

Abstract
Background
While auditory neuropathy spectrum disorder (ANSD) is a heterogeneous disorder and its management quite varies depending upon the etiology, even including self-resolution, OTOF is an important molecular etiology of prelingual ANSD and has emerged as an attractive target for implementation of precision medicine in terms of timing and prognosis prediction of auditory rehabilitation. However, to date, the literature is lacking in the genotype–phenotype relationship of this gene as well as efficient molecular testing strategy in the clinic in many populations and to make things more complicated in Koreans, the most prevalent variant p.Arg1939Gln among Korean ANSD children frequently evaded detection by next generation sequencing (NGS), resulting in delayed genetic diagnosis and late cochlear implantation (CI). The aims of this study are to document the mutational and phenotypic spectrum of OTOF-related ANSD (DFNB9) in the Korean population, further establishing genotype–phenotype correlation and proposing a set of the most commonly found OTOF variants to be screened first.
Methods
Genetic diagnosis through the NGS-based sequencing was made on patients with ANSD in two tertiary hospitals. Genotype and phenotypes of eleven DFNB9 patients were reviewed. For data analysis, Mann–Whitney test and Fisher’s exact test were applied.
Results
This study disclosed four prevalent variants in Koreans: p.Arg1939Gln with an allele frequency of 40.9%, p.Glu841Lys (13.6%), p.Leu1011Pro and p.Arg1856Trp (9.1%). Three novel variants (c.4227 + 5G > C, p.Gly1845Glu, and p.Pro1931Thr) were identified. Interestingly, a significant association of p.Arg1939Gln with worse ASSR thresholds was observed despite consistently no ABR response. Ten of 11 DFNB9 patients received CI for auditory rehabilitation, showing favorable outcomes with more rapid improvement on early-CI group (age at CI ≤ 18 mo.) than late-CI group.
Conclusions
This study included the largest Korean DFNB9 cohort to date and proposed a set of the most frequent four OTOF variants, allowing the potential prioritization of exons during Sanger sequencing. Further, a significant association of p.Arg1939Gln homozygotes with poor residual hearing was observed. We may have to suspect p.Arg1939Gln homozygosity in cases of poor auditory thresholds in ANSD children with putative negative OTOF variants solely screened by NGS. Reciprocal feedback between bench and clinics regarding DFNB9 would complement each other.
Keywords: OTOF, Auditory neuropathy spectrum disorder, Whole exome sequencing, DFNB9, Auditory steady-state response, Cochlear implantation
Background
Auditory neuropathy spectrum disorder (ANSD) is a hearing disorder characterized by impaired transmission of sound signals from the inner ear to the auditory nerve/brain cortex with normally-functioning outer hair cells. Clinically, ANSD is diagnosed by the presence of otoacoustic emissions (OAE) or cochlear microphonics (CM), and the absence or severe impairment of auditory brainstem response (ABR) [1]. Approximately 10% of neonates or infants diagnosed with profound hearing loss suffer from ANSD; in about 50% of them, genetic cause is suspected [2, 3]. OTOF, and PJVK in an autosomal recessive manner, DIAPH3in an autosomal dominant manner and a locus AUNX1 in an X-linked recessive manner have been reported to be associated with non-syndromic ANSD [4–7]. Specifically, OTOF-related ANSD (DFNB9) is assumed to be caused by a pathology confined to the synapse [8–10], proximal to the postsynaptic neuron or auditory nerve, which is the site for electrical stimulation of cochlear implantation (CI). For this reason, and as evidenced by several previous studies showing good outcomes in patients receiving CI before the age of 24 months [11, 12], OTOF-related ANSD patients with early intervention are considered good candidates for CI. However, residual pure-tone hearing, which occurs not infrequently in ANSD children, may lead to a reluctance to perform CI earlier, unless there is a definite molecular etiologic diagnosis. Decision of timing of CI in ANSD is furthermore clinically challenging in that a subset of patients indeed manifest spontaneous hearing recovery with age [13], while ANSD patients with OTOF variant do not recover spontaneously.
In fact, a large size of this gene encompassing 46 exons hindered the screening of the whole gene by Sanger sequencing, causing a total dependence on next generation sequencing (NGS). Some previous studies from Korea relying on NGS, including our own, misconceived that the majority of Korean ANSD is unrelated to DFNB9 [14, 15]. Later, this misconception turned out to be mainly due to frequent capture failures and poor coverage of the last exon of the cochlear isoform of OTOF in the conventional panel sequencing strategy, where the predominant variant of DFNB9 in this population p.Arg1939Gln resides [16]. Resultantly, we reported that some portion of anatomically normal cochlear nerve, prelingual-onset ANSD in Koreans could be due to mutations in OTOF and identified that screening of p.Arg1939Gln, which is a major allele in prelingual-onset Korean ANSD, may facilitate detection of DFNB9. Interestingly p.Arg1939Gln was also recognized as a founder mutation in Japanese ANSD populations [17].
However, the overall mutational spectrum of DFNB9 in Koreans has not been fully established yet, and performing either NGS or Sanger sequencing of all the exons of OTOF for all ANSD children is still costly in a clinical setting. Moreover, the potential correlation between the degree of residual hearing as measured by behavioral audiometry and certain OTOF variants has not been clearly elucidated thus far except for a few temperature sensitive mutant alleles of this gene resulting in elevation of hearing thresholds presumably due to a synaptic dysfunction elicited by fever [18, 19]. Knowing the genotype–phenotype correlation, if any, of this large gene may significantly help to narrow down the candidate variants of OTOF. Since our previous report, we further recruited anatomically normal cochlear nerve, prelingual-onset ANSD subjects and confirmed again a high genetic load of OTOF to Korean prelingual ANSD.
Here, we disclose a mutational and phenotypic spectrum of OTOF-related ANSD in this Korean population and propose a set of commonly found OTOF variants in our population that would allow for the potential prioritization of specific OTOF exons during the screening process. The outcomes of CI were also addressed with respect to the timing of implantation.
