

Abstract
Pathogenic germline DICER1 variants cause a hereditary cancer predisposition syndrome with a variety of manifestations. In addition to conferring increased cancer risks for pleuropulmonary blastoma (PPB) and ovarian sex cord-stromal tumors, particularly Sertoli–Leydig cell tumor, individuals with pathogenic germline DICER1 variants may also develop lung cysts, cystic nephroma, renal sarcoma and Wilms tumor, nodular hyperplasia of the thyroid, nasal chondromesenchymal hamartoma, ciliary body medulloepithelioma, genitourinary embryonal rhabdomyosarcoma and brain tumors including pineoblastoma and pituitary blastoma.
In May 2016, the International PPB Registry convened the inaugural International DICER1 Symposium to develop consensus testing, surveillance and treatment recommendations. Attendees from North America, Europe and Russia provided expert representation from the disciplines of pediatric oncology, endocrinology, genetics, genetic counseling, radiology, pediatric surgery, pathology and clinical research. Recommendations are provided for genetic testing, prenatal management, and surveillance for DICER1-associated pulmonary, renal, gynecologic, thyroid, ophthalmologic, otolaryngologic, central nervous system tumors and gastrointestinal polyps. Risk for most DICER1-associated neoplasms is highest in early childhood and decreases in adulthood. Individual and caregiver education and judicious imaging-based surveillance are the primary recommended approaches. These testing and surveillance recommendations reflect a consensus of expert opinion and current literature. As DICER1 research expands, guidelines for screening and treatment will continue to be updated.
Keywords: DICER1, pleuropulmonary blastoma, Sertoli-Leydig cell tumor, hereditary cancer, genetic counseling
Introduction
Pleuropulmonary blastoma (PPB) (OMIM 601200) is the most common primary lung malignancy in children.(1) This rare, dysembryonic lung neoplasm of childhood was first recognized in 1988.(1) PPB is analogous to other embryonal tumors such as Wilms tumor, hepatoblastoma and neuroblastoma in which the neoplastic process has a morphologic resemblance to the developmental stages in organogenesis.(2, 3) PPB is primarily diagnosed in infants and young children, mainly before 7 years of age.(4) There is compelling clinicopathologic evidence that PPB progresses through successive stages from a purely multicystic lesion (Type I) to a mixed cystic-solid stage (Type II) and solid stage (Type III).(5, 6) The malignant component of PPB derives from lung mesenchyme. In Type I tumors, the malignant component is limited to cyst septa and is most often composed of primitive rhabdomyoblasts. As progression ensues, the tumor cells acquire high grade and often anaplastic features with a variety of patterns from primitive blastemal, embryonal rhabdomyosarcoma to malignant cartilage and/or spindle cell sarcoma.(7) A fourth type of PPB, Type Ir “regressed”, does not possess the primitive cell component of malignant PPB and can be found in individuals of all ages.(4) We hypothesize that these so-called regressed (or non-progressed) PPBs are analogous to some cases of congenital neuroblastoma or nephrogenic rests.
In 1988, building on the original description of 11 cases as a new entity,(3) the International PPB Registry was established to collect and centrally review the pathology from patients with putative PPB, and to collect clinical records and annual follow-up data for these patients. The International PPB Registry is a partnership between Children’s Minnesota, Minneapolis, MN, Washington University School of Medicine, St. Louis, MO, and Children’s National Medical Center, Washington, DC. In 1996, data from the International PPB Registry led to the initial observation of the association between PPB and other neoplasms in patients with PPB and their close relatives.(2) In 2009, the Registry identified pathogenic germline DICER1 variants as the genetic basis of PPB.(8) Over time, the constellation of DICER-associated benign and malignant neoplasms has expanded to include Sertoli-Leydig cell tumor (SLCT) of the ovary, cystic nephroma (CN), renal sarcoma and Wilms tumor (WT), nodular hyperplasia and carcinoma of the thyroid gland, nasal chondromesenchymal hamartoma (NCMH), embryonal rhabdomyosarcoma (ERMS), ciliary body medulloepithelioma (CBME), pituitary blastoma and pineoblastoma.(8–31) In 2011, a second registry, the International Ovarian and Testicular Stromal Tumor (OTST) Registry, was established to enroll individuals with rare ovarian and testicular sex cord-stromal tumors with an initial focus on the relationship between DICER1 and sex cord-stromal tumors. To date, the International PPB Registry and the OTST Registry have enrolled more than 500 and 160 individuals, respectively.
In this paper, we review the biology of DICER1, associated cancer risks and other clinical manifestations. Approaches to the diagnosis of DICER1-associated conditions and risk management recommendations for patients and family members with germline pathogenic variants in DICER1 are presented.
Methodology
Data from the International PPB and OTST Registries were collated to generate a dataset of 682 individuals from 652 families with pathogenic germline variants in DICER1 or clinical history of DICER1-associated conditions. Although not all patients in the registries had germline DICER1 status available for review, the majority of individuals with PPB have germline mutations, and nearly all individuals with DICER1-associated conditions enrolled in the registries were individuals with PPB or ascertained on the basis of a first or second degree relative with PPB. Informed consent was previously obtained. Protocols were reviewed and approved by the relevant human subjects committees. Age at diagnosis was determined for all participants and reviewed in aggregate.
The International PPB Registry staff continuously performs literature searches and reviews pertaining to keywords, “PPB”, “pulmonary blastoma”, “pulmonary rhabdomyosarcoma”, “DICER1”, and other “DICER1-associated conditions” and through May 2017 had identified a total of 608 articles and abstracts which were reviewed and relevant information abstracted. In addition, a May 2017 PubMed literature search using “DICER1” paired with each of the following keywords: “PPB”, “Sertoli-Leydig cell tumor”, “gynandroblastoma”, “juvenile hamartomatous polyp”, “cystic nephroma”, “multinodular goiter”, “thyroid adenoma”, “thyroid carcinoma”, “ciliary body medulloepithelioma”, “botryoid-type embryonal rhabdomyosarcoma”, “nasal chondromesenchymal hamartoma”, “renal sarcoma”, “pituitary blastoma”, and “pineoblastoma” identified a total of 248 articles, 159 of which were examined and relevant information abstracted (Supplementary Table 1). All papers identified had their bibliographies searched manually to recognize further citations of interest and identify individuals with clinical characteristics not encompassed by Registry data (Figure 1). Data from these papers was aggregated and reviewed to broadly assess general age and symptoms at clinical presentation and clinical course for each tumor type.
Figure 1.
QUORUM diagram for screening evidence review.
In May 2016, International PPB Registry data was presented at the inaugural International DICER1 Symposium and reviewed by participants. Participants included pediatric oncologists, geneticists and genetic counselors, endocrinologists, radiologists, pediatric surgeons, pathologists and clinical researchers from the U.S., Canada, Germany, France and Russia. Based on ensuing evidence-based discussions and expert opinion, consensus guidelines were developed as described below. Following the meeting, an additional systematic review of the available literature was carried out using PubMed to ensure no relevant articles were omitted, and revised consensus guidelines were circulated to ensure harmonization.
