

Abstract
The phylum Apicomplexa encompasses deadly pathogens such as malaria and Cryptosporidium. Apicomplexa cell division is mechanistically divergent from that of their mammalian host, potentially representing an attractive source of drug targets. Depending on the species, apicomplexan parasites can modulate the output of cell division, producing two to thousands of daughter cells at once. The inherent flexibility of their cell division mechanisms allows these parasites to adapt to different niches, facilitating their dissemination. Toxoplasma gondii tachyzoites divide using a unique form of cell division called endodyogeny. This process involves a single round of DNA replication, closed nuclear mitosis, and assembly of two daughter cells within a mother. In higher Eukaryotes, the four-subunit chromosomal passenger complex (CPC) (Aurora kinase B (ARKB)/INCENP/Borealin/Survivin) promotes chromosome bi-orientation by detaching incorrect kinetochore–microtubule attachments, playing an essential role in controlling cell division fidelity. Herein, we report the characterization of the Toxoplasma CPC (Aurora kinase 1 (Ark1)/INCENP1/INCENP2). We show that the CPC exhibits dynamic localization in a cell cycle-dependent manner. TgArk1 interacts with both TgINCENPs, with TgINCENP2 being essential for its translocation to the nucleus. While TgINCENP1 appears to be dispensable, interfering with TgArk1 or TgINCENP2 results in pronounced division and growth defects. Significant anti-cancer drug development efforts have focused on targeting human ARKB. Parasite treatment with low doses of hesperadin, a known inhibitor of human ARKB at higher concentrations, phenocopies the TgArk1 and TgINCENP2 mutants. Overall, our study provides new insights into the mechanisms underpinning cell cycle control in Apicomplexa, and highlights TgArk1 as potential drug target.
Electronic supplementary material
The online version of this article (10.1007/s00018-018-2889-6) contains supplementary material, which is available to authorized users.
Keywords: Apicomplexa, Toxoplasma gondii, Cytokinesis, Mitosis, Centrosome, Kinetochore, Centromere, Spindle, Chromosomal passenger complex, Aurora kinase, Endodyogeny, Inner membrane complex
Introduction
Apicomplexan parasites are widespread pathogens of both humans and animals. The phylum includes deadly pathogens such as Plasmodium falciparum, the causative agent of malaria, Toxoplasma gondii, Babesia, Eimeria, and Cryptosporidium [1]. Apicomplexan parasites are all obligate intracellular, and display complex life cycles, alternating among different hosts, and among very different cell types within their hosts. Inside their host cells, apicomplexans undergo several cycles of cell division which allow fast population growth within a short time, causing host cell destruction and effective dissemination of the infection [2].
Apicomplexan cell division can vary widely among species, and even within a single species when infecting different hosts. However, the general mechanism remains conserved with a considerable number of features that distinguish it from mitosis in metazoan cells. Apicomplexan cell division consists of closed mitosis of the nucleus, followed by the assembly of daughter cells [2]. During closed mitosis, the nuclear envelope remains in place, and chromosomes do not condense significantly [3]. Toxoplasma gondii (T. gondii) tachyzoites use endodyogeny to divide. During this process, a single round of DNA replication is followed by the internal assembly of two daughter cells [4].
Mitosis and cytokinesis in eukaryotic cells involve the concerted action of several conserved serine/threonine protein kinases, which include cyclin-dependent kinases (and the cyclic expression of cyclins), Polo-like kinases, Nima-related kinases and Aurora kinases [5, 6]. Unlike other eukaryotes, progression through the cell cycle in Apicomplexa appears to be controlled at the level of the centrosome [7–11]. In addition to functioning as a microtubule organizing center, the apicomplexan centrosome serves as signaling hub which not only nucleates spindle microtubules during mitosis but also the structures required to scaffold emerging daughter cells [2, 11]. Recently, several kinases have been described to play a role in centrosome biology, and/or in coordinating the progression of the cell cycle. T. gondii Nek1 and MAPK1-like are recruited to the centrosome, and are required for daughter cell budding and mitosis [7, 11], TgCDPK7 is critical for centrosomal positioning and integrity [12]; TgArk2 is dispensable but mutants of TgArk3 divide more slowly, taking an average of an additional hour to complete endodyogeny (6.5 vs. 7.7 h) [13]. In addition, parasites express ten homologs of cyclin-dependent kinases, named TgCrks. TgCrk1 is essential for daughter bud assembly, TgCrk2 is involved in the transition from G1 to S phase, TgCrk4 and TgCrk6 assist spindle function and centrosome duplication, and TgCrk9 regulates transcription and gene expression [14–16]. However, kinase activity, targets and precise roles remain understudied. This is also true for other cell cycle control kinase homologs present in the apicomplexan genome. One such example is Aurora kinase homologs.
Aurora kinases control the dynamics of the centrosome and bipolar microtubule spindle formation, as well as, chromosome segregation and cytokinesis [17–19]. During mitosis, chromosomal passenger complex (CPC) is a central player that orchestrates various events, such as chromosome alignment and cytokinesis [20]. In higher eukaryotes, the CPC is formed by Aurora kinase B (ARKB) and three other regulatory subunits; the inner centromere protein (INCENP), Survivin and Borealin [20]. In metaphase, ARKB concentrates in the proximity of centromeres and kinetochores, the sites of attachment of chromosomes to spindle microtubules. There, it ensures proper kinetochore–microtubule attachment by removing improper attachments. To accomplish its job in chromosome bi-orientation, Aurora B phosphorylates centromere-associated proteins, kinetochores and microtubules [20–22]. In telophase, the CPC enriches at the midbody, a bridge that connects dividing cells. There it phosphorylates intermediate filament proteins, as well as, desmin, vimentin and myosin II, to control the timing of abscission, therefore, playing a central role in completing cytokinesis [23–28]. In the present study, we identified putative component of the T. gondii CPC complex; TgArk1 (the homolog of aurora kinase B), TgINCENP1 and TgINCENP2, determined their intracellular location, their mutual interactions, and assessed their function. Finally, we determined the efficacy of an anti-human ARKB on Toxoplasma growth. Altogether, our study will further our understanding on the importance of effectors controlling phosphorylation-dependent events in governing successful cell division in Apicomplexa. In addition, our study highlights TgArk1 as a potential drug target.
Materials and methods
Parasite culture
T. gondii strains were grown in human foreskin fibroblasts (HFF) maintained in Dulbecco’s Modified Eagle’s Medium (DMEM; GIBCO, Invitrogen) supplemented with 5% fetal calf serum and 2 mM glutamine. Selections of transgenic parasites were performed with chloramphenicol for CAT selection [29], pyrimethamine for DHFR-TS selection [30], mycophenolic acid (MPA) and xanthine for HXGPRT selection [31], anhydrotetracycline (ATc) at 1.5 μg/ml for the inducible system [32], and 1 µM Shld-1 for DD-fusion stabilization [33].
Toxoplasma vectors and generation of transgenic and mutant T. gondii parasites
Primers used in this study are listed in Table S2. To tag TgArk1 at the N-terminal end, we used the CRISPR/Cas9 strategy. We designed a guide that would recruit Cas9 to cut 13 base pairs downstream the start codon of TgArk1 and a donor DNA to insert triple HA by double homologous recombination. The guide was cloned by annealing primers 1 and 2 and cloning BsaI site of vector pU6-universal (gift of Sebastian Lourido, USA). The donor DNA sequence containing the HA3 sequence flanked by 200-bp homology arms of TgArk1 gene was generated by IDT services and TOPO cloned. A 500-bp PCR product corresponding to the donor DNA was amplified with KOD polymerase (Novagen) using primers 3/4 and co-transfected with pU6-TgArk1guide-Cas9 plasmid (30 µg) in RH-∆ku80 strain. Plasmid pTub8-DDFKBP-myc-TgArk1WT.HX was designed to insert in the RH strain genome a second or multiple copies of TgArk1 open reading frame under the control of the tubulin promoter. The annotated TgArk1 open reading frame was amplified by PCR and cloned into the NsiI and PacI sites in the pTub8DDFKBPmyc.HX vector [33]. To mutate the aspartic acid (D) residue, primers described in the Table S2 were used in a site-directed mutagenesis reaction using the commercial QuikChange II Site-Directed Mutagenesis Kit (Stratagene) and according to manufacturer’s instructions. The mutated construct (pTub8-DDFKBP-myc-TgArk1D/A.HX) was sequenced along the entire open reading frame (ORF) to confirm the correct sequence. The RH strain was transfected with 100 μg of pTub8-DDFKBP-myc-TgArk1.HX or pTub8-DDFKBP-myc-TgArk1D/A.HX vectors and then subjected to MPA xanthine selection. This allowed us to obtain the following two strains: RH-ark1WT and RH-ark1D/A. To be able to tag the different proteins examined in this study in RH-ark1WT and RH-ark1D/A strains, we interrupted the KU80 gene using the CRISPR/Cas9 technique. Replacement of the KU80 locus by a DHFR cassette has been done in both RH-ark1WT and RH-ark1D/A strains. Two guides named Ku80-1-Cas9 and Ku80-2-Cas9 have been generated by annealing, respectively, primers 36/37 and 38/39 into the pU6-universal plasmid. These two guides target the beginning and the end of the KU80 genomic DNA, respectively. A fragment donor corresponding to the DHFR resistance cassette flanked by 30-bp homology arms of KU80 gene was amplified with KOD polymerase (Novagen) using primers 40 and 41. The PCR product (100 µl) corresponding to the donor fragment and the plasmids containing the two guides (15 µg of each) were co-transfected in RH-ark1WT or RH-ark1D/A strains allowing the removal of the entire KU80 gDNA. Transfected RH-ark1WT and RH-ark1D/A parasites were selected with pyrimethamine and cloned by limit dilution. This operation allowed us to obtain the following strains: RH-ark1WT-Δku80 and RH-ark1D/A-Δku80. Plasmids LIC-CAT-Chromo1/Ctg-HA3, LIC-CAT-CEP250L1/Ctg-HA3, LIC-CAT-Bub3/Ctg-HA3, LIC-CAT-INCENP1/Ctg-HA3 and LIC-CAT-INCENP1/Ctg-GFP were designed to add a sequence coding for (3)HA or a GFP at the endogenous locus of TgChromo1, TgCEP250L1, TgBub3 and TgINCENP1 open reading frames. A 1251-, 1113-, 1315- and 1818-bp fragments corresponding to the 3′ of TgChromo1, TgCEP250L1, TgBub3 and TgINCENP1, respectively were amplified from genomic DNA using primers listed in Table S2 and cloned into LIC-CAT-HA3 or LIC-CAT-GFP vectors [34]. 40 μg of these plasmids was digested by the restriction enzymes cited in supplementary Figs. 2 and 5A, and then transfected in the RH-Δku80 or RH-ark1WT-Δku80 or RH-ark1D/A-Δku80 or Tgincenp1i or Tgincenp2i strains and were subjected to chloramphenicol selection. A 1193-bp fragment corresponding to the 5′ of the TgINCENP1 coding region (downstream of the codon corresponding to the first predicted in-frame methionine residue) was amplified by PCR from T. gondii genomic DNA and then cloned in the DHFR-tetO7-Sag4 plasmid between BglII and NotI restriction sites [35] downstream the DHFR selection marker, tetO7 operator and pSag4 promoter. The final construct was linearized by AatII prior to transfection. Transfected TATi1-Δku80 parasites were selected with pyrimethamine and cloned by limit dilution. Positive clones were verified by PCR to detect the native locus or the single homologous recombination of the inducible vectors in the TgINCENP1 locus. We used the CRISPR/Cas9 strategy to examine the localization of native HA3-TgArk1 kinase in Tgincenp1i mutant parasites treated or not with ATc. Plasmid pT8myc-NtTgINCENP2.HX was designed to replace the endogenous promoter of TgINCENP2 open reading frame with a tubulin promoter and to add a myc-tag at the N-terminal part of TgINCENP2 protein. One thousand and thirteen base pair (TgINCENP2) fragment corresponding to the coding region downstream of the first predicted in-frame start codon (ATG) codon was amplified by PCR from T. gondii genomic DNA and cloned in the pTUB8-mycGFPPftailTy.HX vector. Forty micrograms of the plasmid was digested by AfeI, prior to transfection in the RH-ku80ko strain and was subjected to MPA and xanthine selection. Inducible Tgincenp2i knock-down strain has been generated by adding the tetO7 operator 1 kb upstream of the TgINCENP2 start codon. A guide inducing a double-strand breaks 1 kb upstream of TgINCENP2 has been generated by annealing primers 26 and 27 into the pU6-universal plasmid. A fragment donor corresponding to the DHFR resistance cassette and the tetO7 sequence flanked by 30-bp homology arms of TgINCENP2 5′ UTR was amplified with KOD polymerase (Novagen) using primers 28 and 29. The PCR product corresponding to the donor fragment and the pU6-TgINCENP2guide-Cas9 plasmid (30 µg) were co-transfected in TATi1-Δku80 strain allowing the insertion of DHFR-tetO7 sequence 1 kb upstream of the TgINCENP2 start codon. Transfected TATi1-Δku80 parasites were selected with pyrimethamine and cloned by limit dilution. This operation allowed us to obtain the Tgincenp2i strain. We used the CRISPR/Cas9 strategy to examine the localization of native HA3-TgArk1 kinase in Tgincenp2i mutant parasites treated or not with ATc.