Methods
Subjects and ethical considerations
Among the total 162 families with hereditary hearing loss ascertained from Seoul National University Hospital (SNUH) and Seoul National University Bundang Hospital (SNUBH) between June 2015 and March 2017, 78 families turned out to manifest hearing loss in an autosomal recessive or sporadic fashion. Of these 78 families, we were able to recruit five new families (SB239, SH195, SH230, SH234, and AJ2) with ANSD through rigorous audiological phenotyping [14]. Together with the five previously reported families (SB10, SB22, SB204, SH132, and SH81) [16], these additional families provided a total of ten families. Genotype and phenotype evaluations, including audiometric assessment, were made through molecular genetic diagnosis and medical chart review. Regarding SB239, two family members (SB239-465, 466) are genetically unrelated, thus they were counted distinctly in the genotypic analysis, and their two children (SB239-463, 464) were included only in the phenotype analysis. The pedigrees of participants were described (Fig. 1).
Fig. 1.

Pedigrees of DFNB9 families with molecular genetic diagnosis. Filled symbols represent hearing-impaired individuals, and clear symbols denote those with normal hearing. A diagonal line through a symbol indicates a deceased person and black arrows indicate probands. Designations below each proband represent sex/age. Previously diagnosed families are surrounded by a red border
Molecular genetic diagnosis
Molecular genetic diagnoses of 5 families with 6 probands was made through a screening kit for detection of prevalent 11 deafness-causing variants [20] and Sanger sequencing of previously reported OTOF variants among Koreans including p.Arg1939Gln followed by either targeted exome sequencing (TES), or whole exome sequencing (WES), as previously described [12, 21–24]. In detail, genomic DNA was extracted from the peripheral blood samples or buccal cells using standard protocols (Gentra Puregene Blood Kit, Qiagen, cat. 158389; Venlo, Limburg, 27 Netherlands). First, screening panel sequencing of 11 variants of 5 deafness genes (GJB2, SLC26A4, CDH23, 12S rRNA, TMPRSS3) in SB239-463, 464 or Sanger sequencing of p.Arg1939Gln of OTOF in SH195-443, SH234-547, AJ2-3, and SH230-538 was performed based on the prevalence of deaf genes in the Korean population. If a potential candidate variant(s) of OTOF was detected, then detailed Sanger sequencing of OTOF was performed to find another candidate variant causing a recessive form of DFNB9 (SH234). If no variant was detected in the screening, then TES129 (SH195, SH230) or WES (SB239, AJ2) followed [22]. Candidate variants were narrowed down filtered through a bioinformatics analysis of the data and selected variants were confirmed by Sanger sequencing. The filtered variants were evaluated for pathogenicity using various in silico prediction software as previously described [14, 25].
Audiometric evaluation
Pure-tone audiometry, ABR, OAE, including transient-evoked OAE (TEOAE) or distortion product OAE (DPOAE), and/or Auditory Steady-State Response (ASSR) were performed to assess the hearing status. Because it was difficult to acquire behavioral audiometry from children under 3 years old, ASSR was performed to infer the auditory threshold; ASSR threshold of 500 Hz was used as a reliable factor to correlate with the auditory threshold [26]. Hearing level calculated by the four-tone average (0.5, 1, 2, and 4 kHz) was labeled as mild (26–40 dB), moderate (41–70 dB), severe (71–90 dB), or profound (> 90 dB) and a difference between the left and right ear air conduction threshold > 20 dB at least two frequencies out of 0.5, 1, and 2 kHz was indicated as asymmetric. Speech perception performance was assessed with modified categories of auditory perception (CAP) scores, which evaluates the auditory receptive ability with a nonlinear, hierarchical scale [27], before and after CI in case of CI recipients.
Statistical analysis
The data are expressed as the mean ± SE. Mann–Whitney test was used to compare the means of postoperative CAP scores between early-CI and late-CI groups at each time point, and the mean ASSR threshold at 500 Hz between patients with homozygous p.Arg1939Gln and non-carriers. Fisher’s exact test was used to determine the association of frequency of p.Arg1939Gln allele in accordance with the timing of CI (early and late-CI group). Statistical analyses were performed and graphs were plotted using GraphPad prism 5.0 software (GraphPad Software, La Jolla, CA, USA). A p value of < 0.05 was considered statistically significant.
Results
Mutational spectrum of DFNB9 in Koreans
We analyzed six unrelated DFNB9 subjects, making the total number of unrelated DFNB9 subjects to 11 (Table 1). The molecular genetic approach adopted in our cohort identified eight different variants and one large genomic deletion of the OTOF gene, of which, three variants segregating in three families were novel (Table 1). Specifically, the inhibitory effect of the c.4227 + 5G > C on the splice donor site was suggested by splice site variant prediction tools, which strongly suggested that the c.4227 + 5G > C splicing variant significantly impairs the canonical splice donor site of exon 34.
Table 1.