Genotyping
Over 70% of individuals with PPB have a germline loss-of-function mutation with a second, tumor specific missense mutation in the RNase IIIb domain. Most germline loss-of-function mutations are inherited, but 10–20% appear to arise de novo. Intronic mutations, which disrupt splicing, and large deletions have also been described.(32, 33, 34)
The tumor-specific RNase IIIb missense mutations involve one of 5 “hotspot” codons, E1705, D1709, G1809, D1810 or E1813. Missense mutations at one of these hotspots prevent the proper cleavage of the mature 5p microRNA (miRNA) of the precursor miRNA hairpin.(35) It is proposed that improper cleavage of the 5p miRNA leads to rapid degradation. Lack of mature 5p miRNAs, including the let-7 family, leaves numerous target messenger RNAs (mRNAs) unregulated. Although most children with PPB have a germline loss-of-function mutation and a second tumor specific “hotspot” mutation, approximately 10% exhibit mosaicism. Individuals with mosaic “hotspot” mutations in the RNase IIIb domain may have tumor specific loss-of-function mutations and have earlier onset disease and a higher frequency of multisite disease.(33, 35) We have postulated this severely affected phenotype may simply result from probabilities of generating the characteristic “loss-of-function plus hotspot” two hit typical of a DICER1 syndrome neoplasm. Consider the two scenarios: a child with germline loss-of-function DICER1 mutations vs. a child with mosaicism for a missense mutation in one of the 5 hotspot amino acids. In both of these scenarios, a second mutation in the wild-type DICER1 allele must occur, in the appropriate developmental or physiologic time window, to initiate a DICER1 neoplasm. In the first child, this second somatic mutation must be a base substitution in one of the 36 potential bases in the RNase IIIb domain, a seemingly small target in the genome. In the second child, the second somatic mutation only needs to be a loss-of-function which may be hundreds of times more likely (chromosome loss, gene deletion, truncating mutations, etc).(33) An alternate possibility is that the allele combination of “hotspot plus wildtype” may be tumorigenic on its own.(36, 37) Regardless of the reason, initial observations of phenotype in individuals with mosaicism for “hotspot” mutations may warrant more intensive surveillance than discussed in this guideline.
About 10–15% of individuals with DICER1 tumors appear to have biallelic mutations limited to tumor tissue, or low-level mosaicism for loss-of-function mutations. To date, all the children with suspected tumor limited mutations have had single site disease(33) and it is reasonable to consider that these children may not need intensive surveillance. But without extensive sequencing of multiple tissue types and sites, it can be difficult to differentiate a child with tumor-limited DICER1 mutations from one with low-level mosaicism for a loss-of-function mutation. Thus it is not yet clear if there should be reductions in surveillance for this genetically unique subgroup at this time. At a minimum, individuals with presumed biallelic somatic mutations should be aware of the risk for low-level mosaicism and individuals and their health care providers should be aware of the need to seek care expeditiously if signs or symptoms of DICER1-related conditions develop.
Tumor-based or somatic testing for DICER1 mutations may be useful in a variety of clinical settings including diagnosing tumors which are challenging histologically, confirmation of biallelic somatic mutations, assessment for mosaicism and determining whether new findings represents a metastatic/recurrent or metachronous tumor.
Prevalence, inheritance, penetrance and expressivity
DICER1 is inherited as an autosomal dominant condition with decreased penetrance. Therefore, the children of individuals with a DICER1 pathogenic variant have a 50%, chance of inheriting the mutation. An analysis of the prevalence of pathogenic germline DICER1 variation in the Exome Aggregation Consortium (excluding cases ascertained from The Cancer Genome Atlas) found that approximately 1:2,529 – 1:10,600 individuals in the general population carry a pathogenic or likely pathogenic DICER1 variant.(38) The penetrance of each of the DICER1-associated conditions is not fully understood. Lung cysts, presumably Type I or Type Ir PPB, and thyroid nodules appear to be the most common manifestations in individuals with germline loss-of-function mutations. When available, the penetrance of specific conditions is discussed within the sections below (e.g., thyroid). The penetrance of Type II and Type III PPB and less common conditions, such as pineoblastoma, are the subject of ongoing investigation. While individuals with a germline loss-of-function mutation often have between 0 and 2 sites of disease, as discussed above, individuals with mosaic “hotspot” mutations are more likely to have more than a single site manifestation.
Indications for DICER1 genetic counseling and testing
We recommend genetic counseling and testing for individuals with a personal history of at least one major or two minor indications for testing (Table 1) and/or for individuals with one minor indication and a family history of a major or minor indication. Ideally, genetic testing should be performed on a proband with a DICER1-associated tumor when available. In cases where proband testing is not feasible, testing should be considered for those with a family history of DICER1-associated conditions so that appropriate surveillance can be undertaken. Pre-test and post-test genetic counseling is advised to ensure that patients, and/or their family as in the case of minor children, are fully informed and prepared for any subsequent decisions. Ethical considerations, especially for presymptomatic testing of a minor child, should be discussed and factored into the decision process, as some individuals may choose, and have the right to choose, not to know their/their child’s genetic status. Individuals at 50% risk of a germline pathogenic variant based on family history who do not pursue genetic testing should follow surveillance guidelines as if they have a DICER1 mutation unless/until genetic testing confirms that they did not inherit the familial mutation. Genetic counseling may also help identify the most appropriate test to order on the most appropriate individual(s) in the family and/or help with interpretation, especially with variants of uncertain significance.
Table 1.
Indications for DICER1 testing. Consider germline DICER1 genetic testing in an individual with one major or two minor indications.
| Major: | Minor: |
|---|---|
| −Individuals with PPB (all types) | −Lung cyst(s) in adults |
| −Lung cyst(s) in childhood, especially if multi−septated, multiple or bilateral | −Renal cyst(s)* |
| −Thoracic embryonal rhabdomyosarcoma* | −Wilms tumor |
| −Cystic nephroma | −Multinodular goiter or differentiated thyroid cancer |
| −Genitourinary sarcomas including undifferentiated sarcoma* | −Embryonal rhabdomyosarcoma other than thoracic or gynecologic* |
| −Ovarian Sertoli−Leydig cell tumor | |
| −Gynandroblastoma | −Poorly differentiated neuroendocrine tumor |
| −Uterine cervical or ovarian embryonal rhabdomyosarcoma* | |
| −Genitourinary/gynecologic neuroendocrine tumors | −Undifferentiated sarcoma* |
| −Macrocephaly* | |
| − Multinodular goiter or thyroid cancer in 2 or more 1st degree relatives or in an index patient with a family history consistent with DICER1 syndrome* | −Consider testing for any childhood cancer in constellation with any other minor criteria |
| −Childhood onset multinodular goiter* or differentiated thyroid cancer* | |
| −Ciliary body medulloepithelioma | |
| −Nasal chondromesenchymal hamartoma | |
| −Pineoblastoma | |
| −Pituitary blastoma |
Multinodular goiter, differentiated thyroid cancer (papillary or follicular carcinomas), sarcomas, Wilms tumor, neuroendocrine tumors, renal cysts and macrocephaly may also be associated with other genetic predisposition syndromes. Consider testing for additional hereditary cancer predispositions and/or next generation sequencing panel that includes deletion/duplication of DICER1 and/or other genes indicated by clinical and family history
For individuals with pathogenic germline DICER1 variants, we recommend additional site-specific genetic testing of that variant for all first-degree relatives. Testing should be prioritized in children less than 7 years of age, who are at the greatest risk for any one of the pathologic types of PPB. Second and third degree relatives may also benefit from testing, especially if they have young children.
Recommendations for testing must also consider the role of DICER1 in individual tumor types. For example, although Wilms tumor may be DICER1-associated, current studies suggest that only 1–5% of Wilms tumor is DICER1-associated in contrast to PPB in which pathogenic germline DICER1 variants underlie more than 70% of cases.(33) However, since genetic testing is shifting from single gene to panel testing, it may be appropriate to add DICER1 gene testing if a child with Wilms tumor or another DICER1-related tumor is being tested for other genes.