Proteins detection by western blot
To detect HA3-TgArk1 or DD-Myc-TgArk1WT or DD-Myc-TgArk1D/A or SAG1, GFP or TgINCENP1-GFP or TgINCENP1-HA3 or TgINCENP1i-HA3 or Myc-TgINCENP2 proteins, parasite lysates or eluted proteins were fractionated on 12 and 10% acrylamide gels, respectively, prior to detection. Separated proteins were transferred to nitrocellulose membranes and probed with appropriate antibodies in 5% non-fat milk powder in TNT buffer (50 mM Tris pH 8.0; 150 mM NaCl; 0.05% Tween20). The primary antibodies used for detection and their respective dilutions were: rat anti-HA antibodies (Roche) at 1/300, mouse anti-Myc antibodies (SANTA CRUZ BIOTECHNOLOGY) at 1/100, rabbit polyclonal anti-TgSAG1 at 1/1000 [36], rabbit anti-GFP antibodies (abcam) at 1/1000. Bound secondary conjugated antibodies were visualized using either the ECL system (Amersham Corp) or using alkaline phosphatase kit according to manufacturer’s instructions (Promega).
Fluorescent staining of cells
Briefly, for IFAs of intracellular parasites, infected confluent HFF monolayers were fixed for 20 min in 4% paraformaldehyde in PBS, permeabilized with 0.2% triton X-100, blocked with 10% FCS in PBS, and then incubated with primary antibodies [anti-HA (Roche) 1:100, anti-centrin 1:500 (kindly provided by Dr. Iain Cheeseman), anti-IMC1 1:1000 [37], anti-ISP1 1:1000 [38], anti-NDC80 1:1000 [39], anti-Myc 1:100 (SANTA CRUZ BIOTECHNOLOGY), anti-AMA1 1:1000 [40], anti-GAP45 1:2000 [41], anti-GFP 1:2000 (abcam)] followed by goat-anti-rabbit or goat-anti-mouse or goat-anti-Guinea pig or goat-anti-rat immunoglobulin G (IgG) conjugated to Alexa Fluor 488 or Alexa Fluor 594 (Molecular Probes, Invitrogen). Coverslips were mounted onto microscope slides using Immu-mount (Calbiochem). Samples were observed with a Zeiss Axioimager epifluorescence microscope equipped with an apotome and a Zeiss Axiocam MRmCCD camera driven by the Axiovision software (Zeiss), at the Montpellier RIO imaging facility.
Plaque assay
Fresh monolayers of HFF on circular coverslips were infected with parasites in the presence or absence of 1.5 µg/ml ATc and 1 µM Shld-1 and hesperadin at different concentrations ranging from 15 to 100 nM and DMSO and Barasertib at 800 nM for 7 days. Fixation, staining and visualization were performed as previously described [36].
Electron microscopy
Infected HFF monolayers on coverslips were fixed for 4 h at room temperature with 2.5% glutaraldehyde (EMS) in 0.1 M phosphate buffer pH 7.2, washed in buffer and post-fixed for 1 h in 1% OsO4, washed in water and stained overnight in 2% uranyl acetate. Coverslips were then dehydrated in ethanol series and embedded in Epon (Embed 812, EMS). Ultrathin sections were prepared with a Leica ultracut E microtome, contrasted with 2% uranyl acetate in ethanol and lead citrate and observed with a JEOL 1200E electron microscope.
Semi-quantitative RT-PCR
Total RNA was extracted from T. gondii tachyzoites using the Nucleospin RNA II kit (Macherey-Nagel, 740955.10). RT-PCR was performed with the Superscript III first-strand synthesis kit (Invitrogen, 18080-051). Three hundred nanograms of total RNA as a template were used per RT-PCR reaction, and specific primers of TgINCENP2 (32/33) or TgFYVE1 (34/35) were used. Thirty cycles of PCR were performed.
T. gondii DNA content analysis
Low numbers of RH-Δku80 and RH-ark1D/A-Δku80 tachyzoites were seeded and grown in the presence of Shld-1 for 12 h. The extracellular parasites were removed by 1X PBS wash, and only the intracellular parasites were released from HFFs after scrapping of the monolayer and passage through a 22G needle. Parasites were filtered through glass wool to eliminate cell debris. After 24-h fixation in 70% (v/v) ethanol/30% 1XPBS at 4 °C, DNA was stained with propidium iodide and RNA removed by RNase treatment followed by analysis on a FACS Canto flow cytometer. Data were analyzed using FloJo software.
Intracellular growth and endodyogeny assays
RH-Δku80, RH-ark1WT-Δku80 and RH-ark1D/A-Δku80 parasites were treated for 12 h with or without 1 µM Shld-1, fixed with PFA and stained with anti-TgSAG1. TgINCENP2 mutant parasites (Tgincenp2i) were treated for 24 h with or without 1.5 µg/ml ATc, fixed with PFA and stained with anti-TgSAG1. RH-Δku80 parasites were treated for 24 h with DMSO or with hesperadin drug at 75 nM for 24 h, fixed with PFA and stained with anti-TgSAG1. TgINCENP1 mutant parasites (Tgincenp1i) were pretreated for 48 h with or without 1.5 µg/ml ATc, collected promptly after egress and inoculated onto new HFF monolayers. 24 h later, culture was fixed with PFA and stained with anti-TgSAG1. The numbers of parasites per vacuole were counted for more than 300 vacuoles for each condition. Replication defect was determined by staining of the nascent apical cones of the mother and daughter parasites (anti-ISP1 antibodies). Duplications of the centrosome outer core, centrosome inner core, spindle pole, kinetochore and centromere were assessed using anti-centrin antibodies, anti-HA antibodies that recognize CEP250L1-HA3 protein, anti-GFP antibodies that recognize EB1-GFP protein, anti-NDC80 antibodies and anti-HA antibodies that recognize Chromo1 protein. Data are mean values ± SD from three independent biological experiments. For each condition, 300 parasites or 300 spots were observed.
Sequence alignment
Sequences of Aurora Kinase 1 (TgArk1) from T. gondii (TGME49_210280) was aligned with H. sapiens (UniProtKB-Q96GD4) and X. laevis (UniProtKB-Q6DE08) Aurora kinase B using the online tool Clustal Omega [42]. TgINCENP1 (TGME49_232500) and TgINCENP2 (TGME49_315360) from T. gondii were aligned using the same online tool with H. sapiens INCENP1 (UniProtKB-Q9NQS7) and X. laevis INCENPA (UniProtKB-O13024) to determine the conserved IN-box domains. For both proteins, sequence alignment was performed on the whole sequence to identify the IN-box and the interacting N-lobe for INCENP and ARKB/TgArk1, respectively. The resulting sequence parts were then used to calculate the reported sequence identities.
Identification of TgArk1 substrates and validating the interaction between TgINCENP1-GFP and Myc-TgArk1
Freshly released tachyzoites (around 109 freshly egressed parasites) were harvested, washed in PBS, and lysed in RIPA buffer [50 mM Tris HCl pH 7.4, 1% NP40, 76 mM NaCl, 2 mM EGTA, 10% Glycerol, protease inhibitor cocktail (Roche)] and incubated in ice for 20 min. After centrifugation at 14,000 rpm during 45 min at 4 °C, the supernatants were subjected to immuno-precipitation using anti-GFP lama antibodies or anti-Myc antibodies (GFP-Trap or Myc-Trap Agarose Beads from ChromoTek). Following stringent washing conditions [50 mM Tris HCl pH 7.4, 1% Nonidet P-40, 76 mM NaCl, 2 mM EGTA, 10% Glycerol, protease inhibitor cocktail (Roche)], beads were suspended in loading buffer for SDS-PAGE, and the proteins were either subjected to western blot analysis or submitted to trypsin treatment for mass spectrometry analysis.
Mass spectrometry proteomic analysis
After denaturation at 100 °C in 5% SDS, 5% β-mercapto-ethanol, 1-mM EDTA, 10% glycerol, and 10-mM Tris pH 8 buffer for 3 min, protein samples were fractionated on a 10% acrylamide SDS-PAGE gel. The electrophoretic migration was stopped as soon as the protein sample entered 1 cm into the separating gel. The gel was briefly labeled with Coomassie Blue, and five bands, containing the whole sample, were cut. In gel, digestion of gel slices was performed as previously described [43]. An UltiMate 3000 RSLCnano System (Thermo Fisher Scientific) was used for separation of the protein digests. Peptides were automatically fractionated onto a commercial C18 reversed phase column (75 µm × 250 mm, 2-µm particle, PepMap100 RSLC column, Thermo Fisher Scientific, temperature 35 °C). Trapping was performed during 4 min at 5 μL/min, with solvent A (98% H2O, 2% CAN and 0.1% FA). Elution was performed using two solvents A (0,1% FA in water) and B (0.1% FA in ACN) at a flow rate of 300 nL/min. Gradient separation was 3 min at 3% B, 110 min from 3 to 20% B, 10 min from 20 to 80% B, and maintained for 15 min. The column was equilibrated for 6 min with 3% buffer B prior to the next sample analysis. The eluted peptides from the C18 column were analyzed by Q-Exactive instruments (Thermo Fisher Scientific). The electrospray voltage was 1.9 kV, and the capillary temperature was 275 °C. Full MS scans were acquired in the Orbitrap mass analyzer over m/z 300–1200 range with resolution 35,000 (m/z 200). The target value was 3.00E+06. Fifteen most intense peaks with charge state between 2 and 5 were fragmented in the HCD collision cell with normalized collision energy of 27%, and tandem mass spectrum was acquired in the Orbitrap mass analyzer with resolution 17,500 at m/z 200. The target value was 1.00E+05. The ion selection threshold was 5.0E+04 counts, and the maximum allowed ion accumulation times were 250 ms for full MS scans and 100 ms for tandem mass spectrum. Dynamic exclusion was set to 30 s.
Proteomic data analysis
Raw data collected during nanoLC–MS/MS analyses were processed and converted into *.mgf peak list format with Proteome Discoverer 1.4 (Thermo Fisher Scientific). MS/MS data were interpreted using search engine Mascot (version 2.4.0, Matrix Science, London, UK) installed on a local server. Searches were performed with a tolerance on mass measurement of 0.2 Da for precursor and 0.2 Da for fragment ions, against a composite target decoy database built with TGME49 strain of T. gondii ToxoDB.org database fused with the sequences of recombinant trypsin and a list of classical contaminants. Cysteine carbamidomethylation, methionine oxidation, protein N-terminal acetylation, and cysteine propionamidation were searched as variable modifications. Up to one trypsin, missed cleavage was allowed. For each sample, peptides were filtered out according to the cut-off set for protein hits with one or more peptides taller than nine residues, ion score > 35, identity score > 8, and 1% false positive rate.
Analysis of centrin1, CEP250L1-HA3, EB1-YFP, NDC80 and Chromo1-HA3 labelling
RH-Δku80, RH-ark1WT-Δku80 and RH-ark1D/A-Δku80 parasites were treated for 12 h with or without 1 µM Shld-1, fixed with PFA and stained with anti-centrin1 or anti-HA or anti-GFP or anti-NDC80 antibodies. The intact or stretched centrin1 and CEP250L1-HA3 and EB1-YFP and NDC80 and Chromo1-HA3 staining were quantified using Zeiss Axiovision software. Images were acquired by focusing on each or combined signals in the maximum number of parasites within a field of view and captured on a Zeiss Axio-imager epifluorescence microscope equipped with a 100× oil objective and an AxioCAM MRm camera and processed using Zeiss Axiovision software. Quantification and scoring of the staining were performed by blind observers on centrosome outer core and centrosome inner core, and spindle, kinetochores and centromeres, within focus using ImageJ. Equal parameters for the capture and scoring of images were consistently applied to all samples. Nine hundred images corresponding to each labeling, per condition, were analyzed and plotted in Figs. 4, 5 and 6.
Fig. 4.

Interfering with TgArk1 function decreasingly affects the duplication of spindle pole and centrosome inner core. a Co-staining of TgCEP250L1-HA3 (a marker of the centrosome inner core, red dots) and TgEB1-YFP (spindle pole, green dots) shows under-duplication of both subcellular structures in the ark1D/A-Δku80 strain treated with shield-1 (lower panel). The ark1D/A-Δku80 strain in the absence of shield-1 (top panel) shows a normal pairing of spindle pole/centrosome inner core per nucleus (two spindle pole clusters/two centrosome inner cores). The nuclei are stained with DAPI (in blue). Scale bars represent 2 μm. b, c Quantification of the number of parasites showing a stretched centrosome inner core or spindle pole in three different strains (RH-Δku80, ark1WT-Δku80 and ark1D/A-Δku80) in the presence or absence of shield-1 for 12 h. At least 900 parasites were examined for each condition. Values are mean ± SD for three independent experiments. d, e The average of TgCEP250L1-HA3 containing inner cores or TgEB1-YFP containing spindle poles per parasite, was quantified in 900 parasites revealing a significant reduction of the spindle poles and centrosomes inner cores when ark1D/A-Δku80 parasites were treated in presence of shield-1 (red and green dots). Values are mean ± SD for three independent experiments
Fig. 5.