Mutational spectrum of 11 subjects from 10 families with DFNB9
| Family ID | Variant (OTOF) NM_001287489 NP_001274418 |
State | Prediction algorithm | Conservation score | MAF | Classification of pathogenic variants [31] | References | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mutation taster | PolyPhen-2 | SIFT | PhyloP | GERP++ | Global MAF | KRGDB (n = 1722) | |||||
| SB10-23 | c.5816G > A: p.Arg1939Gln rs201326023 |
Hom | DC | PD | D | 2.261 | 2.28 | T = 0.00003/1 (ExAC) T = 0.0002/1 (1000 Genomes) |
T = 0.001452/5 | Pathogenic (PS4, PM2, PM3 PP1, PP3, PP4) |
[31] |
| SH132-273 | c.5816G > A: p.Arg1939Gln rs201326023 |
Hom | DC | PD | D | 2.261 | 2.28 | T = 0.00003/1 (ExAC) T = 0.0002/1 (1000 Genomes) |
T = 0.001452/5 | Pathogenic | [31] |
| SB22-51 | c.5816G > A: p.Arg1939Gln rs201326023 |
Het | DC | PD | D | 2.261 | 2.28 | T = 0.00003/1 (ExAC) T = 0.0002/1 (1000 Genomes) |
T = 0.001452/5 | Pathogenic | [31] |
| Large genomic deletion Chr2:26710657 ~ 26706557 | Het | NA | NA | NA | NA | NA | Pathogenic (PVS1, PP1, PP4) | [16] | |||
| SB204-398 | c.5816G > A: p.Arg1939Gln rs201326023 |
Het | DC | PD | D | 2.261 | 2.28 | T = 0.00003/1 (ExAC) T = 0.0002/1 (1000 Genomes) |
T = 0.001452/5 | Pathogenic | [31] |
| c.5566C > T: p.Arg1856Trp rs368155547 |
Het | DC | PD | D | 2.963 | 2.84 | A = 0.00004/5 (ExAC) A = 0.00008/1 (GO-ESP) |
A = 0.000871/3 | Pathogenic (PS4, PM2, PM3 PP1, PP3, PP4) |
[10] | |
| SH81-185 | c.5816G > A: p.Arg1939Gln rs201326023 |
Het | DC | PD | D | 2.261 | 2.28 | T = 0.00003/1 (ExAC) T = 0.0002/1 (1000 Genomes) |
T = 0.001452/5 | Pathogenic | [31] |
| c.2521G > A: p.Glu841Lys rs772729658 |
Het | DC | PD | D | 5.523 | 5 | T = 0.00003/3 (ExAC) | ND | Pathogenic (PS4, PM2, PM3 PP1, PP3, PP4) |
[16] | |
| SB239-465 | c.5816G > A: p.Arg1939Gln rs201326023 |
Het | DC | PD | D | 2.261 | 2.28 | T = 0.00003/1 (ExAC) T = 0.0002/1 (1000 Genomes) |
T = 0.001452/5 | Pathogenic | [31] |
| c.3032T > C: p.Leu1011Pro rs80356596 |
Het | DC | PD | D | 5.012 | 4.64 | ND | ND | Pathogenic (PS4, PM2, PM3 PP1, PP3, PP4) |
[32] | |
| SB239-466 | ac.5791C > A: p.Pro1931Thr rs537706054 | Het | DC | PD | D | 5.867 | 5.22 | T = 0.000008/1 (ExAC) | ND | Pathogenic (PM2, PM3, PP1, PP3, PP4) |
This study |
| c.2521G > A: p.Glu841Lys rs772729658 |
Het | DC | PD | D | 5.523 | 5 | T = 0.00003/3 (ExAC) | ND | Pathogenic | [16] | |
| SH195-443 | c.3192C > G: p.Tyr1064Ter rs766819324 |
Hom | DC | NA | NA | 1.937 | 2.78 | C = 0.000008/1 (ExAC) | C = 0.00029/1 | Pathogenic (PVS1, PM2, PP1, PP3, PP4) | [15] |
| SH234-547 | c.5816G > A: p.Arg1939Gln rs201326023 |
Het | DC | PD | D | 2.261 | 2.28 | T = 0.00003/1 (ExAC) T = 0.0002/1 (1000 Genomes) |
T = 0.001452/5 | Pathogenic | [31] |
| c.5566C > T: p.Arg1856Trp rs368155547 |
Het | DC | PD | D | 2.963 | 2.84 | A = 0.00004/5 (ExAC) A = 0.00008/1 (GO-ESP) |
A = 0.000871/3 | Pathogenic | [10] | |
| AJ2-3 |
ac.5534G > A: p.Gly1845Glu dbSNP ID:ND |
Het | DC | PD | D | 5.739 | 4.97 | ND | ND | Pathogenic (PM2, PM3 PP1, PP3, PP4) |
This study |
| c.3032T > C: p.Leu1011Pro rs80356596 |
Het | DC | PD | D | 5.012 | 4.64 | ND | ND | Pathogenic | [32] | |
| SH230-538 | c.2521G > A: p.Glu841Lys rs772729658 |
Het | DC | PD | D | 5.523 | 5 | T = 0.00003/3 (ExAC) | ND | Pathogenic | [16] |
|
ac.4227 + 5G > C rs571671530 |
Het | DC | NA | NA | 1.616 | 3.95 | G = 0.00006/7 (ExAC) G = 0.0002/1 (1000 Genomes) |
ND | Likely pathogenic (PM2, PM3 PP1, PP3, PP4) |
This study | |
Splice site variant prediction tools by ESEfinder, NNSplice, and NetGene2: splice site broken. Normal score (10.34940) and mutant score (6.54650) by ESEfinder, Normal score (0.99) and mutant score (0.50) by NNSplice, Normal score (0.997) and mutant score (0.685) by NetGene2
In silico prediction Algorithm: Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/index.shtml); SIFT (http://sift.jcvi.org/www/SIFT_chr_coords_submit.html) or SIFT-indels2 (http://sift.bii.a-star.edu.sg/www/SIFT_indels2.html)
Conservation tools: GERP++ score in the UCSC Genome Browser (http://genome-asia.ucsc.edu/); PhyloP score from the Mutation Taster (http://www.mutationtaster.org/)
Splice site prediction tools: ESEfinder (http://rulai.cshl.edu/cgi-bin/tools/ESE3/esefinder.cgi?process=home); NNSplice (http://www.fruitfly.org/seq_tools/splice.html); NetGene2 (http://www.cbs.dtu.dk/services/NetGene2/)
ExAC, Exome Aggregation Consortium (http://exac.broadinstitute.org/)
1000 Genomes (https://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/)
KRGDB, Korean Reference Genome DB (http://152.99.75.168/KRGDB/)
Het, heterozygote mutant; Hom, homozygote mutant; DC, disease causing; PD, probably damaging; D, damaging; ND, not detected; NA, not applicable
aNovel variant
Taken together with the data from the previous study, six different kinds of missense variants, one nonsense variant, one splice site variant, and one large deletion were found in our cohort of 11 DFNB9, estimating the contribution of OTOF etiology to be 90.9% (10 DFNB9 pedigrees/11 ANSD families) among confirmed prelingual ANSD patients in our Korean deaf cohorts. Regarding the frequency of OTOF variants, p.Arg1939Gln was most commonly identified among the 22 alleles in 11 DFNB9 subjects, with a frequency of 40.9% (9/22). The variant p.Glu841Lys was the second most frequent (13.6%, 3/22), followed by two variants, p.Leu1011Pro and p.Arg1856Trp, which accounted for 9.1% (2/22) each. The total contribution of these four major alleles was 72.7% among all ANSD patients with OTOF variants. The detected variants in the OTOF gene were distributed ranging from exon 21–46, clustered around C2D, C2E, and C2F domains of otoferlin (Fig. 2).