Testing algorithm
If no mutation is detected via DICER1 sequencing in a germline specimen (e.g., blood, saliva or skin fibroblasts), germline DICER1 deletion/duplication analyses should be performed, as some cases are due to copy-number variants. Some labs offer sequencing and deletion/duplication testing simultaneously. Whenever possible, tumor tissue should be tested concurrently with germline DNA. The presence of two somatic mutations in tumor tissue in the setting of negative germline testing may then help confirm a non-inherited case. The possibility of mosaicism may be considered with testing of other normal tissue, tumor tissue, or future children, if feasible. If the individual has two or more DICER1 neoplasms, sequencing both neoplasms may identify the recurrent variant and classify the individual with subsequent risk assessment based on whether or not the mosaicism is for loss-of-function or a hotspot mutation (increased surveillance for the latter). There is probably a pragmatic limit to how many tissue types should be tested to rule out mosaicism in a child with single site disease. Opportunistic sequencing of non-neoplastic tissue taken at related or unrelated surgeries (such as testing of adjacent normal ovary when salpino-oophorectomy is performed for sex-cord stromal tumor) may be informative. Testing of relatives is not generally indicated for individuals with biallelic somatic mutations in the absence of detectable pathogenic germline DICER1 variants, however, it should be noted that gonadal mosaicism may result in risk to offspring and therefore consideration could be given to offering children of individuals with possible mosaicism site specific testing in the rare event that their germline is involved.
Timing of testing
If testing is initiated due to a known familial DICER1 pathogenic variant, we recommend prioritization of predictive testing of first-degree relatives. For newborns, testing is recommended before 4 months of age so that pulmonary screening can be initiated only in at-risk infants (see Prenatal/Management, below). In these cases, testing only for the known familial mutation would be indicated.
Prenatal management
We recommend third trimester ultrasound (US) for women whose fetuses are at risk for a pathogenic germline DICER1 variant from either the maternal or paternal side to detect large lung cysts which might require early intervention after delivery. Prenatal US also has a higher sensitivity for cystic lung disease than neonatal chest x-ray. We know of no medical indication requiring prenatal DICER1 testing; prenatal US will detect large cysts which only rarely will require immediate intervention. Cysts seen on prenatal US require computed tomography follow-up even if chest x-ray at birth is normal based on experience in other conditions characterized by cystic lung lesions.
Both prenatal and preimplantation genetic counseling are options for determining risk to an embryo/fetus, if waiting until after birth is not desired. Parents who are interested in exploring these options should be referred to preimplantation/prenatal centers. In discussions regarding prenatal testing, a thorough dialogue should include the relatively low penetrance of malignant conditions and high likelihood of a healthy child/adult (one without DICER1-associated conditions or minor findings) even if a DICER1 mutation is inherited. Penetrance for serious conditions is considered low. Less common and more serious manifestations of DICER1 such as Type III PPB are anticipated to be less likely when screening regimens are initiated in early childhood well before the risk period for the development of Type II and III PPB.
Screening recommendations for DICER1-associated conditions by system
Screening recommendations must consider typical age of onset, potential benefits of early detection and risks and availability of screening modalities (e.g., radiation exposure, need for sedation and likelihood of false positives). Individual and family education is strongly recommended (see Table 2). In addition to imaging-based surveillance, individuals and families should be counseled at each visit regarding potential signs and symptoms of DICER1-associated conditions (Table 2). Individuals should also continue to receive appropriate age- and gender-specific preventive screening studies and immunizations.
Table 2.
Suggested signs and symptoms and imaging surveillance by system for individuals with DICER1 pathogenic variants
| System | Signs/Symptoms to consider |
Condition of interest | Screening, Clinical and Radiographic |
|---|---|---|---|
| Lung | Tachypnea, cough, fever, and pain; pneumothorax |
- PPB - Lung cysts -Pulmonary blastoma |
CXR at birth and every 4– 6 months until 8 years of age, every 12 months 8 −12 years of age; consider a CT of chest at 3 − 6 months of age.* Toddlers: if initial CT normal: repeat between 2−1/2 and 3 years of age.* If mutation detected at > 12 years of age, consider baseline CXR or chest CT. |
| Thyroid | Visible or palpable thyroid nodule(s) Persistent cervical lymphadenopathy Hoarseness Dysphagia Neck pain Cough |
− Multinodular goiter; − Differentiated thyroid cancer |
Baseline thyroid US by 8 years of age then every 3 years or with symptoms/findings on physical exam. With anticipated chemotherapy or radiation therapy: baseline US and then annually for 5 years, decreasing to every 2 to 3 years if no nodules are detected |
|
Female reproductive tract |
Hirsutism Virilization Abdominal distension, pain or mass |
− SLCT − Gynandroblastoma − Cervical embryonal rhabdomyosarcoma |
For females beginning at 8 – 10 years of age: pelvic and abdominal US every 6−12 months at least until age 40. End of interval is undetermined but current oldest patient with DICER1− associated SLCT was 61 years of age. Education regarding symptoms strongly recommended. |
| Renal | Abdominal or flank mass and/or pain, hematuria |
− Wilms tumor − Renal sarcoma − Cystic nephroma |
Abdominal US every 6 months until 8 years of age then every 12 months until 12 years of age. If mutation detected at > 12 years of age, consider baseline abdominal US |
| Gastrointestinal | Signs of intestinal obstruction |
− Small intestine polyps |
Education regarding symptoms recommended. |
|
Central nervous system And head and neck (excluding thyroid) |
Headache, emesis, diplopia, decreased ability for upward gaze, altered gait (pineoblastoma); Precocious puberty; Cushing’s syndrome (pituitary blastoma); Decreased visual acuity and leukocoria (CBME); Nasal obstruction (NCMH) |
− Macrocephaly − Pineoblastoma − Pituitary blastoma − CBME − NCMH |
Physical exam. Annual routine dilated ophthalmologic exam (generally unsedated) with visual acuity screening from 3 years of age through at least 10 years of age. Further testing if clinically indicated. Recommend urgent MRI for any symptoms of intracranial pathology. |
Key: PPB = pleuropulmonary blastoma; CXR = chest x−ray; CT = computed tomography; US = ultrasound; SLCT = Sertoli−Leydig cell tumor; CBME = ciliary body medulloepithelioma; NCMH = nasal chondromesenchymal hamartoma.
When CT is performed, techniques to minimize radiation exposure should be employed. As novel magnetic resonance imaging (MRI) techniques are developed that will eventually allow detection of small cystic lesions, transition to non−radiation containing cross sectional imaging should be considered.
Lung
The finding of PPB in siblings first led to the recognition of PPB as a familial disease.(2) This neoplasm remains one of the pathognomonic manifestations of pathogenic germline DICER1 variants and one of the most important causes of DICER1-associated morbidity and mortality. The goal of surveillance imaging in DICER1 is to detect PPB in its earliest, cystic and most curable form (Type I PPB). The 5-year disease-free survival (DFS) and overall survival (OS) for Type I PPB is 82% and 91% respectively.(4) For Type II and Type III the 5-year DFS are 59% and 37% and the 5-year OS is 71% and 53%.(4)
The highest risk for clinically significant PPB is prior to 7 years of age with the earliest PPBs having been seen from 31 weeks gestation.(4) Age of onset for data for all PPB cases confirmed by International PPB Registry central pathology review is shown in Table 3. There is a lack of consensus on the role of chemotherapy in Type I PPB at this time. Chemotherapy and sometimes radiation therapy is required in Types II and III PPB.(4)
Table 3.
