not yet been examined. In this study, we used post-pubertal, non-pregnant mice, 8 weeks of age, which is a common age used to model urogenital diseases [17, 26]. At this age, the estrous cycle does not appear to influence the composition of the vaginal microbiota, but does impact sequencing success (Fig. 3). In line with this observation, previous studies using culture-based quantification have reported increased bacterial abundance in estrus compared to diestrus [21, 27, 45]. We selected vaginal lavage as our sampling method due to precedence in the literature for > 90% bacterial recovery [45, 46] and for inter-sample consistency and collection volume between our cohorts. To overcome the low biomass resulting from vaginal lavage, future studies may consider alternatives such as swabbing or scraping to obtain more biomass from the mucosa. Though physiology and immune cell populations fluctuate throughout the estrous cycle, potential variability in bacterial abundance provides further impetus for synchronization. The precedence for steroid hormone administration to achieve optimal infection of urogenital pathogens in murine models supports this observation [47, 48].
While factors such as mouse strain, facility, and vendor could certainly influence mCSTs as observed with gut microbiota [49, 50], our two separate studies and shipments of mice exhibited remarkable vaginal mCST homogeneity (Additional file 1) with the exception of an emerging Streptococcus-dominated mCST in GBS-infected mice. Future microbiome sequencing of the murine vaginal microbiota extended to other ages, strains, vendors, and vivaria, may reveal the existence of more community states or sub-groups within mCSTs.
Conclusions
This current study is the first to our knowledge to characterize the murine vaginal microbiota throughout estrus using 16S rRNA sequencing. We further demonstrate the influence of endogenous flora on successful colonization and by a human pathogen. This work underscores the importance of continuing to assess the native murine flora in models of human vaginal pathogens.
Methods
Mouse model and sample collection
All animal studies were reviewed and approved by the UC San Diego Animal Care and Use Committee and conducted using accepted veterinary standards. Mice were maintained on a 12 h light/dark cycle, with controlled temperature (19–22 °C) and 40–60% humidity. Mice were fed a commercial diet (2020X, Teklad) and sterile water ad libitum. See Additional file 4 for an overview of experimental design. For the estrous staging study (Study 1), 7-week-old female C57Bl/6 J mice (n = 40) were purchased from the Jackson Laboratory and housed five mice per cage. Mice were allowed to acclimate for one week prior to sample collection. To sample the vaginal microbiota, mice were manually restrained, and lavaged twice with 50 μl of sterile phosphate-buffered saline (PBS) using 200 μl Gel-Loading pipet tips (Fisher Scientific) in a laminar flow hood [26]. To control for environmental contamination, each day that lavage was performed, we used a single container of PBS, open in the hood during the entire sampling time. Every pipette tip was introduced to the same PBS container before lavaging. At the end of sample collection, an aliquot of this PBS was collected and used as a control for sequencing contamination. Twenty-five mice were lavaged every 3 days for 15 days, and fifteen mice were lavaged only twice, 15 days apart. After collecting lavage samples, 3 μl was removed from each sample for estrous cycle staging. Samples were visualized with a Zeiss Observer.D1 microscope at 200X magnification and the estrous cycle stage was identified independently by two individuals with results corroborated. For the GBS pathogen challenge study (Study 2), 7-week-old female C57Bl/6 mice (n = 72) were purchased from the Jackson Laboratory, housed four mice per cage, and allowed to acclimate for 1 week before infection. For GBS inoculation, we utilized a previously described GBS vaginal colonization model [26]. Briefly, mice were first injected IP with 0.5 mg of beta-estradiol suspended in 100 μl sesame oil, and 24 h later, mice were vaginally inoculated with 1 × 107 CFU of GBS COH1 [51] in 10 μl of PBS or PBS only as a control. Vaginal lavage sampling as described above was performed on days 0, 3, 7, 14, and 21 post-inoculation. For tissue collection at days 3, 14, and 21, mice were sacrificed and vagina, cervix, and uterus dissected and homogenized in PBS using 1 mm silica beads (Biospec) and shaken at 6000 rpm for 60 s using a MagNA Lyser (Roche). To quantify bacterial burdens, tissue homogenates were diluted and plated on CHROMagar StrepB (DRG International) to distinguish GBS CFU as light pink or mauve colonies.