Fig. 2.

Protein domain structures of otoferlin with pathogenic variants in our cohort. Upper panel: Otoferlin contains six C2 domains (C2A–C2F). Missense variants are displayed (top) on the domain structure, and nonsense and splice site variants are located below the domain structure. A red bordered area contains three C2 domains encoded by short isoform of OTOF. TM: transmembrane domain. Lower panel: Exon–Intron structure of various OTOF isoforms (NM_001287489, NM_194248, NM_194323, NM_194322, NM_004802) locating each variant detected in the study (Captured from UCSC genome browser (https://genome.ucsc.edu/))
Phenotypic spectrum of DFNB9 in Koreans
Temporal bone CT or internal auditory canal MRI clearly revealed no anatomical abnormalities of the cochlear nerve in the 11 probands. Regarding newborn hearing screening (NBHS), one patient (SH230-538) passed the NBHS even with significant hearing loss because only OAE was performed; the other patient (SH132-273) did not undergo NBHS. All patients, except these two, failed NBHS, facilitating an early detection of hearing loss and leading to a molecular genetic diagnosis of DFNB9, and no fever-induced hearing aggravation was observed.
Detailed audiological results of eleven patients are described in Fig. 3 and Table 2. The results of ABR did not show any noticeable responses on both sides, and OAEs indicated the presence of response in accordance with the definition of ANSD. On the contrary, ASSR recordings demonstrated various results, including asymmetric or low-frequency hearing loss configuration. In detail, one patient (SB10-23) showed asymmetric hearing loss, and another patient (SH230-538) had characteristic low-frequency hearing loss on both sides. The severity ranged mostly from severe to profound only with some exceptions (SB22-51, SB239-463, SH230-538) based on ASSR results. All patients who had no response with ABR test, presented measurable ASSR thresholds, showing the possibility of providing more detailed hearing level and configuration.
Fig. 3.

Audiological phenotypes of affected members in ten families with DFNB9. ABR, OAE, and ASSR results are described in order in each patient. Each number on each side of ABR (right in red and left in blue) represents the stimulus intensity calibrated in dB or nHL, which showed no response in all patients. Auditory thresholds of ASSR at 500 Hz are circled in red and blue, each for right and left sides, respectively
Table 2.
Phenotypic spectrum of 11 subjects (including SB239-463, 464) from 10 families with DFNB9
| Family ID | Genotype variant (OTOF) NM_001287489 NP_001274418 |
Age at first CI (month) | Laterality | Pre-CI assessment | Post-CI CAP score | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ABR (dB) | OAE | ASSR at 500 Hz (dB) | CAP score | 3 m | 6 m | 12 m | 24 m | 36 m | ||||
| SB10-23 | c.5816G > A: p.Arg1939Gln homozygous state |
21 | Bilateral, Sequential |
B) NR | L) response (+) | R) 105 L) 80 |
0 | 1 | 1 | 4 | 6 | 7 |
| SH132-273 | c.5816G > A: p.Arg1939Gln homozygous state |
22 | Bilateral, Sequential |
B) NR | B) response (+) | R) 110 L) 105 |
0 | 2 | 3 | 5 | 7 | |
| SB22-51 | c.5816G > A: p.Arg1939Gln Large genomic deletion (Chr2:26710657 ~ 26706557) |
26 | Bilateral, Sequential |
B) NR | L) response (+) | R) 75 L) 70 |
0 | 1 | 5 | 5 | 7 | |
| SB204-398 | c.5816G > A: p.Arg1939Gln c.5566C > T: p.Arg1856Trp |
13 | Bilateral, Simulatneous |
B) NR | B) response (+) | R) 80 L) 100 |
0 | 2 | 4 | 4 | 5 | |
| SH81-185 | c.5816G > A: p.Arg1939Gln c.2521G > A: p.Glu841Lys |
25 | Bilateral, Sequential |
B) NR | B) response (+) | NA | 0 | 1 | 1 | 4 | 4 | |
| SB239-463 | c.2521G > A: p.Glu841Lys c.3032T > C: p.Leu1011Pro |
14 | Bilateral, Simulatneous |
B) NR | B) response (+) | R) 65 L) 65 |
1 | 3 | 5 | 5 | ||
| SB239-464 | c.5816G > A: p.Arg1939Gln c.2521G > A: p.Glu841Lys |
14 | Bilateral, Simulatneous |
B) NR | B) response (+) | R) 80 L) 80 |
0 | 2 | 4 | 5 | ||
| SH195-443 | c.3192C > G: p.Tyr1064Ter homozygous state |
24 | Bilateral, Simulatneous |
B) NR | B) response (+) | R) 85 L) 100 |
1 | 3 | FU loss | |||
| SH234-547 | c.5816G > A: p.Arg1939Gln c.5566C > T: p.Arg1856Trp |
15 | Bilateral, Simulatneous |
B) NR | B) response (+) | R) 90 L) 85 |
0 | 3 | ||||
| AJ2-3 | c.5534G > A: p.Gly1845Glu c.3032T > C: p.Leu1011Pro |
18 | Bilateral, Sequential |
B) NR | B) response (+) | R) 100 L) 90 |
0 | 4 | 4 | |||
| SH230-538 | c.4227 + 5G > C c.2521G > A: p.Glu841Lys |
Scheduled | NA | B) NR | B) response (+) | R) 75 L) 60 |
0 | |||||
NR, no response; NA, not applicable
We observed a slight genotype–phenotype correlation in one family (SB239), where even the intra-familial phenotypic variations were found. In SB239, there are four family members having DFNB9 (parents are biological parents and genetically unrelated). In detail, SB239-463 (daughter: compound heterozygous state carrying p.Glu841Lys and p.Leu1011Pro) showed better pre-CI CAP score and ASSR hearing thresholds at 500 Hz than her dizygotic twin brother SB239-464 carrying p.Arg1939Gln along with p.Glu841Lys (Fig. 3). Likewise, of their two parents who had received CIs during adulthood, the father (SB239-465) carrying p.Arg1939Gln did not improve in listening skills with auditory rehabilitation, while the mother (SB239-466) without p.Arg1939Gln demonstrated great improvement, showing a stark contrast with her husband under similar clinical settings.