International PPB Registry age range of risk for all centrally confirmed cases of PPB
| Type I | Type II | Type III | Type II/III NOS* |
Type Ir | |
|---|---|---|---|---|---|
|
Number (total = 527) |
144 | 176 | 131 | 19 | 57 |
|
Age range
(months) |
0 – 114 | 5 – 431 | 19 – 184 | 7 – 75 | 3 – 546 |
|
Median
(months) |
7 | 34 | 41 | 36 | 44 |
NOS= not otherwise specified (represents PPB which is at least partially solid and a determination of Type II vs. III PPB cannot be definitely made).
Surveillance recommendations:
We recommend a chest x-ray (CXR) at birth for all children at risk of a pathogenic germline DICER1 variant to screen for any large pulmonary cysts. DICER1 molecular testing should be obtained between birth and 3 months of age. In children who are found to be carriers of a pathogenic germline DICER1 variant, the first chest computed tomography (CT) should be obtained by 9 months of age, preferably between 3 and 6 months of age, since the incidence of Type II and III PPB increases after one year of age.
Due to the radiation dose and possible need for sedation with cross sectional imaging, CXRs are the primary screening modality. In the absence of pulmonary cysts/PPB, most individuals with DICER1 mutations detected in early childhood will undergo only 2 chest CTs. If the first chest CT is normal with no evidence of cysts, we recommend a follow-up low-dose chest CT at approximately 2.5 years of age, prior to the peak incidence of Types II and III PPB. In the absence of concerning radiographic findings, a CXR every 6 months from birth through 7 years of age and then annually from 8 – 12 years of age is recommended. The utility of routine surveillance CXR or CT in adolescence and adulthood is unknown and thus not recommended. Currently, CT offers a higher level of sensitivity for the detection of cystic lung lesions not achievable with CXR alone. When CT is performed, techniques to minimize radiation exposure should be included. As novel magnetic resonance imaging (MRI) techniques are developed that will eventually allow detection of small cystic lesions, transition to non-radiation containing cross sectional imaging should be considered.
When a pulmonary cyst is identified in a young child with a pathogenic germline DICER1 variant or family history of a DICER1-associated condition, it should be assumed to be Type I PPB until proven otherwise. Consideration of cyst resection vs. observation should take into account the patient’s age, clinical history, resectability, additional findings and evidence of growth. Resection of cystic lung lesions in children at highest age related risk of progression (especially under 7 years of age) should be considered. If surgery is undertaken, an approach to avoid rupture and optimize likelihood of clear margins should be emphasized. Consultation should be sought with an experienced pediatric thoracic surgeon, oncologist, radiologist and pathologist. We expect that additional studies will also guide future decisions regarding resection vs. observation.
Pulmonary cysts are often identified in adults with pathogenic germline DICER1 variants and are assumed to be Type Ir PPB. Since the risk for malignancy is low, resection of these lesions in adults is not typically undertaken for oncologic indications, but may be considered based on size and location due to the potential risk of pneumothorax or superinfection.
Further discussion of the management of pulmonary cysts among individuals including adolescents with pathogenic germline DICER1 variants is beyond the scope of this guideline but is available through the International PPB Registry’s website (www.PPBregistry.org) or in the European Very Rare Tumor Group guidelines (www.raretumors-children.eu).
Kidney
Individuals with a DICER1-associated condition may develop cystic nephroma (CN), anaplastic sarcoma of the kidney or Wilms tumor (Fig. 2). A recent analysis showed 2/41 (5%) Wilms tumors are secondary to pathogenic germline DICER1 variants. Progression from CN to renal sarcoma has been described.(16, 39, 40) CN is often treated with resection alone via partial nephrectomy; however, early resection may maximize residual renal function and is optimal in cases at risk for bilateral disease. Wilms tumor may be treated with surgery, usually complete nephrectomy, preceded or followed by chemotherapy with or without radiation according to conventional protocols.(41, 42) Early stage disease is associated with a lower burden of therapy and a more favorable prognosis, thus early diagnosis may improve outcomes and reduce late effects associated with therapy.
Figure 2.
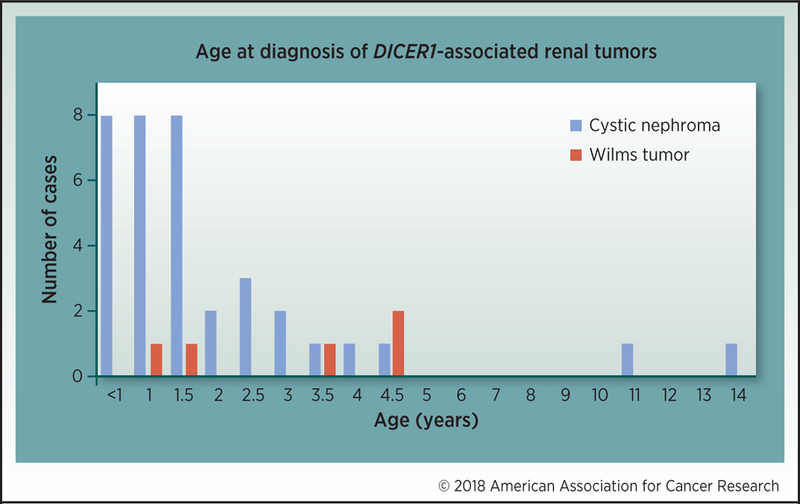
Age and incidence of cystic nephroma (blue) and Wilms tumor (red) in individuals known to the International PPB Registry.
Surveillance recommendations:
We recommend an abdominal ultrasound (US) in infancy at the time of the first chest CT (done for PPB surveillance) and every 6 – 12 months until at least 8 years of age. Annual US after age 8 years may be considered until 12 years of age. More frequent US could also be considered based on separate data in non-DICER1 individuals at risk for Wilms tumor in which US at least every 3–4 months has been recommended.(43) As above, we expect that additional studies will clarify whether a normal US and/or resection of CN in early childhood predicts a lower likelihood of later renal malignancy.
Female reproductive tract
Individuals with pathogenic germline DICER1 variants are at increased risk for Sertoli-Leydig cell tumor (SLCT) and gynandroblastoma. These tumors are commonly associated with hormonal symptoms including virilization or menstrual irregularities in addition to abdominal pain or distention. Per the International Federation of Gynecology and Obstetrics (FIGO), stage IA/IB (intraovarian tumor localized to one or both ovaries, without capsular rupture or positive cytology) disease is typically treated with surgery alone. Higher stage disease requires surgery followed by adjuvant therapy, most often platinum-based chemotherapy.(44) Cervical embryonal rhabdomyosarcoma (ERMS) presenting as a cervical polyp in older children, adolescents and young adults may be discovered in absence of other tumors or before, during or after the recognition of SLCT. Other ovarian or genitourinary tract tumors including undifferentiated sarcoma(32) have been rarely seen.
The age distribution of risk for SLCT and gynandroblastoma risk is wide, with a range from 4 – 61 years of age in the OTST Registry’s current series of girls and women with DICER1-associated ovarian tumors. Most individuals are diagnosed from late childhood through early adulthood with a median of 16.9 years; 95% of individuals are diagnosed by 40 years of age. Thus far, data shows that individuals with germline DICER1 mutations have a lower risk of recurrence of the primary SLCT compared with those with biallelic tumor specific mutations, however, individuals with germline mutations may develop synchronous or metachronous contralateral tumors (see Table 4). Metachronous tumors must be carefully distinguished from recurrent disease when discovered early as FIGO stage IA; the former is associated with a favorable prognosis.(45)
Table 4.