Interfering with TgArk1 function decreasingly affects the duplication of kinetochores and centromeres and their clustering. a Co-staining of TgNDC80 (a kinetochore marker, red dots) and TgChromo1-HA3 (a centromere marker, green dots) shows under-duplication of both subcellular structures and their detachment in the ark1D/A-Δku80 strain treated with shield-1 (lower panels). The ark1D/A-Δku80 strain in the absence of shield-1 (top panel) shows a normal pairing of kinetochore/centromere per nucleus (two kinetochore clusters/two centromeres). The nuclei are stained with DAPI (in blue). Scale bars represent 2 μm. b, c Quantification of the number of parasites showing a stretched kinetochore or centromere in three different strains (RH-Δku80, ark1WT-Δku80 and ark1D/A-Δku80) in the presence or absence of shield-1 for 12 h. At least 900 parasites were examined for each condition. Values are mean ± SD for three independent experiments. d, e The average of TgNDC80 containing kinetochores or TgChromo1-HA3 containing centromeres per parasite was quantified in 900 parasites revealing a significant reduction of the kinetochores and centromeres when ark1D/A-Δku80 parasites were treated in the presence of shield-1 (red and green dots). Values are mean ± SD for three independent experiments. f Quantification of the number of parasites showing clustered or detached kinetochore/centromere pairing in three different strains (RH-Δku80, ark1WT-Δku80 and ark1D/A-Δku80) in the presence or absence of shield-1 for 12 h. At least 900 parasites were examined for each condition. Values are mean ± SD for three independent experiments
Fig. 6.

Interfering with TgArk1 function increasingly affects the duplication of the centrosome outer core and impairs its clustering with the centrosome inner core. a Co-staining of TgCentrin1 (a centrosome outer core marker, red dots) and TgCEP250L1-HA3 (a centrosome inner core marker, green dots) shows their over-duplication and under-duplication, respectively, and their detachment (red arrows) in the ark1D/A-Δku80 strain treated with shield-1 (lower panel). The ark1D/A-Δku80 strain in absence of shield-1 (top panel) shows a normal pairing of both centrosome cores per nucleus (one centrosome outer core clusters/one centrosome inner core; two centrosome outer core clusters/two centrosome inner cores). The nuclei are stained with DAPI (in blue). Scale bars represent 2 μm. b The average of TgCentrin1 containing centrosome outer cores per parasite, was quantified in 900 parasites revealing a significant amplification of the centrosome outer core when ark1D/A-Δku80 parasites were treated in presence of shield-1 (red dots). Values are mean ± SD for three independent experiments. c Co-staining of TgCentrin1 (a centrosome outer core marker, green dots) and TgNDC80 (a kinetochore marker, red dots) shows their over-duplication and under-duplication, respectively, and their detachment (red arrows) in the ark1D/A-Δku80 strain treated with shield-1 (lower panel). The ark1D/A-Δku80 strain in absence of shield-1 (top panel) shows a normal pairing of centrosome outer core/kinetochore per nucleus (two centrosome outer core clusters/one kinetochore; two centrosome outer core clusters/two kinetochores). The nuclei are stained with DAPI (in blue). Scale bars represent 2 μm. d Quantification of the number of parasites showing clustered or detached centrosome outer core/kinetochore pairing in three different strains (RH-Δku80, ark1WT-Δku80 and ark1D/A-Δku80) in the presence or absence of shield-1 for 12 h. At least 900 parasites were examined for each condition. Values are mean ± SD for three independent experiments. e Co-staining of TgCentrin1 (a centrosome outer core marker, red dots) and TgChromo1-HA3 (a centromere marker, green dots) shows their over-duplication and under-duplication, respectively, and their detachment (red arrows) in the ark1D/A-Δku80 strain treated with shield-1 (lower panel). The ark1D/A-Δku80 strain in the absence of shield-1 (top panels) shows a normal pairing of centrosome outer core/centromere per nucleus (two centrosome outer core clusters/one centromere; two centrosome outer core clusters/two centromeres). The nuclei are stained with DAPI (in blue). Scale bars represent 2 μm
Statistics
P values were calculated in Excel using the Student’s t test assuming equal variance, unpaired samples, and using two-tailed distribution. Means and SD were also calculated in Excel.
Results
TgArk1 N-lobe and both TgINCENP IN-box motifs contain several conserved residues known to interact together in other organisms
Among the three aurora kinases expressed in mammalian cells, Aurora kinase B (ARKB) is the most studied because it plays a key role in the separation of chromosomes [20, 44]. Proteins carrying out such a function in Apicomplexa are unidentified. In human cells, ARKB functions in the CPC complex with a core protein called INCENP. INCENP has an IN-box motif at its C-terminus required for its interaction with the N-lobe at the N-terminus of ARKB (Supplementary Fig. 1A) [45, 46]. To identify homologues of human ARKB and INCENP in T. gondii, we searched for conserved residues of the N-lobe and IN-box, respectively, in the T. gondii genome. As a result, we identified an ARKB homologue which we name T. gondii Aurora-related kinase 1 (TGME49_210280), hereon referred to as TgArk1, and two putative TgINCENPs, TgINCENP1 (TGME49_232500) and TgINCENP2 (TGME49_315360) (Supplementary Fig. 1A, 1B and 1C). Sequence alignment shows that TgArk1 N-lobe shares 36% sequence identity with the human ARKB N-lobe (Supplementary Fig. 1C), whereas the two putative IN-box motifs of T. gondii TgINCENP1 and TgINCENP2 have 29 and 26% sequence identity with human INCENP1/IN-box, respectively (Supplementary Fig. 1B). Similar results were obtained when comparing the Xenopus and T. gondii protein sequences. Closer analysis of the overall hydrophobic interfacial residues, based on the extended structural study of the Xenopus ARKB/IN-box apo-complex [47], showed that nearly all these residues are conserved between the studied species, with only one exception. Indeed, the crystal structure of the Xenopus ARKB in complex with INCENP shows that P799, W801 and A802 in the N-terminal part of IN-box pack against an exposed hydrophobic pockets in ARKB formed by G96, L99 and I118, with the side chain of W801 stacking against the side chain of R111 of ARKB [47]. This interface is conserved in TgINCENP2 IN-box/TgArk1 sequences, as P672, W674 and A675 in the IN-box and G217, L220, V239 and R232 in TgArk1, whereas TgINCENP1 lacks this P-WA motif, and instead shows an A-WY one (Supplementary Fig. 1B and 1C).
These results suggest that TgArk1 and both putative TgINCENPs might interact together in a similar manner as the well-characterized mammalian ARKB with their corresponding INCENPs.
We next set out to investigate the localization, functionality and interaction of these putative CPC proteins.
The Aurora-related kinase 1 (TgArk1) expression is cell cycle regulated and is essential for parasite survival
We previously described that three aurora-related kinases are encoded for in the T. gondii genome [13]. We showed that TgArk2 and TgArk3 concentrate at specific subcellular structures linked to parasite division: the mitotic spindle and intra-nuclear mitotic structures (TgArk2), and the outer core of the centrosome and the budding daughter cells cytoskeleton (TgArk3) [13]. Functional analyses showed that TgArk2 was not essential for parasite survival, but TgArk3 plays an important role in the formation of daughter cells. In this study, we describe TgArk1 in detail, whose molecular and functional characterization remains underexplored [13]. To gain insight on the role of TgArk1, we first assessed its localization by fusing it to a triple Hemagglutinin epitope (3HA). We repeatedly failed to generate C-terminally tagged TgArk1 parasites, suggesting that the integrity of this domain may be functionally important. We thus generated a cell line of parasites bearing a knocked-in HA tag on TgArk1’s N-terminus (Supplementary Fig. 2A and 2B). Western blot analysis of whole parasite lysate showed that HA3-TgArk1 migrates as a single band of the expected molecular weight (Fig. 1a). TgSAG1 was used as a loading control (Fig. 1a). Immunofluorescence assays (IFAs) using the inner membrane complex-1 (IMC1) antibody to delineate the cell periphery, and 4,6-diamidino-2-phenylindole (DAPI) to visualize the nuclei, showed that HA3-TgArk1 displays a dynamic localization through the division cycle. In non-dividing parasites, HA3-TgArk1 is found exclusively in the nucleus (Fig. 1b, top panel). In contrast, during endodyogeny, when daughter cells are formed, HA3-TgArk1 is found distributed throughout the parasite (nucleus and cytoplasm) (Fig. 1b, bottom panel). Previous studies of whole genome high-throughput mutagenesis using CRISPR-Cas9 in T. gondii, whereby neutral or positive scores were attributed to dispensable genes, attributed a phenotypic score of -5 in survival fitness to the TgArk1 gene. This negative score strongly suggests that this kinase is likely important for parasite comparative fitness and survival [48]. To asses TgArk1's role in parasite survival, we opted for a point mutation strategy to generate dominant negative version of TgArk1, based on previous studies [49, 50]. Mutation of a lysine (K) residue in the ATP-binding site or of an aspartic acid (D) residue involved in the catalysis of phosphoryl group transfer generate a dead kinase which traps its substrate as it becomes unable to complete the phospho-transfer reaction [51–54]. Protein sequence alignments identified the conserved residues K243 and D338 in TgArk1 which were mutated into an arginine (R) and an alanine (A) residue, respectively. The DD small domain is known to confer instability to proteins in the absence of the folding inducer shield-1 (Shld-1) (Supplementary Fig. 2C) [33]. Transgenic parasites expressing WT (DD-Myc-TgArk1 WT) or TgArk1-dead kinases (DD-Myc-TgArk1K243R and DD-Myc-TgArk1D338A) were generated. The phenotypes exhibited by the DD-Myc-TgArk1K243R and DD-Myc-TgArk1D338A mutants were virtually indistinguishable. Therefore, experimental results presented here correspond to the DD-Myc-TgArk1D338A only, for simplicity. The tight control of protein stabilization of DD-Myc-TgArk1 WT and DD-Myc-TgArk1D338A fusions by Shld-1 was confirmed by western blot and IFA (Fig. 1c and Supplementary Fig. 2D). After adding shield-1 for 12 h, both DD-Myc-TgArk1WT and DD-Myc-TgArk1D338A were detectable by IFAs in the cytoplasm and in the nucleus using anti-Myc antibodies (Supplementary Fig. 2D), and are expressed to comparable levels as shown by western blot (Fig. 1c). The SAG1 protein was used as a loading control (Fig. 1c). Interestingly, stabilization of DD-Myc-TgArk1D338A severely impaired parasite survival, as shown by the reduced ability of these parasites to generate plaques on a host cell monolayer (Fig. 1d). In contrast, the overexpression of DD-Myc-TgArk1WT or shld-1 alone did not result in detectable growth defects (Fig. 1d).
Fig. 1.

Examination of TgArk1 localization during tachyzoite replication, and its ability to inhibit parasite growth when expressed in a controlled manner as a dead kinase in the presence of Shield-1. a Western blot analysis performed on HA3-TgArk1, and RH-Δku80 wild-type parasite lysates probed with an anti-HA antibody. HA3-TgArk1 is found at the expected molecular masses (62 kDa). SAG1 was used as loading control. b Immunofluorescence images reveal that TgArk1 can switch from nucleus to cytoplasm depending on phase during parasite division. Parasites were co-stained with antibodies for HA3-TgArk1 and IMC1 as well as stained with DAPI. Representative images at two cell cycle phases are shown as identified by established cell cycle criteria based on the absence or presence of internal daughters (red = IMC1). Scale bars represent 2 μm. c Controlled expression of the series of TgArk1 kinase by Shld-1. Western blot analysis of parasites grown in the presence and absence of Shld-1 for 12 h using anti-myc antibodies. SAG1 was used as loading control. d Expression of DD-Myc-TgArk1D/A dead-kinase but not DD-Myc-TgArk1 wild type affects parasite growth. Plaque assays were performed on parasite strains expressing DD-Myc-TgArk1WT or DD-Myc-TgArk1D/A. HFF monolayers were infected with parasites in the presence or absence of Shld-1, fixed after 7 days, and stained with Giemsa
Overall, our results indicate that the localization of TgArk1 is dynamic along the cell cycle under the control of its endogenous promoter, and that its function is important for parasite survival.
TgArk1 dominant negative mutants show marked defects in endodyogeny
The defect in plaque formation due to impaired TgArk1 function suggested alterations in parasite survival. For its expected role in cell division, we went on to study the endodyogeny process in the dominant negative mutants. In wild-type parasites, Toxoplasma tachyzoite division is composed of G1, S phase, mitosis (M) and cytokinesis (C), with G2 being absent [55]. Centrosome duplication marks the start of S phase and of DNA replication [56]. Late in S phase, the spindle is assembled between the daughter centrosomes in a trans-nuclear funnel delimited by the nuclear envelope (Fig. 9) [2]. The funnel then opens in the nucleoplasm, allowing chromosome attachment to the mitotic microtubules via the kinetochore protein complexes assembled on the centromeres; the latter is permanently clustered during the cell cycle (Fig. 9) [3, 39]. Concomitantly with chromosome replication and segregation, two daughter buds are nucleated near the centrosomes (Fig. 9) [2]. Daughter cells progressively assemble to complete the formation of fully differentiated parasites [2, 57] leaving virtually no leftover at the end of the process.