Given that only ASSR thresholds at 500 Hz were reported to correlate with behavioral thresholds from ANSD subjects [26], we specifically pursued the issue of genotype–phenotype correlation using the thresholds at 500 Hz. ASSR thresholds at 500 Hz from p.Arg1939Gln homozygotes (n = 2) were higher compared with those from the p.Arg1939Gln non-carriers (p = 0.033) (n = 4), and this significant phenotypic difference was also reproduced for a comparison between p.Arg1939Gln homozygotes and all the others (n = 8) including p.Arg1939Gln single heterozygotes (p = 0.022) although the sample size was small to draw a firm conclusion.
With respect to auditory rehabilitation in our cohort, ten out of eleven patients underwent bilateral CI, either sequentially or simultaneously, and one patient (SH230-538) is scheduled to have one (Table 2). Based on the CAP score, our cohort showed comparable postoperative auditory outcomes with those observed in cochlear implantees with other etiologies [12]. For further analysis, CI recipients were divided into two groups: early (age at CI ≤ 18mo) and late-CI groups (age at CI > 18mo) with potential waiting time for CI in ANSD patients in a clinical setting considered. Longitudinal changes in the auditory performance (CAP scores) were compared between the early (n = 5) and late-CI groups (n = 5) (Fig. 4). The early-CI group showed better outcomes than the late-CI group at post-CI 6mo (p = 0.04), but the difference became minimal by post-CI 12 mo. The p.Arg1939Gln allele tended to be more frequently detected in the late-CI group (60%) than in the early-CI group (30%), but without statistical significance (p = 0.37).
Fig. 4.
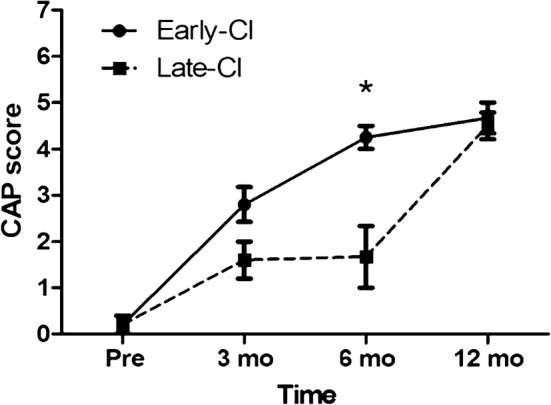
Average CAP scores of CI recipients. Audiologic performances of the two subgroups (early-CI and late-CI) at pre- and post-CI were analyzed longitudinally (p = 0.04 and 0.08 at post-CI 3 mo and 6 mo respectively). CAP is composed of a nonlinear, hierarchical scale with eight categories: (0) Displays no awareness of environmental sounds; (1) awareness of environmental sounds; (2) responds to speech sounds; (3) recognizes environmental sounds; (4) discriminates at least two speech sounds; (5) understands common phrases without lip-reading; (6) understands conversation without lip-reading with a familiar talker; (7) can use telephone with a familiar talker
Discussion
In this study, which included the largest Korean cohort to date, a strong genetic etiology of ANSD in Korean pediatric deaf population was confirmed again, with alterations of OTOF explaining 91% of ANSD patients and we were able to come up with a set of four recurring variants that allow screening of 72.7% of the total OTOF variants detected in Korean ANSD children. Moreover, our study identified three novel OTOF variants, which comprised of two missense variants and one splice site variant. Interestingly, all variants detected in our cohort were distributed in relatively limited parts of the OTOF gene, ranging between exons 21 and 46. It is worth noting that all of the OTOF variants detected in Korean DFNB9 subjects are confined to the common regions shared by both short and long isoforms, affecting both of the two isoforms. We could not detect the pathogenic variants that were present exclusively in the long isoform-specific sequences (from exon 1 through exon 20) from our cohorts. However, in other ethnic populations, variants residing in the sequences specific to the long isoforms are present, clearly establishing the requirement of long isoforms of otoferlin for hearing in the literature [10]. As such, the confined characteristic of Korean OTOF variants to the region shared by both long and short isoforms is contrasted with the dispersed characteristic of Pakistani DFNB9 subjects, in which several private variants are scattered all across the entire long isoforms [10]. It is worth mentioning that a predominantly recurring variant, such as p.Arg1939Gln in Koreans, does not exist in the Pakistani population. Preponderance of a small number of variants only in the limited region clearly offers an advantageous condition from the perspectives of mutation screening strategy, which should also consider the future discovery of other pathogenic OTOF variants in more representative samples of Korean ANSD patients.