International OTST Registry cases with ovarian tumors
| Primary ovarian tumor diagnosis |
Number of cases in individuals with germline or mosaicism for DICER1 |
Median age at diagnosis (years) |
|---|---|---|
| SLCT | 28 | 14.5 |
| Gynandroblastoma | 4 | 16 |
| Sarcoma | 2 | 7.5 |
| PNET | 1 | 14 |
Cervical ERMS, typically a botryoid variant, may present with vaginal bleeding, pain or mass.
Surveillance recommendations:
Patient and parent education regarding symptoms (hormonal symptoms including virilization, recurrent abdominal pain, abdominal mass) is recommended so that imaging evaluation may be performed urgently if clinically indicated. For girls, we recommend pelvic US be performed in conjunction with abdominal US (see above) every 6–12 months and that pelvic US continue throughout adulthood. Our current recommendation is to consider surveillance imaging until at least 40 years of age by which time 95% of SLCTs/gynandroblastomas have been diagnosed, however, the optimal end point of imaging remains undetermined; the OTST Registry’s current oldest patient with DICER1-related SLCT was diagnosed at 61 years of age. Alternatively, adult women and their health care providers may choose a surveillance strategy based on physical examination. In that instance, US should be obtained if any clinical findings suggest gynecologic tumor. The optimal timing of US and/or pelvic examinations and relative sensitivity of these varying surveillance strategies are not yet known.
Thyroid
Epidemiologic analysis of a large cohort of individuals with one or more DICER1-associated condition (and controls) shows that by 20 years of age, the cumulative incidence of multinodular goiter or history of thyroidectomy is 32% in women and 13% in men (vs. 0% in control women and control men), and there is a 16- to 24-fold increased risk of thyroid cancer, compared to the National Cancer Institute’s Surveillance, Epidemiology and End Results program, over a patient’s lifetime.(46) Within pediatrics, thyroid cancer was first described in a series of case reports of children who had received chemotherapy for DICER1-associated tumors prior to 10 years of age.(13, 47, 48) However, more recent reports show that children with DICER1-associated conditions may also develop thyroid cancer even in the absence of a history of chemotherapy or radiation.(49) Follicular variant of papillary cancer and follicular thyroid cancer are the most common histologies seen in individuals with pathogenic germline DICER1 variants. To date, all thyroid tumors associated with pathogenic germline DICER1 variants have displayed indolent behavior, with disease usually confined to the thyroid gland. Thyroidectomy is often curative; however, patients should undergo complete and thorough preoperative evaluation following standard guidelines based on the American Thyroid Association pediatric risk levels.(50) In contrast to multiple endocrine neoplasia type 2, there is no indication for prophylactic thyroidectomy in patients with DICER1-associated conditions secondary to the variable expressivity and the typically indolent nature of most thyroid nodules and cancer in this group.
Surveillance recommendations:
Individuals and families should be counseled regarding the increased risk for thyroid nodules and cancer. There are no prospective studies on the efficacy or timing of screening thyroid US in individuals with germline DICER1 pathogenic variants. We recommend a thyroid US at approximately 8 years of age in children with DICER1-associated conditions, and then every 2 to 3 years, or as needed for worrisome signs or symptoms, including an enlarged thyroid, a thyroid nodule, or persistent cervical lymphadenopathy. For individuals receiving chemotherapy or radiation treatment for a non-thyroid malignancy (e.g., for PPB, pineoblastoma), a baseline thyroid US should be performed at the time of diagnosis, then annually for 5 years after exposure, decreasing to every 2 to 3 years if no lesions are found on initial US exams. All thyroid nodules should undergo fine needle aspiration with stratification of surgery based on the Bethesda System for Reporting Thyroid Cytopathology and the presence or absence of bilateral disease.(51) For patients undergoing thyroid surgery based on cytology, complete US assessment of lateral neck lymph nodes per standard guidelines must be performed prior to surgery to ensure that a lateral neck lymph node dissection is not warranted.(50)
Eye
Individuals with pathogenic germline DICER1 variants are at risk for ciliary body medulloepithelioma (CBME),(22, 52) and in an earlier report the PPB Registry noted four individuals, age 4, 6, 8 and 9 years, with CBME and pathogenic germline DICER1 variants.(22) There is an additional patient in the literature with somatic DICER1 variants, diagnosed at 18 years of age.(53) Most patients presented with visual symptoms or abnormalities on ophthalmologic examination.
Surveillance recommendations:
Individuals and families should be counseled regarding potential symptoms of strabismus, visual acuity changes or leukocoria. We recommend annual routine dilated ophthalmologic exam (generally unsedated) with visual acuity screening from 3 years of age through at least 10 years of age. No imaging studies are indicated in asymptomatic individuals but care providers should be aware of the risk, albeit low, for ophthalmologic DICER1-associated conditions and the need for fundoscopic evaluation and imaging evaluation if clinically indicated.
Head and neck
Individuals with pathogenic germline DICER1 variants are at risk for developing benign nasal chondromesenchymal hamartoma (NCMH).(28) Most individuals with NCMH present with signs of nasal obstruction. We deem the likelihood and clinical relevance of finding this tumor early based on imaging to be low. Thus, at this time, we recommend that individuals, caregivers and providers be aware of the potential for NCMH and that otolaryngologic evaluation be considered for persistent nasal or sinus symptoms in individuals of any age with pathogenic germline DICER1 variants.(54)
Surveillance recommendations:
No imaging studies are indicated in asymptomatic individuals but care providers should be aware of the risk, albeit low, for NCMH and the need for evaluation with endoscopy and potential imaging evaluation if clinically indicated.
Central nervous system
Primary tumors involving the central nervous system (CNS) have also been described among individuals with pathogenic germline DICER1 variants. CNS tumors that are currently considered syndrome components include pituitary blastomas and pineoblastomas.(4, 11, 12, 55, 56) Other rare cases of CNS tumors have been reported in PPB kindreds as well as in DICER1 germline carriers, including sarcomas and medulloblastomas/infratentorial embryonal CNS tumors; larger studies are needed to further confirm these associations (Table 5).(10, 11, 27, 57)
Table 5.
International PPB Registry cases with primary central nervous system (CNS) tumors
|
Primary CNS tumor diagnosis |
Number of cases |
Age at PPB (months) |
Age at CNS tumor (in months from PPB diagnosis) |
| Glioblastoma | 1 | 45 | 92 |
| Meningeal sarcoma | 1 | 34 | 168 |
| Pineoblastoma | 2 | 30, 211 | −4, 64 |
| Primitive neuroectodermal tumor |
2 | 27, 37 | 9, 20 |
| Optic glioma | 1 | 236 | 0 |
Pituitary blastomas have been described in the context of DICER1 syndrome. The largest reported series to date described 13 cases, 9 of which were previously published.(12) The median age at diagnosis in this series was 9 months (range 7 – 24 months), with the most common presenting symptoms including Cushing’s syndrome and/or ophthalmoplegia. Elevated adrenocorticotropic hormone (ACTH) levels with hypercortisolism were also commonly seen. Eleven of 12 cases with germline or tumor material available for analysis demonstrated at least one germline or somatic DICER1 mutation.