Fig. 9.

a The different steps that occur during closed mitosis and IMC formation in wild-type T. gondii parasites. 1—the centrioles duplicate at the posterior pole of the nucleus and initiate the formation of the spindle; the newly formed spindle and the duplicated centrioles migrate back towards the anterior pole of the nucleus; centromeres and kinetochores cluster together facing centrioles; no centrocone is formed at this stage; 2—the nuclear envelope funnel develops between daughter pairs of centrioles; the spindle microtubules forms in the funnel; centromeres duplicate and associate to the nuclear envelope through kinetochores; 3—opening of the middle part of the funnel; kinetochores are connected to mitotic microtubules; the metaphase plate is organized and daughter cells assembly is initiated; 4—formation of the centrocones by closure of the nuclear envelope against kinetochores that reattach to the nuclear envelope; disappearance of mitotic microtubules; transient persistence of spindle microtubules running through pores at the centrocone basis making half spindles in nucleoplasm; 5—centrocones budding off the nuclear envelope attracted into daughter buds; 6—buds elongation and daughter zoite biogenesis towards accomplishment of karyokinesis and cytokinesis. b ark1D/A-Δku80 dominant negative mutants and TgINCENP2 knock-down parasites are stuck at step number 3. 3′—funnel persistence; kinetochores and centromeres remain associated on external face of the nuclear envelope; centrocone markers are spread into the spindle; daughter cells budding is initiated; 4′—(when wild-type parasites have reached the end of the first endodyogeny) centrioles and probably centromeres continue to duplicate; the mother centriole pair leaves the spindle pole while keeping developing the first bud; daughter centriole pair starts initiating a second bud inside the first one; 5′—(when wild-type parasites have reached the second endodyogeny, i.e. 4 parasites) the two centriole pairs duplicate and probably also centromeres. These later lose association with kinetochores that remain associated to the funnel membrane. The new centrioles start initiating buds, producing the Russian dolls figures. Black: centrioles, blue: kinetochores, green: centromeres, yellow: microtubules, red: CEP250L1 (outer face of the nuclear envelope)
We followed the development of TgArk1 dominant negative mutants by electron microscopy and by light microscopy using probes to specifically mark compartments associated with mitosis or zoite genesis.
By transmission electron microscopy (TEM), TgArk1 mutant vacuoles, showed profound ultrastructural anomalies. Large cytoplasmic masses (known as residual bodies, often found in T. gondii when endodyogeny is defective) were found in addition to tachyzoite profiles, and these masses contained both irregularly shaped nuclei and incomplete inner membrane complexes (IMCs) (compare Fig. 2a–c with Fig. 2m). Unusually, many mutant parasites contained multiple daughter IMCs wrapped around one another resembling Russian dolls (compare Fig. 2d–e′ with Fig. 2m). Within the irregularly shaped nuclei, nuclear envelope funnels containing spindle microtubules were often observable (Fig. 2f–i), similar to the early mitotic spindle observed transiently during wild-type mitosis before the formation of centrocones (Figs. 2j–l and 9). In TgArk1 mutant parasites, these funnels are often found opening on centrioles and aberrantly associated with early developing daughter organisms (Fig. 2f–h′). Surprisingly, centrocones were never observed in TgArk1 mutant parasites.
Fig. 2.
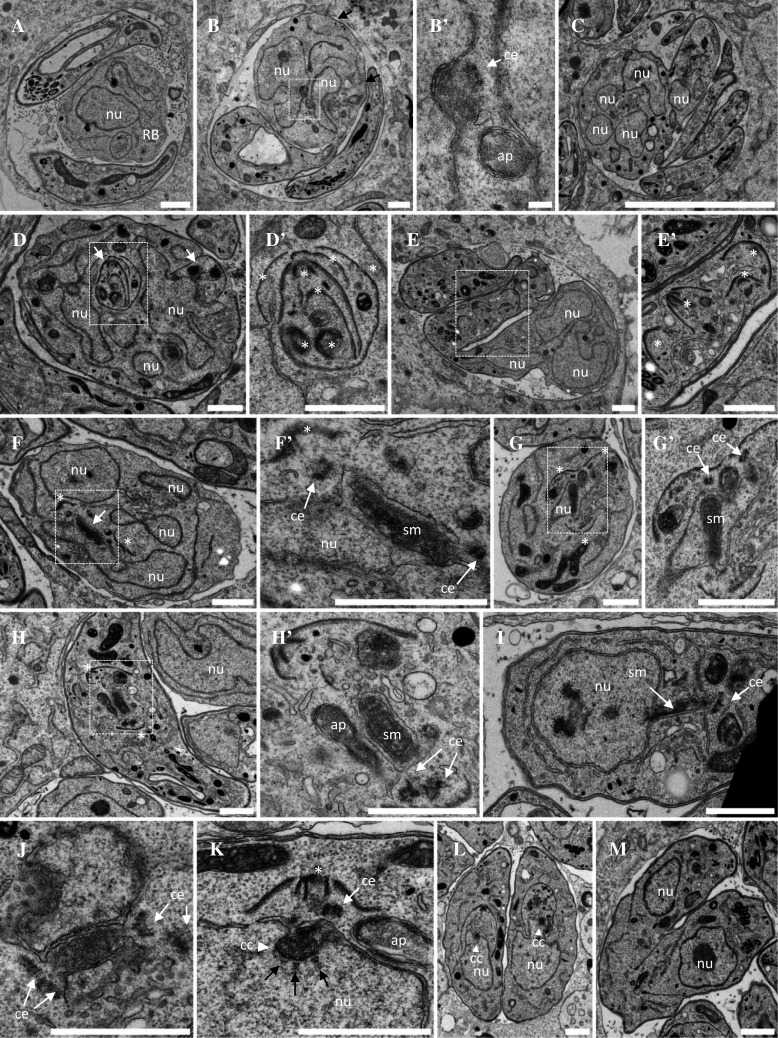
Ultrastructural analysis of ark1D/A-Δku80 dominant negative mutant parasites. a–e′ Representative electron micrographs of parasites treated during 12 h with shield-1 to stabilize the expression of the DD-Myc-TgArk1D/A protein. Parasites showed catastrophic division defects. a Vacuole showing a large residual body containing a nucleus and various organelles. Two parasites show cytoplasmic and apical organelles and large aberrant electron lucent vacuoles but no apparent nucleus. b A parasite shows discontinuous IMC stretches under the plasma membrane (black arrows) and two nuclei in one of which a centriole next to a depression of the nuclear envelope suggests an early step of spindle formation. Higher magnification in b′. c Monster cell showing multiple nucleus sections and three nucleus free tachyzoites. d Monster cell showing multi-lobed nucleus and multiple daughter inner membrane complexes (IMC, white arrows). A detail shown at higher magnification in d′ shows several IMCs nested like Russian dolls (white asterisk). e, e′ Monster cell showing multi-lobed nucleus and some cytoplasmic organelles, connected to another IMC-coated compartment containing multiple daughter apical complexes randomly organized (white asterisks in the enlarged area e′) and various organelles but no apparent nucleus. f Residual body containing several nuclei, one of which harboring a nuclear envelope-bounded funnel containing spindle microtubules (White arrow and enlargement in f′). At both ends of the funnel a centriole is found, associated with a daughter IMC (white asterisks). g Parasite containing multiple daughter IMCs. The nucleus is traversed by a funnel containing spindle microtubules (higher magnification in g′). h, h′ Parasite showing two apical buds (white asterisks) at both ends of a spindle surrounded by a double membrane, but no nucleus can be seen on the same section. i Parasite showing a nucleus harboring a membranous funnel stretched out of the nucleus, containing spindle microtubules. Centrioles are present at the extremity of the funnel. j–m DD-Myc-TgArk1WT-expressing parasites grown in the presence of shield-1. j A funnel containing spindle microtubules is transiently observed after centriole duplication, and before the assembly of daughter cells apical complexes. k Later during mitosis, the mitotic spindle poles turn into centrocones (white arrowhead) with kinetochores aligned next to their base (black arrows). The daughter apical complex has started to assemble (white asterisk). l Early steps of cytokinesis by endodyogeny. Nuclei are pulled inside the body of the assembling daughter cells. m Late stages of cytokinesis. Daughter cells emerging from the body of the mother. All the steps described from J to M are similar to the ones observed in wild-type untreated parasites. nu nucleus, ap apicoplast, ce centriole, sm spindle microtubule, rb residual body. Scale bar = 1 µm
At the end of endodyogeny, each newly formed parasite must package all newly replicated or de novo produced organelles [58]. Visualization of large multi-lobed nuclei by electron microscopy, presumably expelled into a residual body, prompted us to quantify the nuclear segregation defects in the DD-Myc-TgArk1D338A-expressing parasites. To monitor nuclear inheritance during cell division, we performed IFAs labeling the inner membrane complex with anti-TgIMC1, the nuclear protein BUB3-HA3 (anti-HA), or DAPI (Fig. 3a and Supplementary Fig. 3) [37, 59]. In the absence of Shld-1, the majority of parasites contain a single nucleus (Fig. 3a, upper panel). In sharp contrast, approximately 60% of parasites expressing DD-Myc-TgArk1D338A are devoid of nuclei (Fig. 3a, b, lower panels, white asterisk). Consistently, in 40% of the vacuoles containing anucleated parasites, the unsegregated nuclei are found outside the parasites within structures that could correspond to the residual bodies (Fig. 3a, c, intermediate panel, white arrow). Furthermore, DNA content analysis by flow cytometry of single parasites expressing DD-Myc-TgArk1D338A revealed that the DD-Myc-TgArk1D338A-expressing strain shows a dramatic increase in < 1 N DNA content, presumably corresponding to cells who have lost their nucleus (Fig. 3e). Note that wild-type cells normally exhibit a 1 N peak corresponding to G1 cells, a 1.8 N peak corresponding to S-phase parasites, and a 2 N peak corresponding to M-phase cells (Fig. 3d).
Fig. 3.

Interfering with TgArk1 function results in a nuclear segregation defect. a The overexpression of DD-Myc-TgArk1D/A kinase results in formation of nucleus-deficient parasites. IFA performed on intracellular parasites of the ark1D/A-Δku80 strain shows an IMC marker (TgIMC1, red), a nucleus marker (TgBUB3-HA3, green, the two upper panels) and nuclei stained by DAPI (lower panel). In the presence of shield-1 for 12 h, nucleus-deficient parasites appear, whereas normal nucleus segregation is observed in the absence of shield-1 for ark1D/A-Δku80 strain, as expected. A white asterisk indicates the parasites lacking nuclei. In the intermediate panel, a nucleus is observed outside the parasites, in the residual body. b Quantification of the number of nucleus-deficient parasites in three different strains (RH-Δku80, ark1WT-Δku80 and ark1D/A-Δku80) in the presence or absence of shield-1 for 12 h. The percentage of parasites displaying nucleus segregation defect was determined for at least 300 parasites. The results shown are from three independent experiments. c Quantification of the number of vacuoles showing nuclei in residual bodies in three different strains (RH-Δku80, ark1WT-Δku80 and ark1D/A-Δku80) ± shield-1 for 12 h. At least 300 vacuoles were examined for each condition. Values are mean ± SD for three independent experiments. d Flow cytometric analysis of ethanol (EtOH)-fixed wild-type and mutant parasites stained with propidium iodide after RNase treatment. Parental (RH-Δku80) and dominant negative mutant (ark1D/A-Δku80) parasites were grown in the presence of shield-1 for 12 h. The 1 N population was considered in the G1 phase, while the 1.8–2 N population is considered in the S/M phase of the cell cycle. ark1D/A-Δku80 dominant negative mutant parasites showed an important increase in their sub-1 N aneuploidy population that is consistent with nuclei loss
Normally, two daughter cells are formed inside the mother cell (Supplementary Fig. 4a, upper panel). By TEM, we observed that the DD-Myc-TgArk1D338A-expressing cells exhibited an atypical assembly of multiple daughter cell IMCs within one mother cell, resembling Russian dolls. To monitor the formation of daughter cells in TgArk1 mutant parasites, we labeled the parasites with anti-ISP1 antibodies, which specifically label one of the first membranous components laid down during the daughter cell formation process [38]. In DD-Myc-TgArk1D338A-expressing cells division was often asymmetric giving rise to odd numbers of parasites (Supplementary Fig. 4A and 4B). Scoring the number of parasites per vacuole, we observed a decrease in the number of vacuoles containing two and four parasites, and an increase in vacuoles containing an odd number of parasites, such as three and five (Supplementary Fig. 4B). Moreover, DD-Myc-TgArk1D338A-expressing cells staining with anti-IMC1 protein (a marker of the inner membrane complex), we detected interruptions of the parasite’s pellicle (Supplementary Fig. 4C, green arrows), which is consistent with TEM observations (Fig. 2b).