In terms of auditory phenotypes, ASSR showed diverse configurations and thresholds in spite of no response in the ABR test throughout our entire prelingual ANSD cohort. The variable degree of reduced neurotransmitter release in each variant or each patient might be a cause of various ASSR results [8]. However, these ASSR results need to be interpreted cautiously given the characteristics of ASSR. ASSR has its merits for evaluating hearing in children with hearing threshold beyond the limit of ABR, and it could also measure more frequency-specific hearing threshold than ABR [28]. The variance in ASSR results compared with the consistency of no response in ABR recordings may suggest hidden residual hearing or frequency-specific hearing threshold not evaluated by ABR. However, its reliability in evaluating middle-frequency hearing is relatively low; hence, ASSR alone may be insufficient, requiring a combination with other hearing assessment tools, including ABR. In this study, the ASSR threshold only at 500 Hz was used to infer the behavioral threshold [26]. This prompted us to extend the analysis to include all ASSR thresholds at 500 Hz of our DFNB9 cohort and evaluate whether there is any auditory phenotype-genotype correlation among DFNB9 subjects using this parameter. The elucidation of this correlation, if any, might in turn help to suspect certain OTOF variants, which may facilitate molecular diagnosis of DFNB9 and eventually ensure early CI for better outcomes. Inspired by the intra-familial phenotypic difference from family 239, we speculated that there might be some differences in the pathogenic potential between those with and without p.Arg1939Gln that causes this kind of variations. Previously, more severe phenotype with p.Arg1939Gln than other non-truncating variants was suggested, which could support the possible genotype–phenotype correlation that contributed to the intra-familial phenotypic differences [17]. In our study, significantly worse ASSR thresholds at 500 Hz of patients with homozygous p.Arg1939Gln compared with those with non-p.Arg1939Gln homozygotes and those with non-p.Arg1939Gln carriers were observed although the interpretation should be made cautiously due to the small sample size. However, at least we could say that in case of ANSD subjects with null residual hearing, we may have to think about the p. Arg1939Gln homozygous phenotype, which frequently escapes capture in NGS. The exclusion of DFNB9 by just relying on the NGS results may be risky in these cases.
Interestingly, the p.Arg1939Gln allele tended to be more frequently detected in the late-CI group (60%) than in early-CI group (30%). This tendency might have been partly due to the frequent capture failures through NGS in screening the last exon carrying p. Arg1939Gln although no statistical inferences can be made due to the small sample size. Many surgeons tend to delay CI in ANSD children, at least until the child is 18 months old, unless they are given a definite molecular diagnosis of DFNB9. If the molecular genetic diagnosis is made earlier, it may be possible to perform CI sooner, rather than later. Given this, the elucidation of a set of frequently occurring OTOF variants that can be easily screened by Sanger sequencing in Koreans may significantly contribute to earlier CI in DFNB9 children, which comprised a majority of ANSD children in Koreans.
In line with the previous report [11], the CI outcomes of our DFNB9 cohorts also demonstrated favorable results, with small variance, according to the timing of implantation (early vs. late) although small sample size undermined the conclusion. This phenomenon might be partly due to the fact that the late-CI group also received CI around 24 months, which was still regarded as the acceptable period for proper auditory development [29].
Then, why is the p.Arg1939Gln allele associated with poor ASSR thresholds? No possible explanations were proposed yet for this frequent allele as well as other DFNB9 alleles. OTOF encodes the multi-C2 domain protein otoferlin, whose alterations are associated with human prelingual deafness DFNB9 [4]. Otoferlin was reported to play a key role in synaptic sound encoding by promoting exocytosis in the inner hair cells and priming the vesicle in the presynaptic area [30]. There have been previous reports about temperature sensitive elevation of hearing thresholds in humans presumably caused by a synaptic dysfunction elicited by fever [18, 19]. In a recent study comparing mice to humans, mice with homozygous Otof mutation (OtofI515T/I515T) were less susceptible to heat than humans with the same variants, and temperature sensitivity of the phenotype observed in humans was assumed to be related with alterations in arginine-rich RXR motifs, which were located in positions 1244–1263 only in the human reference sequences (NP_001274418), and not in mice, causing heat instability of otoferlin [30]. According to the article, there seems to be heat sensitivity within otoferlin itself, which could worsen with mutations that weaken intermolecular interactions—like the Ile515Thr mutation that was predicted to exchange a hydrophobic residue in the hydrophobic core of the C2C domain against a more hydrophilic residue. In our study, no variant seems to be associated with such a RXR motif described previously [30], and no fever-induced deafness was observed. However, temperature sensitivity observed in some DFNB9 needs further evaluation and analysis and an in vivo or in vitro study using molecular biology tools will help us elucidating the mechanism underlying the genotype–phenotype correlation in the near future.
Conclusions
Here, we report four major variants p.Arg1939Gln, p.Glu841Lys, p.Leu1011Pro, and p.Arg1856Trp—responsible for 73% of our Korean DFNB9 cohort. We believe that these variants can be implemented into the genetic screening of ANSD patients in Korea. Sanger sequencing of exons 21 through 46 could also be considered as a next tier of mutation screening in Korean patients with prelingual ANSD, especially when there is a lack of conclusive results of OTOF genotypes from screening the four major OTOF variants and/or when NGS technologies are not available. Early genetic diagnosis of OTOF-related ANSD could allow for greater accuracy in counseling of auditory rehabilitation and sooner clinical decision to lead to better auditory outcomes. Our study may suggest that encountering poorer ASSR thresholds in potential DFNB9 candidates might be more preferentially related to p.Arg1939Gln homozygotes, of which genotype specific to East Asians have been frequently undiagnosed solely by NGS technologies. The intimate reciprocal interaction and feedback between genotypes and auditory phenotype related to DFNB9 may complement the drawbacks of the existing molecular diagnostic tests that target DFNB9, which in turn, would result in earlier implantation and better CI outcomes.
Authors’ contributions
BJK, JHJ, Y-C and BYC designed the study, analyzed data and wrote the paper; JHH, ARK, CL and NKDK collected and analyzed data; H-P, DYO, SL and MYK performed experiments, collected data, and designed figures; Y-P and BYC reviewed the manuscript. All authors read and approved the final manuscript.
Acknowledgements
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its additional file.
Consent for publication
Each person provided signed informed consent for publication of the results of the study.
Ethics approval and consent to participate
The study conformed to the ethical guidelines of the World Medical Association Declaration of Helsinki. All steps in this study were approved by the institutional review boards of SNUH (IRBY-H-0905-041-281) and SNUBH (IRB-B-1007-105-402), and written informed consent was obtained from all subjects or guardians in case of children.
Funding
This research was supported by a Grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (HI15C1632, and HI17C0952 to B. Y. Choi), and Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2015R1D1A1A01058225 to B.Y. Choi and 2018R1D1A1B07046159 to B.J.Kim).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- ANSD
auditory neuropathy spectrum disorder
- NGS
next generation sequencing
- CI
cochlear implantation
- OAE
otoacoustic emissions
- CM
cochlear microphonics
- ABR
auditory brainstem response
- TES
targeted exome sequencing
- WES
whole exome sequencing
- PTA
pure-tone audiometry
- ASSR
auditory steady-state response
- CAP
categories of auditory perception
- NBHS
newborn hearing screening
Contributor Information
Bong Jik Kim, Email: cellokimbj@gmail.com.