Pineoblastomas may also be seen among individuals with pathogenic germline DICER1 variants. Pineoblastomas may present with obstructive hydrocephalus and symptoms of increased intracranial pressure, gaze palsy (“Parinaud syndrome”), or endocrine dysfunction (most commonly precocious puberty). After the initial association of pineoblastoma with DICER1 mutations was described, in a subsequent series, 3 of 18 individuals without previously known genotypes were found to carry pathogenic germline DICER1 variants.(11, 55) Loss of heterozygosity (LOH) of the wildtype allele was seen among 4 tumors. Interestingly, 2 pituitary blastoma cases also involving LOH of the wildtype allele have been described, suggesting the possibility of a distinct mechanism of second-hit among these intracranial neoplasms.(12, 55)
In one study, 42% of 67 individuals with a pathogenic germline DICER1 variant were additionally found to be macrocephalic (occipital head circumference > 2 standard deviation) compared with 12% of 43 family controls.(58) This difference persisted after adjusting for differences in height. No microcephaly was observed in the DICER1-associated conditions cohort. The etiology of the macrocephaly is unknown but is hypothesized to be related to dysregulated cellular growth.(59)
Surveillance recommendations:
Given the rarity of pituitary blastoma and pineoblastoma even within DICER1-associated conditions, the role of surveillance imaging by MRI for these tumors among asymptomatic individuals remains controversial. Education regarding signs and symptoms of intracranial tumors (including cortisol excess, increased intracranial pressure, neurologic changes, vomiting, lethargy etc.) is encouraged, with urgent brain MRI suggested for any new presenting symptoms.
Gastrointestinal polyps
Juvenile-type polyps in the duodenum, jejunum and ileum have been described in very young children with PPB as well as in family members.(18, 57) Generally these have been seen in individuals from birth to 10 years of age. This can result in intestinal obstruction and surgery may be required.(18)
Surveillance recommendations:
No systematic imaging studies or endoscopy are indicated in asymptomatic individuals, but care providers should be aware of the risk, albeit low, for intestinal DICER1-associated symptoms and the need for imaging evaluation and/or endoscopy if clinically indicated.
Conclusion
As knowledge of DICER1 and its clinical implications increases, facilitating larger studies of patients with pathogenic germline DICER1 variants, criteria for genetic testing and/or clinical diagnosis will continue to be refined. Similarly, the management guidelines for cancer/tumor screening and risk reduction in patients who have pathogenic germline DICER1 variants will continue to be updated (Table 2).
Most individuals with pathogenic germline DICER1 variants are healthy, or have only minor DICER1-associated conditions. The most severe manifestations of pathogenic germline DICER1 variants tend to present in early childhood with adulthood characterized by good health. Thyroid nodules are common though often asymptomatic. Thyroid cancer may be seen in children and adults, and in general is associated with a similarly good prognosis as sporadic non-DICER1-associated differentiated thyroid carcinoma. Ovarian tumors are curable with surgery alone when found in their earliest stage although the wide age range of risk makes US-based screening cumbersome. Given the overall good prognosis for most individuals with pathogenic germline DICER1 variants, the risks and potential benefits of lifelong screening must be carefully balanced.
In addition to consideration of imaging surveillance, the authors also wish to stress the importance of family and health care provider education regarding the potential for heritable risk and potential signs and symptoms or lack thereof. Some of the manifestations described above, particularly Types II and III PPB, may evolve quickly and recent normal imaging should not sway health care providers from consideration of tumor risk in the setting of new or concerning symptoms.
The proposed screening guidelines are offered based on available information; however, we expect that as more individuals are followed from early childhood, additional information will allow for revision and optimization of these guidelines. The clinical utility and cost/benefit analysis of this screening regimen is a subject of ongoing study, and participation in collaborative research will likely support or guide the modification of this regimen over time. International collaboration is encouraged for all individuals with DICER1 pathogenic variants or DICER1-associated conditions to allow further revision of these guidelines and to help reduce the morbidity and mortality of individuals with pathogenic variants in DICER1.
Supplementary Material
Acknowledgements
The authors wish to thank John R. (Jack) Priest, MD and Jan Watterson-Sheaffer, BA, for their work to establish the International PPB Registry and their contributions to the field of PPB and DICER1- associated conditions. The authors also wish to thank the many treating physicians, genetic counselors, and patients and families who collaboratively support the International PPB Registry, and the Pine Tree Apple Tennis Classic whose volunteers, players and donors have provided more than 30 years of continuous support of PPB research.
Financial Support: The International Pleuropulmonary blastoma (PPB) and Ovarian and Testicular Stromal Tumor (OTST) Registries are supported by the Pine Tree Apple Tennis Classic. The PPB Registry has also been funded by the Children’s Minnesota Foundation, the Mendon F. Schutt Foundation and the Randy Shaver Cancer Research and Community Fund. The OTST Registry has also been funded by St. Baldrick’s Foundation, Hyundai Hope on Wheels and the Randy Shaver Cancer Research and Community Fund. This work is supported by National Institutes of Health grant National Cancer Institute (NCI) R01CA143167 (DAH, YM, KAS) and The Parson’s Foundation (DAH). DTS is supported by the German Childhood Cancer Foundation. DRS is supported by the Intramural Research Program of the Divisions of Cancer Epidemiology and Genetics of the NCI.
Footnotes
Conflict of Interest Disclosure Statement: The authors have no conflicts of interest to disclose.
References
- 1.Dishop MK, Kuruvilla S. Primary and metastatic lung tumors in the pediatric population: a review and 25-year experience at a large children’s hospital. Arch Pathol Lab Med 2008. July;132(7):1079–103. [DOI] [PubMed] [Google Scholar]
- 2.Priest JR, Watterson J, Strong L, Huff V, Woods WG, Byrd RL, et al. Pleuropulmonary blastoma: a marker for familial disease. J Pediatr 1996. February;128(2):220–4. [DOI] [PubMed] [Google Scholar]
- 3.Manivel JC, Priest JR, Watterson J, Steiner M, Woods WG, Wick MR, et al. Pleuropulmonary blastoma. The so-called pulmonary blastoma of childhood. Cancer 1988. October 15;62(8):1516–26. [DOI] [PubMed] [Google Scholar]