As in higher eukaryotes, the centrosome in Toxoplasma is the main microtubule organizing center (MTOC) involved in coordinating mitosis and cytokinesis [10]. In T. gondii, the centrosome has been described as a bipartite structure (comprising outer and inner cores) that critically regulate the dynamics of the budding and nuclear cycles, respectively [11]. The inner centrosome core is known to engage the intra-nuclear spindle [60, 61] ensuring the fidelity of nuclear genome inheritance [11]. Using parasite strains in which inner core and spindle proteins are tagged with 3HA (CEP250L1-HA3) (Supplementary Fig. 3) or YFP (EB1-YFP), respectively, as markers [11, 62], we examined the duplication of both subcellular structures in parental RH strain, DD-Myc-TgArk1WT expressing or DD-Myc-TgArk1D338A expressing, in the presence or absence of Shld-1. Normally, CEP250L1 duplicates associated with the nucleus in the later stages of S phase or early mitosis. In wild-type parasites, CEP250L1-HA3 and EB1-YFP perfectly co-localize, consistent with the fact that the inner core of the centrosome is associated with spindle microtubules (Fig. 4a). In the DD-Myc-TgArk1D338A-expressing cells, co-labeling of these structures revealed that the two exhibit altered morphology during duplication. Instead of localizing to discrete puncta, they seem to stretch and remain associated with a single parasite nucleus within a vacuole roughly 50% of the times (Fig. 4a, lower panel, Fig. 4b, c). Consistently, the average number of structures associated with each parasite is approximately 1 in DD-Myc-TgArk1D338A-expressing cells, while all other control strains, which normally duplicate these structures during cell division, show an average number of structures of approximately 1.3 per cell (Fig. 4d, e). Note that only 25% of a parasite population is dividing at a time in an asynchronous population.
It has been shown that the outer core duplicates before the inner core with both cores remaining in close apposition on the same axis closer to the apical part of the parasite nuclear envelope (Fig. 6a, upper panel) [11]. To further investigate the role of TgArk1 in centrosome biology, we investigated the relative dynamics of duplication of the outer and inner core domains in DD-Myc-TgArk1D338A-expressing cells.
For this purpose, we performed co-localization experiments between TgCentrin1 (an outer core centrosome marker) and TgCEP250L1-HA3. IFAs revealed that unlike the inner core which seems to stretch and remain elongated but unseparated, the outer core retains its ability to duplicate into discrete foci (Fig. 6a, lower panel). On average, we found three times more outer core foci per DD-Myc-TgArk1D338A-expressing parasite than per control parasites (Fig. 6b). Consistently, the majority of outer cores of the centrosome physically disaggregate from the inner cores (Fig. 6a, lower panel, red arrows).
In T. gondii, centromeres, the DNA regions where kinetochore components assemble, are clustered in close proximity to the nuclear envelope, and the centrosome, at all stages of the cell cycle [3]. TgChromo1 is a DNA-binding protein which localizes specifically to peri-centromeric heterochromatin [63]. In G1, TgChromo1-HA3 labels a distinct focus while in S and M-phases two foci are detectable (corresponding to the duplicated chromosomes) (Fig. 5a, upper panel and supplementary Fig. 3) [63]. TgNDC80, a component of the T. gondii kinetochore, follows a similar cell cycle-dependent localization pattern akin to that of TgChromo1 (Fig. 5a, upper panel) [39]. Using TgNDC80 and TgChromo-1 as specific markers to detect kinetochores and centromeres, respectively, we investigated their dynamics in TgArk1 dominant negative mutant parasites. We found that while in all controls the average of both kinetochores and peri-centromeric staining closely match the number of centrosomes found per cell (1.3), in DD-Myc-TgArk1D338A-expressing cells, the average number of each structure is close to 1, and are stretched in ~ 60 and ~ 35% of the analyzed parasites, respectively (Fig. 5a, intermediate panel; Fig. 5b–e). Only in very few parasites (~ 14%) we have observed a duplication of each of these subcellular structures. Otherwise, the majority of mutant parasites possess a single detectable labeled structure; ~ 60% of the kinetochores labeled by TgNDC80 staining, and ~ 35% of the peri-centromeric chromatin labeled by staining TgChromo1, appear to be “stretched”, respectively. Importantly, in addition to an altered relative stoichiometry among structures which are normally segregated together, about 50% of the mutant parasites suffer from a clear physical separation between peri-centromeres and kinetochores (Fig. 5a, lower panel; Fig. 5f). NDC80 was detected on the lateral side of the nucleus, whereas TgChromo1 gathered as a rounded mass in the center of the nucleus. The same kind of spatial detachment was observed between the centrosome outer core and kinetochores, or centromeres (Fig. 6c–e; red arrows).
Taken together with TEM observations, these data suggest that TgArk1-mutant parasites are notably defective in the persistence of the early forms of the spindle, which persist within the nuclear envelope funnel, not opening to allow chromosome partition between daughter nuclei and blocking karyokinesis. The assembly of daughter buds, however, presumably proceeds unhampered leading to nucleus-less tachyzoites, then to accumulation of multiple daughter IMCs and undivided nuclear masses. The centrin1-labeled structures (centrosomes) that drive the budding [9] continue to duplicate, whereas all other mitotic markers presumably get stalled, suggesting a disconnection between nuclear mitosis and daughter cells budding. Noteworthy, in most mutant vacuoles, multiple daughter cell buds were abortive, showing only early buds containing nascent IMCs. This may suggest that daughter cell buds stop their development, and that the centrosome initiates a new round of budding leading to the formation of Russian dolls. Taken together, our data show that, in TgArk1 mutant parasites, the centrioles are still able to duplicate and to initiate the budding process, while the mitotic apparatus is rendered dysfunctional.
TgINCENP1 interacts with TgArk1 but it is dispensable for parasite survival
A structural study of the Xenopus ARKB/IN-box apo-complex showed that only one out of the three residues important for the interaction between INCENP and ARKB is conserved in the IN-box of TgINCENP1 (Supplementary Fig. 1B) [47]. In contrast, the three residues involved in the interaction between INCENP and ARKB in Xenopus are conserved in the IN-box of TgINCENP2 (Supplementary Fig. 1B). This suggested to us that TgArk1 could be carrying out its functions through interactions with TgINCENP2 only. To test this, we studied the interactions between TgArk1, TgINCENP1 and TgINCENP2. We first set out to localize TgINCENP1. For this, we generated a knock-in cell line expressing triple HA C-terminally tagged TgINCENP1 (Supplementary Fig. 5A). We determined that TgINCENP1 localizes to the nucleus. Careful examination of its pattern of expression revealed that TgINCENP1 is detectable only in non-dividing parasites, and is undetectable in parasites whose daughter cells are being assembled (Supplementary Fig. 5B). To test whether TgArk1 and TgINCENP1 interact, we performed GFP-trap pull-down assays to capture GFP-tagged TgINCENP1 in parasites co-expressing Myc-TgArk1. As a control, we performed the same experiment in a cell line expressing both Myc-TgArk1 and GFP alone. Western blot analysis using anti-GFP or anti-Myc antibodies revealed the presence of TgINCENP1-GFP and GFP in the eluted fractions (Supplementary Fig. 5C, left). A single band at the expected size of Myc-TgArk1 was co-precipitated with TgINCENP1-GFP but not with GFP alone (Supplementary Fig. 5C, right).
To investigate the role of TgINCENP1, we generated a conditional knock-down cell line (Tgincenp1i), by replacing the endogenous promoter with an Anhydrotetracycline (ATc)-repressible promoter, as described previously (Supplementary Fig. 6A) [12, 35]. Single homologous integration of the inducible cassette at the TgINCENP1 locus was monitored by PCR (Supplementary Fig. 6B). To study the regulation of TgINCENP1i protein, a C-terminal triple HA epitope-tag was inserted by single homologous recombination at the TgINCENP1 locus in the Tgincenp1i strain (Supplementary Fig. 6A). Under the control of the repressible promoter, TgINCENP1i-HA3 protein was detectable in the cytoplasm and in the nucleus of parasites (Supplementary Fig. 6C). Total protein extracts of the TgINCENP1-HA3 and TgINCENP1i-HA3 (± ATc) transgenic parasites were analyzed by western blot using an anti-HA antibody; SAG1 was used as a loading control (Supplementary Fig. 6D). The endogenous TgINCENP1-HA3 protein levels are comparable to the level of expression of TgINCENP1i-HA3 under the control of the repressible promoter (Supplementary Fig. 6D). Importantly, upon addition of ATc to the growth media for 48 h, the expression of TgINCENP1i-HA3 is no longer detectable (Supplementary Fig. 6C and 6D). Tgincenp1i parasites treated with ATc were able to lyse their host cells virtually identically to untreated controls, as determined by plaque assay (Supplementary Fig. 7A). Depletion of TgINCENP1 did not affect parasite growth or replication, and more importantly, it did not affect the nuclear localization of TgArk1 during the G1 phase (Supplementary Fig. B, 7C and 7D). These results suggest that while a physical interaction exists between TgINCENP1 and TgArk1, the latter does not seem to be dependent on the former neither for functional competency nor for proper subcellular localization.
TgINCENP2 governs the localization of TgArk1 and its loss of function phenocopies ark1D/A-Δku80 mutant parasites
The lack of phenotype following the depletion of TgINCENP1 may suggest a functional redundancy between the two TgINCENPs. To test this hypothesis, we undertook the functional characterization of TgINCENP2. First we tried to localize the endogenous TgINCENP2 protein by fusing it to 3-HA tags on the C-terminal side. This strategy did not allow us to localize it since the endogenous expression of TgINCENP2 is extremely low. Detection of TgINCENP2 was possible when the endogenous promoter of the TgINCENP2 gene was replaced with the constitutive tubulin promoter (Supplementary Fig. 8A). We failed to maintain the transgenic parasites that overexpress the Myc-TgINCENP2 protein in culture thus suggesting that increasing its endogenous expression level, or alternatively altering a putative cell cycle-regulated expression, may be detrimental to the parasite. Nevertheless, this strategy allowed us to transiently assess TgINCENP2’s localization during the cell cycle and to detect it by western blot (Supplementary Fig. 8B and 8C). Similarly to TgArk1, TgINCENP2 seems to localize at the nucleus in G1, and dispersedly in the whole parasites when daughter cells assemble (Supplementary Fig. 8B). To explore the function of TgINCENP2, we first tried, albeit unsuccessfully, to exchange its endogenous promotor with a tetracycline-repressible promoter [12, 32, 35]. To overcome this problem, we inserted the seven repeats of the tetracycline-operon (tetO7) upstream the endogenous TgINCENP2 promoter via CRISPR-Cas9 genome editing (Supplementary Fig. 9A). This strategy does not interfere neither with TgINCENP2's endogenous expression levels nor timing. Correct double homologous integration of the DHFR-tetO7 sequence at the TgINCENP2 locus was confirmed by polymerase chain reaction (Supplementary Fig. 9B). Knock-down of TgINCENP2 expression levels were corroborated by reverse transcription PCR (RT-PCR) analysis (Supplementary Fig. 9C). The very-low endogenous expression level of TgINCENP2 and the toxic effect generated when over-expressed prevented us from studying its interaction with TgArk1. However, loss of TgINCENP2 results in a dramatic effect on parasite growth as determined by plaque assays (Fig. 7a). Strikingly, TgINCENP2 knock-down parasites showed pronounced changes in TgArk1 localization. Depletion of TgINCENP1 protein does not affect the localization or pattern of expression of TgArk1. In contrast, depletion of TgINCENP2 modifies the localization of TgArk1 whereby it accumulates in vesicular structures in the cytoplasm instead of localizing to the nucleus or throughout the parasite. Nonetheless, it remains detectable by IFA (Fig. 7b). Strikingly, TEM analysis revealed that TgINCENP2's knock down phenocopies the TgArk1 mutant; parasites are unable to properly segregate their nuclei and to coordinate the formation of two daughter cells (Fig. 7c–e). Aberrant divisions were frequently observed in these parasites. For instance, cells with large poly-lobed nuclei, nuclei expelled in the residual body, and multiple budding were observed (Fig. 7c–e). Persistence of the spindle microtubule funnel and absence of intra-nuclear spindles were also noticed (compare Fig. 7f, f′ with Fig. 7g, g′). Consistently, we observed a coinciding set of phenotypes by IFA, for both TgArk1 and TgINCENP2 mutants, with regards to the lack of nucleus segregation, defects in mitotic subcellular markers, duplication and clustering, abnormalities in daughter cells formation (presence of odd numbers of daughter cell structures in vacuoles) and the over-duplication of the centrosome outer core (Supplementary Figs. 10 and 11). In summary, the Tgincenp2i mutant cell line phenocopies ark1D/A-Δku80 mutant parasites, suggesting that both proteins may co-depend for proper functioning in the same biological processes during endodyogeny in T. gondii.
Fig. 7.