16S rRNA gene amplicon sequencing
Sample processing was performed following the Earth Microbiome Project [52] DNA extraction and 16S sequencing protocol, detailed on the EMP website: http://www.earthmicrobiome.org/protocols-and-standards/16s/. In brief, lavage sample DNA was extracted using the 96-well MoBio Powersoil DNA kit. Barcoded 515F-806R primers targeting the V4 region of the 16S rRNA gene were used to for 16S amplification, and the resulting V4 amplicons were sequenced at UCSD Institute for Genomic Medicine (IGM) on an Illumina MiSeq.
Sequencing analysis
Raw 16S sequencing data was demultiplexed in Qiita [53] and processed using Deblur [54]. Microbiome data analysis, including feature table filtering, rarefaction, alpha diversity, beta diversity, and taxonomic assignments, was performed with QIIME 2 [55] v 2017.10. Taxonomic assignments used the naive bayes sklearn classifier in QIIME 2 trained on the 515F/806R region of Greengenes 13_8 99% OTUs. As many of the samples were low biomass, DNA contaminants from sequencing reagents and kits had a substantial impact on the dataset. Negative controls that went through the entire pipeline, from DNA extraction to sequencing, were used to catalog these contaminants. Sequences that appeared in negative controls were removed from the lavage samples, excluding a sample-abundant Lactobacillus sequence that was believed to be well to well contamination. Mitochondria and chloroplast 16S sequences were also removed.
Community state types were assigned using the hierarchical clustering with Ward’s linkage (SciPy) of Euclidean distances calculated on a table rarefied to 1500 sequences per sample and validity of clusters assessed with Silhouette Coefficient (sklearn). Data visualizations were generated with the python packages seaborn [56], matplotlib [57], and EMPeror [58] was used to create PCoA plots.
Statistical analysis was performed using SciPy [59] to perform the Mann-Whitney U for comparing pathogen CFU between mCSTs and Bray-Curtis distances between PBS and GBS mice at individual timepoints, and to perform spearman correlations for log ratios and Bray-Curtis distance. PERMANOVA implemented in QIIME 2 was used to assess statistical significance of beta diversity clusters.
Additional files
Community States are Consistent between Datasets. Heatmap displaying mCST and sequencing data from both studies. (PDF 570 kb)
Rarefaction Curves for GBS Challenge Experiment. Observed OTU Rarefaction curves. (PDF 588 kb)
GBS CFU is only recovered in mCST VI mice on Day 21. Displays GBS CFU in tissues by mCST for days 14 and 21. (PDF 280 kb)
Murine Study Design. Overview of experimental design for both studies. (PDF 372 kb)
Acknowledgements
We thank the UCSD vivarium staff for maintaining mice, and are grateful to Gregory Humphrey, Carolina Carpenter, Lisa Marotz, and Jason Munguia for technical support and Yoshiki Vazquez-Baeza for analysis advice.
Funding
This research was supported by a Seed Grant from the UC San Diego Center for Microbiome Innovation by NIH grant (HL107150) to VN. AV was supported by the UCSD Chancellor’s Research Excellence Scholarship, AC is the recipient of an A.P. Giannini Foundation Postdoctoral Fellowship, AR is the recipient of a UC President’s Postdoctoral Fellowship and also a San Diego IRACDA Fellow supported by National Institutes of Health (NIH)/NIGMS K12 GM06852 Award, and KP is the recipient of a UC Chancellor’s Postdoctoral Fellowship. Funding bodies played no roles in the design of the study and collection, analysis, and interpretation of data or in writing the manuscript.