- 4.Messinger YH, Stewart DR, Priest JR, Williams GM, Harris AK, Schultz KA, et al. Pleuropulmonary blastoma: a report on 350 central pathology-confirmed pleuropulmonary blastoma cases by the International Pleuropulmonary Blastoma Registry. Cancer 2015. January 15;121(2):276–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hill DA, Dehner LP. A cautionary note about congenital cystic adenomatoid malformation (CCAM) type 4. Am J Surg Pathol 2004. April;28(4):554–5; author reply 5. [DOI] [PubMed] [Google Scholar]
- 6.MacSweeney F, Papagiannopoulos K, Goldstraw P, Sheppard MN, Corrin B, Nicholson AG. An assessment of the expanded classification of congenital cystic adenomatoid malformations and their relationship to malignant transformation. Am J Surg Pathol 2003. August;27(8):1139–46. [DOI] [PubMed] [Google Scholar]
- 7.Hill DA, Jarzembowski JA, Priest JR, Williams G, Schoettler P, Dehner LP. Type I pleuropulmonary blastoma: pathology and biology study of 51 cases from the international pleuropulmonary blastoma registry. Am J Surg Pathol 2008. February;32(2):282–95. [DOI] [PubMed] [Google Scholar]
- 8.Hill DA, Ivanovich J, Priest JR, Gurnett CA, Dehner LP, Desruisseau D, et al. DICER1 mutations in familial pleuropulmonary blastoma. Science 2009. August 21;325(5943):965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bahubeshi A, Bal N, Frio TR, Hamel N, Pouchet C, Yilmaz A, et al. Germline DICER1 mutations and familial cystic nephroma. Journal Med Genet 2010. December;47(12):863–6. [DOI] [PubMed] [Google Scholar]
- 10.Boman F, Hill DA, Williams GM, Chauvenet A, Fournet JC, Soglio DB, et al. Familial association of pleuropulmonary blastoma with cystic nephroma and other renal tumors: a report from the International Pleuropulmonary Blastoma Registry. J Pediatr 2006. December;149(6):850–4. [DOI] [PubMed] [Google Scholar]
- 11.de Kock L, Sabbaghian N, Druker H, Weber E, Hamel N, Miller S, et al. Germ-line and somatic DICER1 mutations in pineoblastoma. Acta Neuropathol 2014. October;128(4):583–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Kock L, Sabbaghian N, Plourde F, Srivastava A, Weber E, Bouron-Dal Soglio D, et al. Pituitary blastoma: a pathognomonic feature of germ-line DICER1 mutations. Acta Neuropathol 2014. July;128(1):111–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Kock L, Sabbaghian N, Soglio DB, Guillerman RP, Park BK, Chami R, et al. Exploring the association Between DICER1 mutations and differentiated thyroid carcinoma. J Clin Endocrinol Metab 2014. June;99(6):E1072–7. [DOI] [PubMed] [Google Scholar]
- 14.Delahunt B, Thomson KJ, Ferguson AF, Neale TJ, Meffan PJ, Nacey JN. Familial cystic nephroma and pleuropulmonary blastoma. Cancer 1993. February 15;71(4):1338–42. [DOI] [PubMed] [Google Scholar]
- 15.Doros L, Yang J, Dehner L, Rossi CT, Skiver K, Jarzembowski JA, et al. DICER1 mutations in embryonal rhabdomyosarcomas from children with and without familial PPB-tumor predisposition syndrome. Pediatr Blood Cancer 2012. September;59(3):558–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doros LA, Rossi CT, Yang J, Field A, Williams GM, Messinger Y, et al. DICER1 mutations in childhood cystic nephroma and its relationship to DICER1-renal sarcoma. Mod Pathol 2014. September;27(9):1267–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hill DA, Doros L, Schultz KA, Stewart DR, Bauer AJ, Williams G, et al. DICER1-related disorders In: Pagon RA, Adam MP, Ardinger HH, et al. , editors. GeneReviews® [Internet] Seattle: University of Washington, Seattle; 2014. Available from: http://www.ncbi.nlm.nih.gov. [Google Scholar]
- 18.Foulkes WD, Bahubeshi A, Hamel N, Pasini B, Asioli S, Baynam G, et al. Extending the phenotypes associated with DICER1 mutations. Hum Mutat 2011. December;32(12):1381–4. [DOI] [PubMed] [Google Scholar]
- 19.Heravi-Moussavi A, Anglesio MS, Cheng SW, Senz J, Yang W, Prentice L, et al. Recurrent somatic DICER1 mutations in nonepithelial ovarian cancers. New Engl J Med 2012. January 19;366(3):234–42. [DOI] [PubMed] [Google Scholar]
- 20.Lopez-Andreu JA, Ferris J, Esquembre C, Verdeguer A, Gomez J, Castel V. Familial cystic nephroma and pleuropulmonary blastoma. Cancer 1993. November 1;72(9):2792–3. [DOI] [PubMed] [Google Scholar]
- 21.McDermott MB, Ponder TB, Dehner LP. Nasal chondromesenchymal hamartoma: an upper respiratory tract analogue of the chest wall mesenchymal hamartoma. . Am J Surg Pathol 1998;22(4):425–33. [DOI] [PubMed] [Google Scholar]
- 22.Priest JR, Williams GM, Manera R, Jenkinson H, Brundler MA, Davis S, et al. Ciliary body medulloepithelioma: four cases associated with pleuropulmonary blastoma--a report from the International Pleuropulmonary Blastoma Registry. BritJ Ophthalmol 2011. July;95(7):1001–5. [DOI] [PubMed] [Google Scholar]
- 23.Rio Frio T, Bahubeshi A, Kanellopoulou C, Hamel N, Niedziela M, Sabbaghian N, et al. DICER1 mutations in familial multinodular goiter with and without ovarian Sertoli-Leydig cell tumors. JAMA 2011. January 5;305(1):68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schultz KA, Pacheco MC, Yang J, Williams GM, Messinger Y, Hill DA, et al. Ovarian sex cord-stromal tumors, pleuropulmonary blastoma and DICER1 mutations: a report from the International Pleuropulmonary Blastoma Registry. Gynecol Oncol 2011. August;122(2):246–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schultz KAP, Williams GM, Stewart DR, Rosenberg PS, Doros LA, Dehner LP, et al. Recurrent or progressive Type II and Type III pleuropulmonary blastoma (PPB) are associated with poor outcome: A report from the International PPB Registry [abstract]. J Clin Oncol 2015;33(15S):10014. [Google Scholar]
- 26.Shaheen IS, Fitzpatrick M, Brownlee K, Bhuskute N, Elliott M, Powis M, et al. Bilateral progressive cystic nephroma in a 9-month-old male infant requiring renal replacement therapy. Pediatr Nephrol 2010. April 23;25(9):1755–8. [DOI] [PubMed] [Google Scholar]
- 27.Slade I, Bacchelli C, Davies H, Murray A, Abbaszadeh F, Hanks S, et al. DICER1 syndrome: clarifying the diagnosis, clinical features and management implications of a pleiotropic tumour predisposition syndrome. J Med Genet 2011. April;48(4):273–8. [DOI] [PubMed] [Google Scholar]
- 28.Stewart DR, Messinger Y, Williams GM, Yang J, Field A, Schultz KA, et al. Nasal chondromesenchymal hamartomas arise secondary to germline and somatic mutations of DICER1 in the pleuropulmonary blastoma tumor predisposition disorder. Hum Genet 2014. November;133(11):1443–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Witkowski L, Mattina J, Schonberger S, Murray MJ, Choong CS, Huntsman DG, et al. DICER1 hotspot mutations in non-epithelial gonadal tumours. Br J Cancer 2013. November 12;109(10):2744–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu MK, Sabbaghian N, Xu B, Addidou-Kalucki S, Bernard C, Zou D, et al. Biallelic DICER1 mutations occur in Wilms tumours. J Pathol 2013. June;230(2):154–64. [DOI] [PubMed] [Google Scholar]
- 31.Bisogno G, Brennan B, Orbach D, Stachowicz-Stencel T, Cecchetto G, Indolfi P, et al. Treatment and prognostic factors in pleuropulmonary blastoma: an EXPeRT report. Eur J Cancer 2014. January;50(1):178–84. [DOI] [PubMed] [Google Scholar]