TgINCENP2 is essential for parasite replication and dictates the correct targeting of TgArk1. a Plaque assays were carried out by infecting HFF monolayers with TATi1-Δku80 or Tgincenp2i for 7 days ± ATc. The HFF were stained with Giemsa. b Immunofluorescence assays of HA3-TgArk1 in intracellular Tgincenp2i parasites grown without or in the presence of ATc for 24 h. Endogenous TgArk1 was detected using anti-HA antibodies. The parasite pellicle was visualized using anti-GAP45 antibodies. Nuclei were stained with DAPI. Scale bars represent 2 μm. c–e Ultrastructural analysis of TgINCENP2-depleted parasites. c Vacuole containing two tachyzoites and an aberrant parasite with a multi-lobed nucleus. The cellular architecture is lost and the plasma membrane is only partially associated with an inner membrane complex (IMC, white arrows). A few organelles are randomly dispersed in the cytosol. d Vacuole showing a residual body containing a nucleus that can be distinguished from a parasite by the absence of IMC. The four tachyzoites connected to the residual body have an IMC and contain mitochondria and apical organelles, but no nucleus can be seen on this section. e Vacuole containing aberrant endodyogeny profiles, with tachyzoites containing encapsulated successive apical complexes (Russian dolls, white asterisks) and large residual bodies containing multiple nucleus sections. f–g′ Mitosis either in control or in TgINCENP2-depleted parasites. f Control parasites (TATi1-Δku80 treated with ATc): wild-type mitosis. Nascent IMC of daughter cells are indicated with white stars. The bud on the left shows one centriole (white arrow) located at the tip of the centrocone (higher magnification shown in f′, with the kinetochores aligned underneath (black arrows); the spindle microtubules are still present in between kinetochores. The other bud should show the same structure in another section plane. g Mutant parasites. The apical complexes of two developing daughter cells are visible (white stars). The invaginated nuclear envelope delineates an intra-nuclear funnel extending between the centrioles (higher magnification in g′)
Identification of putative TgArk1 substrates in T. gondii
TgArk1 substrates are unknown in T. gondii. To uncover the putative substrates of TgArk1 in T. gondii, we performed a Myc-Trap pull-down experiment using anti-Myc antibodies on lysates derived from parasites expressing either DD-Myc-TgArk1WT or DD-Myc-TgArk1D/A followed by mass spectrometry identification of the immuno-precipitated proteins (Supplementary Table 1 and Supplementary Excel file). An anti-Myc immuno-precipitation on protein extracts from the parental strain (RH) was used as control. For mass spectrometry analysis, we selected the proteins identified in both experiments but not in the control strain, and those present in the control with a single peptide and at the same time in both experiments with at least three unique peptides in one experiment out of two (Supplementary Table 1 and Supplementary Excel file). All the common proteins identified in one of the two experiments and in the negative control, or absent from the control but found in one out of two experiments, were excluded (Supplementary Excel file). Our results revealed that TgArk1 may phosphorylate cytoskeleton-associated proteins, factors involved in DNA biology, nuclear pore proteins and known cell cycle regulators (Supplementary Table 1 and Supplementary Excel file). Note that these results are preliminary, and need further validation.
Hesperadin efficiently acts in vitro on the growth of T. gondii
Hesperadin is an indolinone inhibitor of Aurora B [64, 65]. Its sulphonamide group extends beyond the ATP-pocket and into the adjacent hydrophobic pocket [66]. Given the homology between ARKB and TgArk1, and the importance of TgArk1 on parasite cell division, we reasoned that Hersperadin could possibly impair parasite growth by interfering with TgArk1’s function. We tested the activity of hesperadin against Toxoplasma cell growth by in vitro plaque assays (Fig. 8a). Parasites were grown in the presence of increasing concentrations, up to 100 nM of hesperadin, for 6 days (Fig. 8a, b). Plaque size decreased proportionally to the increasing concentrations of hesperadin (Fig. 8a, b). Hesperadin effectively inhibited T. gondii-infected cell growth from concentrations ranging 30 nM. A drastic decrease on the ability of parasites to lyse human cells was observed in the presence of 50, 75 and 100 nM (Fig. 8a, b). In the presence of hesperadin drug, parasite division was often asymmetric giving rise to odd numbers of parasites and leads to cells devoid of nuclei (Fig. 8c, d, white asterisk). Scoring the number of parasites per vacuole, we observed a decrease in the number of vacuoles containing 4, 8 and 16 parasites, and an increase in vacuoles containing two cells and an odd number of parasites, such as 3, 5 and 6 (Fig. 8c, d). These data suggest that low doses of Aurora kinase B inhibitor are sufficient to kill T. gondii parasites cultured in vitro. To further characterize the drug-induced phenotype, we treated parasites with 75 nM drug, and determined its impact on the different subcellular structures involved in endodyogeny. Our analysis revealed that hesperadin induces the same effect on nuclear mitosis and daughter cell formation as do the interference with both TgArk1 and TgINCENP2 function by genetic manipulation (Supplementary Figs. 12 and 13). Despite several attempts, we could not obtain an active recombinant TgArk1 to test its sensitivity to hesperadin in vitro. These observations suggest that TgArk1, as is the case for ARKB, may be active only in complex with TgINCENPs and probably following several rounds of phosphorylation reactions [20]. Interestingly, when we tested the impact of hesperadin at 75 nM on a strain that overexpresses TgArk1 (endogenous TgArk1 + DD-Myc-TgArk1WT) in the presence of shield-1, we found that the strain becomes less sensitive to the drug. Scoring the number of parasites per vacuole, we observed a decrease in the number of vacuoles containing one, three, five and six parasites, and an increase in vacuoles containing 4, 8 and 16 cells (Supplementary Fig. 14). Our data suggest that hesperadin may target the CPC complex in Toxoplasma, and reinforce the thesis that the CPC complex plays a central role in maintaining the homeostasis of cell division in T. gondii.
Fig. 8.

Hesperadin drug inhibits the in vitro growth of T. gondii at low concentrations. a Plaque assays were carried out by infecting HFF monolayers with RH-Δku80 strain in the presence of DMSO, barasertib at 800 nM and hesperadin drug at various concentrations ranging from 15 to 100 nM for 7 days. The HFF were stained with Giemsa. b The area of 30 plaques formed in the presence of each compound tested at single dose or at different concentrations was measured using ImageJ software. Values are means ± standard deviations. c Intracellular growth of RH-Δku80 strain cultivated in the presence of DMSO or hesperadin drug at different concentrations (15, 30, 40, 50 and 75 nM for 24 h). The percentages of vacuoles containing varying numbers of parasites are represented on the y-axis. Values are mean ± SD for three independent experiments. d The treatment of parasites in the presence of hesperadin drug affects their intracellular growth and results in formation of nucleus-deficient parasites. IFA performed on intracellular parasites of the RH-Δku80 strain shows an IMC marker (TgIMC1, red) and nuclei stained by DAPI. In the presence of hesperadin drug for 24 h, nucleus-deficient parasites appear, whereas normal nucleus segregation is observed in the presence of DMSO for RH-Δku80 strain, as expected. White asterisks indicate the parasites lacking nuclei
Discussion
Coccidia divide their nucleus by closed mitosis, with unusual features. One of such is a complex mode of generating the mitotic spindle. The spindle assembles within a nuclear envelope-bounded funnel extending between two daughter pairs of centrioles. This is followed by the disruption of the envelope in the central part of the funnel, which in turn enables the interaction between the centromeres of the chromosomes with kinetochores that bind to the mitotic spindle, the formation of the metaphase plate and anaphase (Fig. 9). This very restricted opening of the nuclear envelope is transient, and rapidly followed by the closure of the envelope at each pole of the spindle to delineate unusual specialized structures known as the centrocones. Centrocones are shaped by the nuclear membrane, flanked by the kinetochores on the nucleoplasmic side, and the centrosome on the cytoplasmic side (Fig. 9) [67, 68]. Concomitantly, internal assembly of daughter cells is temporally and spatially synchronized with the segregation of uncondensed chromosomes within an intact nucleus. In this context, one of the longstanding questions in the Apicomplexa field is how T. gondii tachyzoites regulate and coordinate the different peculiar events that occur during their cell division. The dynamics of chromosome segregation were a complete black box, until a few years back when centromeres were first identified, and it was observed that they remain permanently associated to the nuclear periphery [3]. A number of studies followed, demonstrating that the peculiar bi-modal construction of the apicomplexan centrosome, and its associated structures and regulatory kinases, all synergized to achieve the compartmentalized regulation of the events occurring during cell division [7–14, 39, 62]. Our study builds onto this knowledge through the functional characterization of the CPC complex in T. gondii: a complex which is required for proper chromosome segregation in higher eukaryotes but had not been studied in Apicomplexa. We thus identified homologs of Aurora Kinase B and its binding partner INCENP; TgArk1, TgINCENP1, and TgINCENP2 in T. gondii.
In mammals, Aurora kinase B and INCENP localize to the centromeres during prophase and metaphase, the central mitotic spindle in anaphase, the spindle midzone/cleavage furrow in telophase and to the mid-body during cytokinesis [18]. The localization of the CPC complex at the centromere is directly related to its role in the bi-orientation of chromosomes on the mitotic spindle, a step required for subsequent chromatid separation [20]. It has been shown that the N-terminus of INCENP and its SAH (single alpha-helix) domain tether the human CPC complex to the inner centromere and to spindle microtubules, respectively [69]. We show that Toxoplasma TgArk1 and TgINCENP2 differentially localized in a cell cycle-dependent manner. In non-dividing parasites, both proteins are found in the nucleus. In dividing parasites, they are found dispersed in the nucleus and cytoplasm. TgINCENP1 is expressed only in non-dividing parasites whereby it is exclusively detected in the nucleus. Intriguingly, we have failed to identify, in the N-terminal part of the two TgINCENPs, the motifs which are involved in the attachment of the CPC complex to either the centromeres or the microtubules. Moreover, it seems that the Apicomplexa do not encode Survivin and Borealin homologues. These observations may explain the absence of accumulation of TgArk1, TgINCENP1 and TgINCENP2 at the centromeres throughout the cell cycle. Thus, the functional specialization of this atypical CPC complex in T. gondii may have been repurposed to accomplish specific functions related to the particular mode of division of this parasite and to accommodate a closed mitosis.
Our electron microscopy findings rather point at a role of TgArk1 in the timely opening of the nuclear membrane enveloping the early spindle, in turn, enabling timely interactions among chromosomes, kinetochores and mitotic microtubules, with centrosomes (Fig. 9). Indeed, this hypothesis is supported by the persistence of long stretches of coinciding “centrosomal inner core”, spindle, kinetochore and centromere markers, in the nucleus. Consistently, persisting early spindle microtubule funnels that do not evolve into bonafide mitotic spindle thus blocking nuclear division were frequently observed by TEM. As in normal conditions the transient funnel gives rise to centrocones, it is not so surprising that when centrocones do not form, their components adopt the shape of the funnel. This can easily be assumed for EB1, a bonafide marker of spindle microtubules, and may appear more surprising for the “centrosome inner core” marker CEP250L1. These findings may suggest that the inner core of the centrosome is in fact part of the centrocone complex. However, centrocones are rather complex organelles. The marker originally used for its localization (TgMORN1) is detected at the base of the organelle by immuno-electron microscopy (IEM) [60]. One could, therefore, suggest that CEP250L1 may be located in a more distal part of the centrocone which would explain its behavior in ark1D/A-Δku80 mutant parasites. The persistence of the coincidence of the kinetochore signal with the spindle markers along the funnel can be understood whereby previous studies have shown the alignment of kinetochores along the early spindle funnel [67], as if “waiting” for the opening of the funnel to connect with the spindle, and also when the association of these kinetochores (together with the centromeres) is known to persist all along the cell cycle near the centriole pairs even in the absence of microtubules, strongly suggesting that kinetochores are mostly associated with the inner surface of the nuclear envelope. Therefore, when the funnel persists and probably elongates more than usual, this association becomes even more obvious. These observations might suggest an important role provided by TgArk1-mediated phosphorylation in opening the nuclear envelope to organize the timely formation of the mitotic spindle. Collectively, this points to a mechanistic model in which TgArk1 may be required for progression from prophase/prometaphase into metaphase. A number of recent studies suggest that ARKB promotes the formation of a passage in the nuclear envelope to link microtubules to kinetochore proteins [70–72]. Our preliminary efforts to identify putative substrates of TgArk1 unraveled three newly assigned parasite proteins (TgNup37, TgNup503 and TgNup115) belonging to the nuclear pore complex (Supplementary Table 1 and Supplementary Excel file, proteins highlighted in green) [73]. Phospho-peptide identification by mass spectrometry suggests that both TgNup503 and TgNup115 are both phosphorylated (Supplementary Table 1). Whether these proteins are truly phosphorylated by TgArk1 and whether their phosphorylation has a direct functional link with the opening of the nuclear envelope to give way to the mitotic spindle remain to be elucidated.