Availability of data and materials
Sequencing Data used in this study is available in EBI under the accession number PRJEB25733, and is accessible at https://www.ebi.ac.uk/ena/data/view/PRJEB25733.
Abbreviations
- CFU
Colony forming units
- CST
Community state type
- GBS
Group B Streptococcus
- mCST
Murine community state type
- OTU
Operational taxonomic units
- PBS
Phosphate-buffered saline
Authors’ contributions
Conceived and designed experiments: AV, KAP. Performed experiments: AV, AC, AR, KAP. Analyzed and interpreted results: AV, RK, KAP. Contributed reagents/materials/analyses/discussion/manuscript edits: AC, AR, RK, VN. Drafted the manuscript: AV, KAP. All authors read and approved the final manuscript.
Ethics approval and consent to participate
All animal studies were reviewed and approved by the University of California San Diego Animal Care and Use Committee, protocol number S00227 M, and conducted using accepted veterinary standards.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Alison Vrbanac, Email: avrbanac@ucsd.edu.
Angelica M. Riestra, Email: ariestra@ucsd.edu
Alison Coady, Email: acoady@ucsd.edu.
Rob Knight, Email: robknight@ucsd.edu.
Victor Nizet, Email: vnizet@ucsd.edu.
Kathryn A. Patras, Phone: (858) 534-9760, Email: kpatras@ucsd.edu
References
- 1.Ravel J, Gajer P, Abdo Z, Schneider GM, Koenig SSK, McCulle SL, et al. Vaginal microbiome of reproductive-age women. Proc Natl Acad Sci U S A. 2011;Suppl 1:4680–4687. doi: 10.1073/pnas.1002611107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gajer P, Brotman RM, Bai G, Sakamoto J, Schütte UME, Zhong X, et al. Temporal dynamics of the human vaginal microbiota. Sci Transl Med. 2012;4:132ra52. doi: 10.1126/scitranslmed.3003605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brotman RM, Shardell MD, Gajer P, Fadrosh D, Chang K, Silver MI, et al. Association between the vaginal microbiota, menopause status, and signs of vulvovaginal atrophy. Menopause. 2014;21:450–458. doi: 10.1097/GME.0b013e3182a4690b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosen GH, Randis TM, Desai PV, Sapra KJ, Ma B, Gajer P, et al. Group B Streptococcus and the vaginal microbiota. J Infect Dis. 2017;216:744–751. doi: 10.1093/infdis/jix395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Srinivasan S, Hoffman NG, Morgan MT, Matsen FA, Fiedler TL, Hall RW, et al. Bacterial communities in women with bacterial vaginosis: high resolution phylogenetic analyses reveal relationships of microbiota to clinical criteria. PLoS One. 2012;7:e37818. doi: 10.1371/journal.pone.0037818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Donders G, Van Calsteren K, Bellen G, Reybrouck R, Van den Bosch T, Riphagen I, et al. Predictive value for preterm birth of abnormal vaginal flora, bacterial vaginosis and aerobic vaginitis during the first trimester of pregnancy. BJOG An Int J Obstet Gynaecol. 2009;116:1315–1324. doi: 10.1111/j.1471-0528.2009.02237.x. [DOI] [PubMed] [Google Scholar]
- 7.DiGiulio DB, Callahan BJ, McMurdie PJ, Costello EK, Lyell DJ, Robaczewska A, et al. Temporal and spatial variation of the human microbiota during pregnancy. Proc Natl Acad Sci. 2015;112:11060–11065. doi: 10.1073/pnas.1502875112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Low N, Chersich MF, Schmidlin K, Egger M, Francis SC, H HM, van de Wijgert J, et al. Intravaginal Practices, Bacterial Vaginosis, and HIV Infection in Women: Individual Participant Data Meta-analysis. PLoS Med. 2011;8:e1000416. doi: 10.1371/journal.pmed.1000416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klatt NR, Cheu R, Birse K, Zevin AS, Perner M, Noël-Romas L, et al. Vaginal bacteria modify HIV tenofovir microbicide efficacy in African women. Science. 2017;356:938–945. doi: 10.1126/science.aai9383. [DOI] [PubMed] [Google Scholar]