- 32.Schultz KA, Harris A, Messinger Y, Sencer S, Baldinger S, Dehner LP, et al. Ovarian tumors related to intronic mutations in DICER1: a report from the international ovarian and testicular stromal tumor registry. Fam Cancer 2016. January;15(1):105–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brenneman M, Field A, Yang J, Williams G, Doros L, Rossi C, et al. Temporal order of RNase IIIb and loss-of-function mutations during development determines phenotype in DICER1 syndrome: a unique variant of the two-hit tumor suppression model. F1000Res 2015;4:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sabbaghian N, Srivastava A, Hamel N, Plourde F, Gajtko-Metera M, Niedziela M, et al. Germ-line deletion in DICER1 revealed by a novel MLPA assay using synthetic oligonucleotides. Eur J Hum Genet 2014. April;22(4):564–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pugh TJ, Yu W, Yang J, Field AL, Ambrogio L, Carter SL, et al. Exome sequencing of pleuropulmonary blastoma reveals frequent biallelic loss of TP53 and two hits in DICER1 resulting in retention of 5p-derived miRNA hairpin loop sequences. Oncogene 2014. November 06;33(45):5295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Amatruda JF, Chen KS. Referee Report For: Temporal order of RNase IIIb and loss-of-function mutations during development determines phenotype in DICER1 syndrome: a unique variant of the two-hit tumor suppression model [version 1; referees: 2 approved with reservations]. F1000Res 2015;4:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rakheja D, Chen KS, Liu Y, Shukla AA, Schmid V, Chang TC, et al. Somatic mutations in DROSHA and DICER1 impair microRNA biogenesis through distinct mechanisms in Wilms tumours. Nat Commun 2014. September 05;2:4802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim J, Field A, Schultz KAP, Hill DA, Stewart DR. The prevalence of DICER1 pathogenic variation in population databases. Int J Cancer 2017. July 27. [DOI] [PMC free article] [PubMed]
- 39.Wu MK, Cotter MB, Pears J, McDermott MB, Fabian MR, Foulkes WD, et al. Tumor progression in DICER1-mutated cystic nephroma-witnessing the genesis of anaplastic sarcoma of the kidney. Hum Pathol 2016. July;53:114–20. [DOI] [PubMed] [Google Scholar]
- 40.Wu MK, Goudie C, Druker H, Thorner P, Traubici J, Grant R, et al. Evolution of Renal Cysts to Anaplastic Sarcoma of Kidney in a Child With DICER1 Syndrome. Pediatr Blood Cancer 2016;63(7):1272–5. [DOI] [PubMed] [Google Scholar]
- 41.Dome J, Huff V. Wilms tumor overview. In: GeneReviews at GeneTests: Medical Genetics Information Resource (database online) Seattle: Copyright, University of Washington, Seattle: 1997–2010. Available at http://www.genetests.org.; 1993. [[Accessed 09/16/2010]]; Available from: http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=wilms-ov. [Google Scholar]
- 42.Group SRTS. Paediatric renal tumours: perspectives from the SIOP-RTSG. Nat Rev Urol 2017. January;14(1):3–4. [DOI] [PubMed] [Google Scholar]
- 43.Scott RH, Walker L, Olsen OE, Levitt G, Kenney I, Maher E, et al. Surveillance for Wilms tumour in at-risk children: pragmatic recommendations for best practice. Arch Dis Child 2006. December;91(12):995–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schneider DT, Orbach D, Cecchetto G, Stachowicz-Stencel T, Brummel B, Brecht IB, et al. Ovarian Sertoli Leydig cell tumours in children and adolescents: an analysis of the European Cooperative Study Group on Pediatric Rare Tumors (EXPeRT). Eur J Cancer 2015. March;51(4):543–50. [DOI] [PubMed] [Google Scholar]
- 45.Schultz KAP, Harris AK, Finch M, Dehner LP, Brown JB, Gershenson DM, et al. DICER1-related Sertoli-Leydig cell tumor and gynandroblastoma: Clinical and genetic findings from the International Ovarian and Testicular Stromal Tumor Registry. Gynecol Oncol 2017. (4C):7. [DOI] [PMC free article] [PubMed]
- 46.Khan NE, Bauer AJ, Schultz KA, Doros L, Decastro RM, Ling A, et al. Quantification of thyroid cancer and multinodular goiter risk in the DICER1 syndrome: a family-based cohort study. J Clin Endocrinol Metab 2017. February 02. [DOI] [PMC free article] [PubMed]
- 47.Oue T, Inoue M, Kubota A, Kuwae Y, Kawa K. Pediatric thyroid cancer arising after treatment for pleuropulmonary blastoma. Pediatr Blood Cancer 2008. April;50(4):901–2. [DOI] [PubMed] [Google Scholar]
- 48.Rome A, Gentet JC, Coze C, Andre N. Pediatric thyroid cancer arising as a fourth cancer in a child with pleuropulmonary blastoma. Pediatr Blood Cancer 2008. May;50(5):1081. [DOI] [PubMed] [Google Scholar]
- 49.Rutter MM, Jha P, Schultz KA, Sheil A, Harris AK, Bauer AJ, et al. DICER1 Mutations and Differentiated Thyroid Carcinoma: Evidence of a Direct Association. J Clin Endocrinol Metab 2016. January;101(1):1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Francis GL, Waguespack SG, Bauer AJ, Angelos P, Benvenga S, Cerutti JM, et al. Management Guidelines for Children with Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 2015. July;25(7):716–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Smith M, Pantanowitz L, Khalbuss WE, Benkovich VA, Monaco SE. Indeterminate Pediatric Thyroid Fine Needle Aspirations: A Study of 68 Cases. Acta Cytol 2013;57(4):341–8. [DOI] [PubMed] [Google Scholar]
- 52.Kramer GD, Arepalli S, Shields CL, Shields JA. Ciliary body medulloepithelioma association with pleuropulmonary blastoma in a familial tumor predisposition syndrome. J Pediatr Ophthalmol Strabismus 2014. July 16;51 Online:e48–50. [DOI] [PubMed] [Google Scholar]
- 53.Durieux E, Descotes F, Nguyen AM, Grange JD, Devouassoux-Shisheboran M. Somatic DICER1 gene mutation in sporadic intraocular medulloepithelioma without pleuropulmonary blastoma syndrome. Hum Pathol 2015. May;46(5):783–7. [DOI] [PubMed] [Google Scholar]
- 54.de Castro CG Jr., de Almeida SG, Gregianin LJ, Loss JF, Rivero LF, Schwartsmann G, et al. High-dose chemotherapy and autologous peripheral blood stem cell rescue in a patient with pleuropulmonary blastoma. J Pediatr Hematol Oncol 2003. January;25(1):78–81. [DOI] [PubMed] [Google Scholar]
- 55.Sabbaghian N, Hamel N, Srivastava A, Albrecht S, Priest JR, Foulkes WD. Germline DICER1 mutation and associated loss of heterozygosity in a pineoblastoma. J Med Genet 2012. July;49(7):417–9. [DOI] [PubMed] [Google Scholar]
- 56.Wildi-Runge S, Bahubeshi A, Carret AS, Crevier L, Robitaille Y, Kovacs K, et al. New Phenotype in the Familial DICER1 Tumor Syndrome: Pituitary Blastoma Presenting at Age 9 Months [Abstract]. Endocr Rev 2011;32(03):P1–777. [Google Scholar]
- 57.Priest JR, Williams GM, Hill DA, Dehner LP, Jaffe A. Pulmonary cysts in early childhood and the risk of malignancy. Pediatr Pulmonol 2009. January;44(1):14–30. [DOI] [PubMed] [Google Scholar]
- 58.Khan NE, Bauer AJ, Doros L, Schultz KAP, Decastro RM, Harney LA, et al. Macrocephaly associated with the DICER1 syndrome. Genet Med 2016. 07/21/online. [DOI] [PMC free article] [PubMed]
- 59.Klein S, Lee H, Ghahremani S, Kempert P, Ischander M, Teitell MA, et al. Expanding the phenotype of mutations in DICER1: mosaic missense mutations in the RNase IIIb domain of DICER1 cause GLOW syndrome. J Med Genet 2014. May;51(5):294–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.