Surprisingly, in ark1D/A-Δku80 mutant parasites, we noticed dissociation between centromeres and kinetochores, highlighting the potential importance of TgArk1-mediated phosphorylation in maintaining the spatio-temporal proximity and/or crosstalk between centromeres and kinetochores. However, we cannot exclude that DNA duplication also proceeds without mitotic partition, the disjunction often observed between centromeres and kinetochores strongly suggesting such malfunction (Fig. 9). Unfortunately, we cannot test this hypothesis because our FACS DNA analysis data likely missed all the monster cells that contained big nuclei, and thus may contain multiple genome copies, probably due to a particular fragility of these structures (IMC absent or partial, disorganization or absence of subpellicular microtubules). ARKB is also known to play an important role in chromosome condensation and cohesion [74], and regulates the G1/S progression of the cell cycle by phosphorylating substrates, together with cyclinA [75]. Our preliminary searches for potential TgArk1 substrates pointed us to a number of proteins (many of them are predicted to be phosphorylated) involved in DNA biology that need to be investigated in the future (Supplementary Table 1 and Supplementary Excel file, proteins highlighted in blue). In addition, we identified cyclin TgPHO80 (also predicted to be heavily phosphorylated), as a potential substrate of TgArk1 (Supplementary Table 1 and Supplementary Excel file, protein highlighted in red). This cyclin is involved in G1/S progression in T. gondii [14].
ARKB is also involved in the terminal stages of the cytokinesis necessary for the separation of the daughter cells [76–78]. We showed, that the initiation of daughter cells IMC biogenesis does not require TgArk1, but further elongation of the IMC is probably blocked in the mutant, leading to an accumulation of abortive daughter cells organized in the form of Russian dolls, as observed by TEM. The form of Russian dolls could be explained by the continuous amplification of the centrosomes that nucleate each an SFA (striated fiber assemblin) fiber that gives rise to one daughter cell [9] and by their disorganization because they are probably no longer located at the poles of the spindles. On the other hand, the role of phosphorylation events in the formation of daughter cells in T. gondii has not been studied. Interestingly, we identified a number of proteins (many of them are predicted to be phosphorylated) as potential TgArk1 substrates (supplementary Table 1 and Supplementary Excel file, proteins highlighted in yellow).
Overall, the phenotype obtained in our study when interfering with the expression of TgArk1 underline dissociation between mitosis and daughter budding during T. gondii endodyogeny. That disturbance has been described by others in T. gondii when manipulating the expression of T. gondii proteins homologous to regulators of mitosis in higher eukaryotes [8, 11, 13, 14, 16, 79]. Yet, the disjunction between nuclear division and zoite differentiation in Apicomplexa is rather the rule than the exception in the phylum. In schizogony, a process whereby a number of successive mitosis proceed in an undifferentiated cytoplasmic mass, up to a last round where a fully differentiated zoite develop in association with each daughter nucleus. T. gondii endodyogeny, therefore, represents an unusual case of obligatory association between the two processes [2, 9]. What is interesting in our mutants is that the centrosomes normally located at each spindle pole keep dividing, partly loosing connection with the mitotic markers. And while nuclear divisions get stuck, early zoite internal budding proceeds repetitively leading to incomplete IMC scaffold encased in one another (Fig. 9). Consequently, our findings show clearly that the clock regulating initiation of daughter cells keeps running at normal pace when the nuclear division process fails. This supports the model proposed by others that traditional “checkpoints” present in higher eukaryotes are absent in Apicomplexa, therefore, allowing greater “flexibility” in cell cycle progression. Rather than a disadvantage, this added flexibility has been proposed as an adaptive advantage, because it allows apicomplexan parasites to adapt their mode of cell division depending on their niche-specific needs. For example, T. gondii itself has the ability to divide by endodyogeny, but can also scale up its form of division and switch to endopolygeny when infecting the cat specifically.
It has been shown that the inner centromere protein (INCENP) acts as a scaffold protein regulating CPC localization and activity [69]. Depletion of INCENP abolished the localization of Aurora kinase B and the formation of the entire Aurora B/INCENP/Survivin complex [54, 80]. Despite the localization of TgINCENP1 to the nucleus and its interaction with TgArk1, our reverse genetic experiments indicate that TgINCENP1 is not essential for mitosis and parasite development. The apparent lack of involvement of TgINCENP1 in mitotic control is intriguing. We speculate that the function of TgINCENP1 during mitosis may overlap with TgINCENP2. Because during endopolygeny multiple rounds of nuclear division occur prior to daughter cell budding, we speculate that the functional redundancy between INCENPs in T. gondii may be beneficial when the DNA content exceeds 2 N per cell [2]. Unlike TgINCENP1, TgINCENP2 plays important roles in mitosis and cytokinesis. Interestingly, we have shown that the proper localization of TgArk1 is dictated by TgINCENP2, confirming and extending previous studies. In the absence of TgINCENP2, TgArk1 remains in the cytoplasm of parasites associated with vesicular structures. This observation suggests that the mis-targeting of TgArk1 prevents it from reaching its substrates and explains the similar phenotypes obtained in the two mutants. Alternatively, the formation of the complex may be required for functional competency of TgArk1 or substrate targeting.
Human ARKB is a prime target in cancer patients by a group of interesting and accessible chemical compounds. In this study, we tested the anti-parasitic activity of two promising inhibitors: barasertib and hesperadin [81–83]. Surprisingly only hesperadin has shown efficacy on T. gondii in vitro. This drug severely impairs parasite growth at minimal doses. Treatment with hesperadin phenocopies the observed effect in ark1D/A-Δku80 mutant parasites, strongly suggesting that TgArk1 is hesperadin's target in T. gondii. The fact that parasites are more sensitive to hesperadin, when compared to mammalian cells, may suggest that the drug associates with other key residues than those described in ARKB, thus more effectively paralyzing the activities of TgArk1. Also perhaps parasites are more sensitive to hesperadin because they divide faster. Nonetheless, the active site of TgArk1 shares 50% similarity to human aurora kinase B, and further work will likely be required to improve the selectivity of this drug for parasite kinase inhibition. On the other hand, the lack of activity of Barasertib on T. gondii can be explained by the fact that the residues involved in the interaction between human ARKB and Barasertib are not conserved in TgArk1. Our work points at TgArk1 as a potential drug target, and pave the way for further drug optimization to treat apicomplexan-caused diseases. Moreover, our work highlights the value of repurposing drugs/compounds originally developed to treat other diseases for the study and/or treatment of microbial infections.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We wish to thank Dominique Soldati-Favre, Vern Carruthers, Lilach Sheiner, Sebastian Lourido, Boris Striepen, Markus Meissner, Peter Bradley, Mathieu Gissot, Christian Doerig, Con Beckers, Iain Cheeseman, Jose Garcia-Bustos, Henri Vial and Marjorie Bienvenu for their kind gift of cell lines, hesperadin drug, plasmids or antibodies, for advices and technical assistance. We also thank the “Montpellier Ressources imagerie” platform, and the electron microscopy facility of the University of Montpellier (MEA), for their assistance in light and electron microscopy experiments, respectively. Dr. Wassim DAHER, Dr. Juliette Morlon-Guyot and Dr. Maryse LEBRUN are INSERM researchers. This work was made possible through core support from the Fondation pour la Recherche Médicale (Equipe FRMDEQ20170336725), the Labex Parafrap (ANR-11-LABX-0024), and National Institute of Health Grants AI110690, AI110638, and AI128136.
Abbreviations
- Ark1
Aurora-related kinase 1
- INCENP
Inner centromere protein
- TATi-1
Trans-activator Trap identified
- IMC
Inner membrane complex
- Nuf2
Nuclear filamentous 2
- EB1
Microtubule-associated protein RP/EB family member
- CEP
Centrosomal protein
Compliance with ethical standards
Conflict of interest
The authors declare that there are no conflicts of interest.
References
- 1.Suarez CE, Bishop RP, Alzan HF, Poole WA, Cooke BM. Advances in the application of genetic manipulation methods to apicomplexan parasites. Int J Parasitol. 2017;47:701–710. doi: 10.1016/j.ijpara.2017.08.002. [DOI] [PubMed] [Google Scholar]
- 2.Francia ME, Striepen B. Cell division in apicomplexan parasites. Nat Rev Microbiol. 2014;12:125–136. doi: 10.1038/nrmicro3184. [DOI] [PubMed] [Google Scholar]
- 3.Brooks CF, Francia ME, Gissot M, Croken MM, Kim K, Striepen B. Toxoplasma gondii sequesters centromeres to a specific nuclear region throughout the cell cycle. Proc Natl Acad Sci USA. 2011;108:3767–3772. doi: 10.1073/pnas.1006741108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gavin MA, Wanko T, Jacobs L. Electron microscope studies of reproducing and interkinetic Toxoplasma. J Protozool. 1962;9:222–234. doi: 10.1111/j.1550-7408.1962.tb02610.x. [DOI] [PubMed] [Google Scholar]
- 5.Nigg EA. Mitotic kinases as regulators of cell division and its checkpoints. Nat Rev Mol Cell Biol. 2001;2:21–32. doi: 10.1038/35048096. [DOI] [PubMed] [Google Scholar]
- 6.Zitouni S, Nabais C, Jana SC, Guerrero A, Bettencourt-Dias M. Polo-like kinases: structural variations lead to multiple functions. Nat Rev Mol Cell Biol. 2014;15:433–452. doi: 10.1038/nrm3819. [DOI] [PubMed] [Google Scholar]
- 7.Chen CT, Gubbels MJ. The Toxoplasma gondii centrosome is the platform for internal daughter budding as revealed by a Nek1 kinase mutant. J Cell Sci. 2013;126:3344–3355. doi: 10.1242/jcs.123364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Courjol F, Gissot M. A coiled-coil protein is required for coordination of karyokinesis and cytokinesis in Toxoplasma gondii. Cell Microbiol. 2018;20:e12832. doi: 10.1111/cmi.12832. [DOI] [PubMed] [Google Scholar]
- 9.Francia ME, et al. Cell division in Apicomplexan parasites is organized by a homolog of the striated rootlet fiber of algal flagella. PLoS Biol. 2012;10:e1001444. doi: 10.1371/journal.pbio.1001444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morlon-Guyot J, Francia ME, Dubremetz JF, Daher W. Towards a molecular architecture of the centrosome in Toxoplasma gondii. Cytoskeleton (Hoboken) 2017;74:55–71. doi: 10.1002/cm.21353. [DOI] [PubMed] [Google Scholar]
- 11.Suvorova ES, Francia M, Striepen B, White MW. A novel bipartite centrosome coordinates the apicomplexan cell cycle. PLoS Biol. 2015;13:e1002093. doi: 10.1371/journal.pbio.1002093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morlon-Guyot J, Berry L, Chen CT, Gubbels MJ, Lebrun M, Daher W. The Toxoplasma gondii calcium-dependent protein kinase 7 is involved in early steps of parasite division and is crucial for parasite survival. Cell Microbiol. 2014;16:95–114. doi: 10.1111/cmi.12186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berry L, et al. The conserved apicomplexan Aurora kinase TgArk3 is involved in endodyogeny, duplication rate and parasite virulence. Cell Microbiol. 2016;18:1106–1120. doi: 10.1111/cmi.12571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alvarez CA, Suvorova ES. Checkpoints of apicomplexan cell division identified in Toxoplasma gondii. PLoS Pathog. 2017;13:e1006483. doi: 10.1371/journal.ppat.1006483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deshmukh AS, Mitra P, Kolagani A, Gurupwar R. Cdk-related kinase 9 regulates RNA polymerase II mediated transcription in Toxoplasma gondii. Biochim Biophys Acta. 2018;1861:572–585. doi: 10.1016/j.bbagrm.2018.02.004. [DOI] [PubMed] [Google Scholar]
- 16.Naumov A, Kratzer S, Ting LM, Kim K, Suvorova ES, White MW. The Toxoplasma centrocone houses cell cycle regulatory factors. MBio. 2017;8:e00579-17. doi: 10.1128/mBio.00579-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andrews PD, Knatko E, Moore WJ, Swedlow JR. Mitotic mechanics: the auroras come into view. Curr Opin Cell Biol. 2003;15:672–683. doi: 10.1016/j.ceb.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 18.Carmena M, Earnshaw WC. The cellular geography of aurora kinases. Nat Rev Mol Cell Biol. 2003;4:842–854. doi: 10.1038/nrm1245. [DOI] [PubMed] [Google Scholar]