- 10.Rathod SD, Krupp K, Klausner JD, Arun A, Reingold AL, Madhivanan P. Bacterial vaginosis and risk for trichomonas vaginalis infection: a longitudinal analysis. Sex Transm Dis. 2011;38:882–886. doi: 10.1097/OLQ.0b013e31821f91a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martin DH, Zozaya M, Lillis RA, Myers L, Nsuami MJ, Ferris MJ. Unique vaginal microbiota that includes an unknown mycoplasma-like organism is associated with trichomonas vaginalis infection. J Infect Dis. 2013;207:1922–1931. doi: 10.1093/infdis/jit100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tamarelle J, de Barbeyrac B, Le Hen I, Thiébaut A, Bébéar C, Ravel J, et al. Vaginal microbiota composition and association with prevalent Chlamydia trachomatis infection: a cross-sectional study of young women attending a STI clinic in France. Sex Transm Infect. 2018;2017:053346. doi: 10.1136/sextrans-2017-053346. [DOI] [PubMed] [Google Scholar]
- 13.Wiesenfeld HC, Hillier SL, Krohn MA, Landers DV, Sweet RL. Bacterial vaginosis is a strong predictor of Neisseria gonorrhoeae and chlamydia trachomatis infection. Clin Infect Dis. 2003;36:663–668. doi: 10.1086/367658. [DOI] [PubMed] [Google Scholar]
- 14.Denton PW, Estes JD, Sun Z, Othieno FA, Wei BL, Wege AK, et al. Antiretroviral pre-exposure prophylaxis prevents vaginal transmission of HIV-1 in humanized BLT mice. PLoS Med. 2008;5:e16. doi: 10.1371/journal.pmed.0050016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Patras KA, Rösler B, Thoman ML, Doran KS. Characterization of host immunity during persistent vaginal colonization by group B Streptococcus. Mucosal Immunol. 2015;8:1339–1348. doi: 10.1038/mi.2015.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rahman D, Mistry M, Thavaraj S, Naglik JR, Challacombe SJ. Murine model of concurrent oral and vaginal Candida albicans colonisation. Methods Mol Biol. 2012;845:527–535. doi: 10.1007/978-1-61779-539-8_38. [DOI] [PubMed] [Google Scholar]
- 17.Yano J, Fidel PL Jr. Protocols for vaginal inoculation and sample collection in the experimental mouse model of candida vaginitis. J Vis Exp. 2011;58:3382. [DOI] [PMC free article] [PubMed]
- 18.Meysick KC, Garber GE. Interactions between trichomonas vaginalis and vaginal flora in a mouse model. J Parasitol. 1992;78:157–160. doi: 10.2307/3283708. [DOI] [PubMed] [Google Scholar]
- 19.Sierra L-J, Brown AG, Barilá GO, Anton L, Barnum CE, Shetye SS, et al. Colonization of the cervicovaginal space with Gardnerella vaginalis leads to local inflammation and cervical remodeling in pregnant mice. PLoS One. 2018;13:e0191524. doi: 10.1371/journal.pone.0191524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Su H, Parnell M, Caldwell HD. Protective efficacy of a parenterally administered MOMP-derived synthetic oligopeptide vaccine in a murine model of chlamydia trachomatis genital tract infection: serum neutralizing IgG antibodies do not protect against chlamydial genital tract infection. Vaccine. 1995;13:1023–1032. doi: 10.1016/0264-410X(95)00017-U. [DOI] [PubMed] [Google Scholar]