Jeong Hun Jang, Email: jhj@ajou.ac.kr.
Jin Hee Han, Email: flyswan@hanmail.net.
Hye-Rim Park, Email: Happyrim11@gmail.com.
Doo Yi Oh, Email: dooyi9@gmail.com.
Seungmin Lee, Email: gdklom76@gmail.com.
Min Young Kim, Email: mn911@hanmail.net.
Ah Reum Kim, Email: amorkimar@naver.com.
Chung Lee, Email: spinelyc@gmail.com.
Nayoung K. D. Kim, Email: nayoung.kd.kim@samsung.com
Woong-Yang Park, Email: woongyang@gmail.com.
Yun-Hoon Choung, Phone: +82-(0)31-219-5263, Email: yhc@ajou.ac.kr.
Byung Yoon Choi, Phone: +82-(0)31-787-7406, Email: choiby2010@gmail.com.
References
- 1.Starr A, Picton TW, Sininger Y, Hood LJ, Berlin CI. Auditory neuropathy. Brain. 1996;119(Pt 3):741–753. doi: 10.1093/brain/119.3.741. [DOI] [PubMed] [Google Scholar]
- 2.Rance G, Beer DE, Cone-Wesson B, Shepherd RK, Dowell RC, King AM, Rickards FW, Clark GM. Clinical findings for a group of infants and young children with auditory neuropathy. Ear Hear. 1999;20:238–252. doi: 10.1097/00003446-199906000-00006. [DOI] [PubMed] [Google Scholar]
- 3.Raveh E, Buller N, Badrana O, Attias J. Auditory neuropathy: clinical characteristics and therapeutic approach. Am J Otolaryngol. 2007;28:302–308. doi: 10.1016/j.amjoto.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 4.Yasunaga S, Grati M, Cohen-Salmon M, El-Amraoui A, Mustapha M, Salem N, El-Zir E, Loiselet J, Petit C. A mutation in OTOF, encoding otoferlin, a FER-1-like protein, causes DFNB9, a nonsyndromic form of deafness. Nat Genet. 1999;21:363–369. doi: 10.1038/7693. [DOI] [PubMed] [Google Scholar]
- 5.Delmaghani S, del Castillo FJ, Michel V, Leibovici M, Aghaie A, Ron U, Van Laer L, Ben-Tal N, Van Camp G, Weil D, et al. Mutations in the gene encoding pejvakin, a newly identified protein of the afferent auditory pathway, cause DFNB59 auditory neuropathy. Nat Genet. 2006;38:770–778. doi: 10.1038/ng1829. [DOI] [PubMed] [Google Scholar]
- 6.Schoen CJ, Emery SB, Thorne MC, Ammana HR, Sliwerska E, Arnett J, Hortsch M, Hannan F, Burmeister M, Lesperance MM. Increased activity of Diaphanous homolog 3 (DIAPH3)/diaphanous causes hearing defects in humans with auditory neuropathy and in Drosophila. Proc Natl Acad Sci USA. 2010;107:13396–13401. doi: 10.1073/pnas.1003027107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang QJ, Li QZ, Rao SQ, Lee K, Huang XS, Yang WY, Zhai SQ, Guo WW, Guo YF, Yu N, et al. AUNX1, a novel locus responsible for X linked recessive auditory and peripheral neuropathy, maps to Xq23-27.3. J Med Genet. 2006;43:e33. doi: 10.1136/jmg.2005.037929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Santarelli R, del Castillo I, Cama E, Scimemi P, Starr A. Audibility, speech perception and processing of temporal cues in ribbon synaptic disorders due to OTOF mutations. Hear Res. 2015;330:200–212. doi: 10.1016/j.heares.2015.07.007. [DOI] [PubMed] [Google Scholar]
- 9.Pangrsic T, Reisinger E, Moser T. Otoferlin: a multi-C2 domain protein essential for hearing. Trends Neurosci. 2012;35:671–680. doi: 10.1016/j.tins.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 10.Choi BY, Ahmed ZM, Riazuddin S, Bhinder MA, Shahzad M, Husnain T, Riazuddin S, Griffith AJ, Friedman TB. Identities and frequencies of mutations of the otoferlin gene (OTOF) causing DFNB9 deafness in Pakistan. Clin Genet. 2009;75:237–243. doi: 10.1111/j.1399-0004.2008.01128.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu CC, Hsu CJ, Huang FL, Lin YH, Lin YH, Liu TC, Wu CM. Timing of cochlear implantation in auditory neuropathy patients with OTOF mutations: our experience with 10 patients. Clin Otolaryngol. 2018;43:352–357. doi: 10.1111/coa.12949. [DOI] [PubMed] [Google Scholar]
- 12.Park JH, Kim AR, Han JH, Kim SD, Kim SH, Koo JW, Oh SH, Choi BY. Outcome of cochlear implantation in prelingually deafened children according to molecular genetic etiology. Ear Hear. 2017;38:e316–e324. doi: 10.1097/AUD.0000000000000437. [DOI] [PubMed] [Google Scholar]
- 13.Harrison RV, Gordon KA, Papsin BC, Negandhi J, James AL. Auditory neuropathy spectrum disorder (ANSD) and cochlear implantation. Int J Pediatr Otorhinolaryngol. 2015;79:1980–1987. doi: 10.1016/j.ijporl.2015.10.006. [DOI] [PubMed] [Google Scholar]
- 14.Jin YJ, Park J, Kim AR, Rah YC, Choi BY. Identification of a novel splice site variant of OTOF in the Korean nonsyndromic hearing loss population with low prevalence of the OTOF mutations. Int J Pediatr Otorhinolaryngol. 2014;78:1030–1035. doi: 10.1016/j.ijporl.2014.03.033. [DOI] [PubMed] [Google Scholar]
- 15.Bae SH, Baek JI, Lee JD, Song MH, Kwon TJ, Oh SK, Jeong JY, Choi JY, Lee KY, Kim UK. Genetic analysis of auditory neuropathy spectrum disorder in the Korean population. Gene. 2013;522:65–69. doi: 10.1016/j.gene.2013.02.057. [DOI] [PubMed] [Google Scholar]