- 21.Noguchi K, Tsukumi K, Urano T. Qualitative and quantitative differences in normal vaginal flora of conventionally reared mice, rats, hamsters, rabbits, and dogs. Comp Med. 2003;53:404–412. [PubMed] [Google Scholar]
- 22.Cowley HM, Heiss GS. Changes in vaginal bacterial Flora during the Oestrous cycle of the mouse. Microb Ecol Health Dis. 1991;4:229–235. doi: 10.3109/08910609109140147. [DOI] [Google Scholar]
- 23.Jašarević E, Howerton CL, Howard CD, Bale TL. Alterations in the vaginal microbiome by maternal stress are associated with metabolic reprogramming of the offspring gut and brain. Endocrinology. 2015;156:3265–3276. doi: 10.1210/en.2015-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barfod K, Roggenbuck M, Hansen L, Schjørring S, Larsen S, Sørensen S, et al. The murine lung microbiome in relation to the intestinal and vaginal bacterial communities. BMC Microbiol. 2013;13:303. doi: 10.1186/1471-2180-13-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Caligioni CS. Assessing reproductive status/stages in mice. Curr Protoc Neurosci. 2009;Appendix 4:Appendix 4I. doi: 10.1002/0471142301.nsa04is48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Patras KA, Doran KS. A murine model of group B Streptococcus vaginal colonization. J Vis Exp. 2016;117:54708. [DOI] [PMC free article] [PubMed]
- 27.Patras KA, Wang N-Y, Fletcher EM, Cavaco CK, Jimenez A, Garg M, et al. Group B Streptococcus CovR regulation modulates host immune signalling pathways to promote vaginal colonization. Cell Microbiol. 2013;15:1154–1167. doi: 10.1111/cmi.12105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baker JA, Lewis EL, Byland LM, Bonakdar M, Randis TM, Ratner AJ. Mucosal vaccination promotes clearance of Streptococcus agalactiae vaginal colonization. Vaccine. 2017;35:1273–1280. doi: 10.1016/j.vaccine.2017.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vornhagen J, Armistead B, Santana-Ufret V, Gendrin C, Merillat S, Coleman M, et al. Group B streptococcus exploits vaginal epithelial exfoliation for ascending infection. J Clin Invest. 2018;128:1985–1999. doi: 10.1172/JCI97043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cook LCC, Hu H, Maienschein-Cline M, Federle MJ. A vaginal tract signal detected by the group B Streptococcus SaeRS system elicits transcriptomic changes and enhances murine colonization. Infect Immun. 2018;86:e00762–17. [DOI] [PMC free article] [PubMed]
- 31.Sheen TR, Jimenez A, Wang N-Y, Banerjee A, van Sorge NM, Doran KS. Serine-rich repeat proteins and pili promote Streptococcus agalactiae colonization of the vaginal tract. J Bacteriol. 2011;193:6834–6842. doi: 10.1128/JB.00094-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patras Kathryn A, Derieux Jaclyn, Al-Bassam Mahmoud M, Adiletta Nichole, Vrbanac Alison, Lapek John D, Zengler Karsten, Gonzalez David J, Nizet Victor. Group B Streptococcus Biofilm Regulatory Protein A Contributes to Bacterial Physiology and Innate Immune Resistance. The Journal of Infectious Diseases. 2018;218(10):1641–1652. doi: 10.1093/infdis/jiy341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carey AJ, Tan CK, Mirza S, Irving-Rodgers H, Webb RI, Lam A, et al. Infection and cellular defense dynamics in a novel 17 -estradiol murine model of chronic human group B Streptococcus genital tract colonization reveal a role for Hemolysin in persistence and neutrophil accumulation. J Immunol. 2014;192:1718–1731. doi: 10.4049/jimmunol.1202811. [DOI] [PubMed] [Google Scholar]
- 34.Carson HJ, Lapoint PG, Monif GRG. Interrelationships within the bacterial Flora of the female genital tract. Infect Dis Obstet Gynecol. 1997;5:303–309. doi: 10.1155/S1064744997000525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Andrews WW, Schelonka R, Waites K, Stamm A, Cliver SP, Moser S. Genital tract methicillin-resistant Staphylococcus aureus. Obstet Gynecol. 2008;111:113–118. doi: 10.1097/01.AOG.0000298344.04916.11. [DOI] [PubMed] [Google Scholar]
- 36.Farley MM, Harvey C, Stull T, Smith JD, Schuchat A, Wenger JD, et al. A population-based assessment of invasive disease due to group B Streptococcus in nonpregnant adults. N Engl J Med. 1993;328:1807–1811. doi: 10.1056/NEJM199306243282503. [DOI] [PubMed] [Google Scholar]
- 37.Skoff TH, Farley MM, Petit S, Craig AS, Schaffner W, Gershman K, et al. Increasing burden of invasive group B streptococcal disease in nonpregnant adults, 1990–2007. Clin Infect Dis. 2009;49:85–92. doi: 10.1086/599369. [DOI] [PubMed] [Google Scholar]
- 38.MacPhee RA, Miller WL, Gloor GB, McCormick JK, Hammond J-A, Burton JP, et al. Influence of the vaginal microbiota on toxic shock syndrome toxin 1 production by Staphylococcus aureus. Appl Environ Microbiol. 2013;79:1835–1842. doi: 10.1128/AEM.02908-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.De Gregorio PR, Tomas MSJ, Terraf MCL, Nader-Macias MEF. In vitro and in vivo effects of beneficial vaginal lactobacilli on pathogens responsible for urogenital tract infections. J Med Microbiol. 2014;63:685–696. doi: 10.1099/jmm.0.069401-0. [DOI] [PubMed] [Google Scholar]
- 40.Ortiz L, Ruiz F, Pascual L, Barberis L. Effect of two probiotic strains of lactobacillus on in vitro adherence of listeria monocytogenes, Streptococcus agalactiae, and Staphylococcus aureus to vaginal epithelial cells. Curr Microbiol. 2014;68:679–684. doi: 10.1007/s00284-014-0524-9. [DOI] [PubMed] [Google Scholar]
- 41.Ruíz FO, Gerbaldo G, García MJ, Giordano W, Pascual L, Barberis IL. Synergistic effect between two Bacteriocin-like inhibitory substances produced by lactobacilli strains with inhibitory activity for Streptococcus agalactiae. Curr Microbiol. 2012;64:349–356. doi: 10.1007/s00284-011-0077-0. [DOI] [PubMed] [Google Scholar]
- 42.De Gregorio PR, Juárez Tomás MS, Leccese Terraf MC, Nader-Macías MEF. Preventive effect of Lactobacillus reuteri CRL1324 on group B Streptococcus vaginal colonization in an experimental mouse model. J Appl Microbiol. 2015;118:1034–1047. doi: 10.1111/jam.12739. [DOI] [PubMed] [Google Scholar]
- 43.Ho M, Chang Y-Y, Chang W-C, Lin H-C, Wang M-H, Lin W-C, et al. Oral lactobacillus rhamnosus GR-1 and lactobacillus reuteri RC-14 to reduce group B Streptococcus colonization in pregnant women: a randomized controlled trial. Taiwan J Obstet Gynecol. 2016;55:515–518. doi: 10.1016/j.tjog.2016.06.003. [DOI] [PubMed] [Google Scholar]
- 44.Olsen P, Williamson M, Traynor V, Georgiou C. The impact of oral probiotics on vaginal group B streptococcal colonisation rates in pregnant women: a pilot randomised control study. Women Birth. 2018;31:31–37. doi: 10.1016/j.wombi.2017.06.012. [DOI] [PubMed] [Google Scholar]
- 45.Larsen B, Markovetz AJ, Galask RP. Relationship of vaginal cytology to alterations of the vaginal microflora of rats during the estrous cycle. Appl Environ Microbiol. 1977;33:556–562. doi: 10.1128/aem.33.3.556-562.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Larsen B, Markovetz AJ, Galask RP. Quantitative alterations in the genital microflora of female rats in relation to the estrous cycle. J Infect Dis. 1976;134:486–489. doi: 10.1093/infdis/134.5.486. [DOI] [PubMed] [Google Scholar]