- 16.Chang MY, Kim AR, Kim NK, Lee C, Park WY, Choi BY. Refinement of molecular diagnostic protocol of auditory neuropathy spectrum disorder: disclosure of significant level of etiologic homogeneity in Koreans and its clinical implications. Medicine (Baltimore) 2015;94:e1996. doi: 10.1097/MD.0000000000001996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsunaga T, Mutai H, Kunishima S, Namba K, Morimoto N, Shinjo Y, Arimoto Y, Kataoka Y, Shintani T, Morita N, et al. A prevalent founder mutation and genotype-phenotype correlations of OTOF in Japanese patients with auditory neuropathy. Clin Genet. 2012;82:425–432. doi: 10.1111/j.1399-0004.2012.01897.x. [DOI] [PubMed] [Google Scholar]
- 18.Varga R, Avenarius MR, Kelley PM, Keats BJ, Berlin CI, Hood LJ, Morlet TG, Brashears SM, Starr A, Cohn ES, et al. OTOF mutations revealed by genetic analysis of hearing loss families including a potential temperature sensitive auditory neuropathy allele. J Med Genet. 2006;43:576–581. doi: 10.1136/jmg.2005.038612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marlin S, Feldmann D, Nguyen Y, Rouillon I, Loundon N, Jonard L, Bonnet C, Couderc R, Garabedian EN, Petit C, Denoyelle F. Temperature-sensitive auditory neuropathy associated with an otoferlin mutation: deafening fever! Biochem Biophys Res Commun. 2010;394:737–742. doi: 10.1016/j.bbrc.2010.03.062. [DOI] [PubMed] [Google Scholar]
- 20.Han KH, Kim AR, Kim MY, Ahn S, Oh SH, Song JH, Choi BY. Establishment of a flexible real-time polymerase chain reaction-based platform for detecting prevalent deafness mutations associated with variable degree of sensorineural hearing loss in Koreans. PLoS ONE. 2016;11:e0161756. doi: 10.1371/journal.pone.0161756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim BJ, Kim AR, Park G, Park WY, Chang SO, Oh SH, Choi BY. Targeted exome sequencing of deafness genes after failure of auditory phenotype-driven candidate gene screening. Otol Neurotol. 2015;36:1096–1102. doi: 10.1097/MAO.0000000000000747. [DOI] [PubMed] [Google Scholar]
- 22.Kim BJ, Kim AR, Lee C, Kim SY, Kim NK, Chang MY, Rhee J, Park MH, Koo SK, Kim MY, et al. Discovery of CDH23 as a significant contributor to progressive postlingual sensorineural hearing loss in Koreans. PLoS ONE. 2016;11:e0165680. doi: 10.1371/journal.pone.0165680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim BJ, Kim AR, Han JH, Lee C, Oh DY, Choi BY. Discovery of MYH14 as an important and unique deafness gene causing prelingually severe autosomal dominant nonsyndromic hearing loss. J Gene Med. 2017;19:e2950. doi: 10.1002/jgm.2950. [DOI] [PubMed] [Google Scholar]
- 24.Han KH, Oh DY, Lee S, Lee C, Han JH, Kim MY, Park HR, Park MK, Kim NKD, Lee J, et al. ATP1A3 mutations can cause progressive auditory neuropathy: a new gene of auditory synaptopathy. Sci Rep. 2017;7:16504. doi: 10.1038/s41598-017-16676-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim BJ, Han JH, Park HR, Kim MY, Kim AR, Oh SH, Park WY, Oh DY, Lee S, Choi BY. A clinical guidance to DFNA22 drawn from a Korean cohort study with an autosomal dominant deaf population: a retrospective cohort study. J Gene Med. 2018;20:e3019. doi: 10.1002/jgm.3019. [DOI] [PubMed] [Google Scholar]
- 26.Jafari Z, Malayeri S, Ashayeri H, Farahani MA. Adults with auditory neuropathy: comparison of auditory steady-state response and pure-tone audiometry. J Am Acad Audiol. 2009;20:621–628. doi: 10.3766/jaaa.20.10.4. [DOI] [PubMed] [Google Scholar]
- 27.Archbold S, Lutman ME, Marshall DH. Categories of auditory performance. Ann Otol Rhinol Laryngol Suppl. 1995;166:312–314. [PubMed] [Google Scholar]
- 28.Park SK, Kim BJ, Hur DG, Lee KJ, Lee JH, Oh SH, Kim CS, Chang SO. The validity of auditory steady-state response as frequency-specific threshold test in infant-toddlers. Korean J Audiol. 2006;10:105–111. [Google Scholar]
- 29.Sharma A, Cardon G. Cortical development and neuroplasticity in Auditory neuropathy spectrum disorder. Hear Res. 2015;330:221–232. doi: 10.1016/j.heares.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Strenzke N, Chakrabarti R, Al-Moyed H, Muller A, Hoch G, Pangrsic T, Yamanbaeva G, Lenz C, Pan KT, Auge E, et al. Hair cell synaptic dysfunction, auditory fatigue and thermal sensitivity in otoferlin Ile515Thr mutants. EMBO J. 2016;35:2519–2535. doi: 10.15252/embj.201694564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Varga R, Kelley PM, Keats BJ, Starr A, Leal SM, Cohn E, Kimberling WJ. Non-syndromic recessive auditory neuropathy is the result of mutations in the otoferlin (OTOF) gene. J Med Genet. 2003;40:45–50. doi: 10.1136/jmg.40.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tekin M, Akcayoz D, Incesulu A. A novel missense mutation in a C2 domain of OTOF results in autosomal recessive auditory neuropathy. Am J Med Genet A. 2005;138:6–10. doi: 10.1002/ajmg.a.30907. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its additional file.