- 47.Islam EA, Shaik-Dasthagirisaheb Y, Kaushic C, Wetzler LM, Gray-Owen SD. The reproductive cycle is a pathogenic determinant during gonococcal pelvic inflammatory disease in mice. Mucosal Immunol. 2016;9:1051–1064. doi: 10.1038/mi.2015.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pal S, Hui W, Peterson EM, De la Maza LM. Factors influencing the induction of infertility in a mouse model of chlamydia trachomatis ascending genital tract infection. J Med Microbiol. 1998;47:599–605. doi: 10.1099/00222615-47-7-599. [DOI] [PubMed] [Google Scholar]
- 49.Nguyen TLA, Vieira-Silva S, Liston A, Raes J. How informative is the mouse for human gut microbiota research? Dis Model Mech. 2015;8:1–16. doi: 10.1242/dmm.017400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ericsson AC, Davis JW, Spollen W, Bivens N, Givan S, Hagan CE, et al. Effects of vendor and genetic background on the composition of the fecal microbiota of inbred mice. PLoS One. 2015;10:e0116704. doi: 10.1371/journal.pone.0116704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wilson CB, Weaver WM. Comparative susceptibility of group B streptococci and Staphylococcus aureus to killing by oxygen metabolites. J Infect Dis. 1985;152:323–329. doi: 10.1093/infdis/152.2.323. [DOI] [PubMed] [Google Scholar]
- 52.Thompson LR, Sanders JG, McDonald D, Amir A, Ladau J, Locey KJ, et al. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature. 2017;551:457–463. doi: 10.1038/nature24621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Qiita. https://qiita.ucsd.edu/. Accessed 23 Mar 2018.
- 54.Amir A, McDonald D, Navas-Molina JA, Kopylova E, Morton JT, Zech Xu Z, et al. Deblur rapidly resolves single-nucleotide community sequence patterns. mSystems. 2017;2:e00191–16. [DOI] [PMC free article] [PubMed]
- 55.Kuczynski J, Stombaugh J, Walters WA, González A, Caporaso JG, Knight R. Using QIIME to analyze 16S rRNA gene sequences from microbial communities. Curr Protoc Bioinforma. 2011;36(1). 10.1002/0471250953.bi1007s36. [DOI] [PMC free article] [PubMed]
- 56.seaborn: statistical data visualization — seaborn 0.8.1 documentation. http://seaborn.pydata.org/. Accessed 23 Mar 2018.
- 57.Hunter JD. Matplotlib: a 2D graphics environment. Comput Sci Eng. 2007;9:90–95. doi: 10.1109/MCSE.2007.55. [DOI] [Google Scholar]
- 58.Vázquez-Baeza Y, Pirrung M, Gonzalez A, Knight R. EMPeror: a tool for visualizing high-throughput microbial community data. Gigascience. 2013;2:16. doi: 10.1186/2047-217X-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.SciPy.org — SciPy.org. https://www.scipy.org/. Accessed 23 Mar 2018.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Community States are Consistent between Datasets. Heatmap displaying mCST and sequencing data from both studies. (PDF 570 kb)
Rarefaction Curves for GBS Challenge Experiment. Observed OTU Rarefaction curves. (PDF 588 kb)
GBS CFU is only recovered in mCST VI mice on Day 21. Displays GBS CFU in tissues by mCST for days 14 and 21. (PDF 280 kb)
Murine Study Design. Overview of experimental design for both studies. (PDF 372 kb)
Data Availability Statement
Sequencing Data used in this study is available in EBI under the accession number PRJEB25733, and is accessible at https://www.ebi.ac.uk/ena/data/view/PRJEB25733.