- 19.Hochegger H, Hegarat N, Pereira-Leal JB. Aurora at the pole and equator: overlapping functions of Aurora kinases in the mitotic spindle. Open Biol. 2013;3:120185. doi: 10.1098/rsob.120185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carmena M, Wheelock M, Funabiki H, Earnshaw WC. The chromosomal passenger complex (CPC): from easy rider to the godfather of mitosis. Nat Rev Mol Cell Biol. 2012;13:789–803. doi: 10.1038/nrm3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lampson MA, Cheeseman IM. Sensing centromere tension: Aurora B and the regulation of kinetochore function. Trends Cell Biol. 2011;21:133–140. doi: 10.1016/j.tcb.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu D, Lampson MA. Regulation of kinetochore-microtubule attachments by Aurora B kinase. Biochem Soc Trans. 2009;37:976–980. doi: 10.1042/BST0370976. [DOI] [PubMed] [Google Scholar]
- 23.Goto H, Yasui Y, Kawajiri A, Nigg EA, Terada Y, Tatsuka M, Nagata K, Inagaki M. Aurora-B regulates the cleavage furrow-specific vimentin phosphorylation in the cytokinetic process. J Biol Chem. 2003;278:8526–8530. doi: 10.1074/jbc.M210892200. [DOI] [PubMed] [Google Scholar]
- 24.Kawajiri A, Yasui Y, Goto H, Tatsuka M, Takahashi M, Nagata K, Inagaki M. Functional significance of the specific sites phosphorylated in desmin at cleavage furrow: Aurora-B may phosphorylate and regulate type III intermediate filaments during cytokinesis coordinatedly with Rho-kinase. Mol Biol Cell. 2003;14:1489–1500. doi: 10.1091/mbc.e02-09-0612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kondo T, Isoda R, Ookusa T, Kamijo K, Hamao K, Hosoya H. Aurora B but not rho/MLCK signaling is required for localization of diphosphorylated myosin II regulatory light chain to the midzone in cytokinesis. PLoS One. 2013;8:e70965. doi: 10.1371/journal.pone.0070965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Landino J, Ohi R. The timing of midzone stabilization during cytokinesis depends on myosin II activity and an interaction between INCENP and actin. Curr Biol. 2016;26:698–706. doi: 10.1016/j.cub.2016.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maciejowski J, et al. Mps1 regulates kinetochore-microtubule attachment stability via the Ska complex to ensure error-free chromosome segregation. Dev Cell. 2017;41(143–156):e6. doi: 10.1016/j.devcel.2017.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou X, et al. Phosphorylation of CENP-C by Aurora B facilitates kinetochore attachment error correction in mitosis. Proc Natl Acad Sci USA. 2017;114:E10667–E10676. doi: 10.1073/pnas.1710506114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim K, Soldati D, Boothroyd JC. Gene replacement in Toxoplasma gondii with chloramphenicol acetyltransferase as selectable marker. Science. 1993;262:911–914. doi: 10.1126/science.8235614. [DOI] [PubMed] [Google Scholar]
- 30.Donald RG, Roos DS. Stable molecular transformation of Toxoplasma gondii: a selectable dihydrofolate reductase-thymidylate synthase marker based on drug-resistance mutations in malaria. Proc Natl Acad Sci USA. 1993;90:11703–11707. doi: 10.1073/pnas.90.24.11703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Donald RG, Carter D, Ullman B, Roos DS. Insertional tagging, cloning, and expression of the Toxoplasma gondii hypoxanthine–xanthine–guanine phosphoribosyltransferase gene. Use as a selectable marker for stable transformation. J Biol Chem. 1996;271:14010–14019. doi: 10.1074/jbc.271.24.14010. [DOI] [PubMed] [Google Scholar]
- 32.Meissner M, Brecht S, Bujard H, Soldati D. Modulation of myosin A expression by a newly established tetracycline repressor-based inducible system in Toxoplasma gondii. Nucleic Acids Res. 2001;29:E115. doi: 10.1093/nar/29.22.e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Herm-Gotz A, Agop-Nersesian C, Munter S, Grimley JS, Wandless TJ, Frischknecht F, Meissner M. Rapid control of protein level in the apicomplexan Toxoplasma gondii. Nat Methods. 2007;4:1003–1005. doi: 10.1038/nmeth1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huynh MH, Carruthers VB. Tagging of endogenous genes in a Toxoplasma gondii strain lacking Ku80. Eukaryot Cell. 2009;8:530–539. doi: 10.1128/EC.00358-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sheiner L, Demerly JL, Poulsen N, Beatty WL, Lucas O, Behnke MS, White MW, Striepen B. A systematic screen to discover and analyze apicoplast proteins identifies a conserved and essential protein import factor. PLoS Pathog. 2011;7:e1002392. doi: 10.1371/journal.ppat.1002392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Daher W, Plattner F, Carlier MF, Soldati-Favre D. Concerted action of two formins in gliding motility and host cell invasion by Toxoplasma gondii. PLoS Pathog. 2010;6:e1001132. doi: 10.1371/journal.ppat.1001132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mann T, Beckers C. Characterization of the subpellicular network, a filamentous membrane skeletal component in the parasite Toxoplasma gondii. Mol Biochem Parasitol. 2001;115:257–268. doi: 10.1016/S0166-6851(01)00289-4. [DOI] [PubMed] [Google Scholar]
- 38.Beck JR, Rodriguez-Fernandez IA, de Leon JC, Huynh MH, Carruthers VB, Morrissette NS, Bradley PJ. A novel family of Toxoplasma IMC proteins displays a hierarchical organization and functions in coordinating parasite division. PLoS Pathog. 2010;6:e1001094. doi: 10.1371/journal.ppat.1001094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Farrell M, Gubbels MJ. The Toxoplasma gondii kinetochore is required for centrosome association with the centrocone (spindle pole) Cell Microbiol. 2014;16:78–94. doi: 10.1111/cmi.12185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lamarque MH, et al. Plasticity and redundancy among AMA-RON pairs ensure host cell entry of Toxoplasma parasites. Nat Commun. 2014;5:4098. doi: 10.1038/ncomms5098. [DOI] [PubMed] [Google Scholar]
- 41.Frenal K, Polonais V, Marq JB, Stratmann R, Limenitakis J, Soldati-Favre D. Functional dissection of the apicomplexan glideosome molecular architecture. Cell Host Microbe. 2010;8:343–357. doi: 10.1016/j.chom.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 42.Sievers F, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miguet L, et al. Proteomic analysis of malignant B-cell derived microparticles reveals CD148 as a potentially useful antigenic biomarker for mantle cell lymphoma diagnosis. J Proteome Res. 2009;8:3346–3354. doi: 10.1021/pr801102c. [DOI] [PubMed] [Google Scholar]
- 44.Manic G, Corradi F, Sistigu A, Siteni S, Vitale I. Molecular regulation of the spindle assembly checkpoint by kinases and phosphatases. Int Rev Cell Mol Biol. 2017;328:105–161. doi: 10.1016/bs.ircmb.2016.08.004. [DOI] [PubMed] [Google Scholar]
- 45.Ainsztein AM, Kandels-Lewis SE, Mackay AM, Earnshaw WC. INCENP centromere and spindle targeting: identification of essential conserved motifs and involvement of heterochromatin protein HP1. J Cell Biol. 1998;143:1763–1774. doi: 10.1083/jcb.143.7.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krenn V, Musacchio A. The Aurora B kinase in chromosome bi-orientation and spindle checkpoint signaling. Front Oncol. 2015;5:225. doi: 10.3389/fonc.2015.00225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sessa F, Mapelli M, Ciferri C, Tarricone C, Areces LB, Schneider TR, Stukenberg PT, Musacchio A. Mechanism of Aurora B activation by INCENP and inhibition by hesperadin. Mol Cell. 2005;18:379–391. doi: 10.1016/j.molcel.2005.03.031. [DOI] [PubMed] [Google Scholar]
- 48.Sidik SM, et al. A genome-wide CRISPR screen in Toxoplasma identifies essential apicomplexan genes. Cell. 2016;166(1423–1435):e12. doi: 10.1016/j.cell.2016.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murata-Hori M, Wang YL. The kinase activity of aurora B is required for kinetochore-microtubule interactions during mitosis. Curr Biol. 2002;12:894–899. doi: 10.1016/S0960-9822(02)00848-5. [DOI] [PubMed] [Google Scholar]
- 50.Shao H, Ma C, Zhang X, Li R, Miller AL, Bement WM, Liu XJ. Aurora B regulates spindle bipolarity in meiosis in vertebrate oocytes. Cell Cycle. 2012;11:2672–2680. doi: 10.4161/cc.21016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Adams JA, Taylor SS. Divalent metal ions influence catalysis and active-site accessibility in the cAMP-dependent protein kinase. Protein Sci. 1993;2:2177–2186. doi: 10.1002/pro.5560021217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cosmelli D, Antonelli M, Allende CC, Allende JE. An inactive mutant of the alpha subunit of protein kinase CK2 that traps the regulatory CK2beta subunit. FEBS Lett. 1997;410:391–396. doi: 10.1016/S0014-5793(97)00625-X. [DOI] [PubMed] [Google Scholar]
- 53.Fentress SJ, et al. Phosphorylation of immunity-related GTPases by a Toxoplasma gondii-secreted kinase promotes macrophage survival and virulence. Cell Host Microbe. 2010;8:484–495. doi: 10.1016/j.chom.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Honda R, Korner R, Nigg EA. Exploring the functional interactions between Aurora B, INCENP, and survivin in mitosis. Mol Biol Cell. 2003;14:3325–3341. doi: 10.1091/mbc.e02-11-0769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gubbels MJ, White M, Szatanek T. The cell cycle and Toxoplasma gondii cell division: tightly knit or loosely stitched? Int J Parasitol. 2008;38:1343–1358. doi: 10.1016/j.ijpara.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 56.Radke JR, Striepen B, Guerini MN, Jerome ME, Roos DS, White MW. Defining the cell cycle for the tachyzoite stage of Toxoplasma gondii. Mol Biochem Parasitol. 2001;115:165–175. doi: 10.1016/S0166-6851(01)00284-5. [DOI] [PubMed] [Google Scholar]
- 57.Anderson-White B, Beck JR, Chen CT, Meissner M, Bradley PJ, Gubbels MJ. Cytoskeleton assembly in Toxoplasma gondii cell division. Int Rev Cell Mol Biol. 2012;298:1–31. doi: 10.1016/B978-0-12-394309-5.00001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nishi M, Hu K, Murray JM, Roos DS. Organellar dynamics during the cell cycle of Toxoplasma gondii. J Cell Sci. 2008;121:1559–1568. doi: 10.1242/jcs.021089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gissot M, Hovasse A, Chaloin L, Schaeffer-Reiss C, Van Dorsselaer A, Tomavo S. An evolutionary conserved zinc finger protein is involved in Toxoplasma gondii mRNA nuclear export. Cell Microbiol. 2017;19:e12644. doi: 10.1111/cmi.12644. [DOI] [PubMed] [Google Scholar]
- 60.Gubbels MJ, Vaishnava S, Boot N, Dubremetz JF, Striepen B. A MORN-repeat protein is a dynamic component of the Toxoplasma gondii cell division apparatus. J Cell Sci. 2006;119:2236–2245. doi: 10.1242/jcs.02949. [DOI] [PubMed] [Google Scholar]
- 61.Hu K. Organizational changes of the daughter basal complex during the parasite replication of Toxoplasma gondii. PLoS Pathog. 2008;4:e10. doi: 10.1371/journal.ppat.0040010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen CT, et al. Compartmentalized Toxoplasma EB1 bundles spindle microtubules to secure accurate chromosome segregation. Mol Biol Cell. 2015;26:4562–4576. doi: 10.1091/mbc.e15-06-0437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gissot M, Walker R, Delhaye S, Huot L, Hot D, Tomavo S. Toxoplasma gondii chromodomain protein 1 binds to heterochromatin and colocalises with centromeres and telomeres at the nuclear periphery. PLoS One. 2012;7:e32671. doi: 10.1371/journal.pone.0032671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hu Y, Zhang J, Musharrafieh R, Hau R, Ma C, Wang J. Chemical genomics approach leads to the identification of hesperadin, an aurora B kinase inhibitor, as a broad-spectrum influenza antiviral. Int J Mol Sci. 2017;18:1929. doi: 10.3390/ijms18091929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shamsipour F, Hosseinzadeh S, Arab SS, Vafaei S, Farid S, Jeddi-Tehrani M, Balalaie S. Synthesis and investigation of new Hesperadin analogues antitumor effects on HeLa cells. J Chem Biol. 2014;7:85–91. doi: 10.1007/s12154-014-0111-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hauf S, et al. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J Cell Biol. 2003;161:281–294. doi: 10.1083/jcb.200208092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dubremetz JF. Ultrastructural study of schizogonic mitosis in the coccidian, Eimeria necatrix (Johnson 1930) J Ultrastruct Res. 1973;42:354–376. doi: 10.1016/S0022-5320(73)90063-4. [DOI] [PubMed] [Google Scholar]
- 68.Dubremetz JF, Elsner YY. Ultrastructural study of schizogony of Eimeria bovis in cell cultures. J Protozool. 1979;26:367–376. doi: 10.1111/j.1550-7408.1979.tb04639.x. [DOI] [PubMed] [Google Scholar]
- 69.Samejima K, Platani M, Wolny M, Ogawa H, Vargiu G, Knight PJ, Peckham M, Earnshaw WC. The inner centromere protein (INCENP) coil is a single alpha-helix (SAH) domain that binds directly to microtubules and is important for chromosome passenger complex (CPC) localization and function in mitosis. J Biol Chem. 2015;290:21460–21472. doi: 10.1074/jbc.M115.645317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Karg T, Warecki B, Sullivan W. Aurora B-mediated localized delays in nuclear envelope formation facilitate inclusion of late-segregating chromosome fragments. Mol Biol Cell. 2015;26:2227–2241. doi: 10.1091/mbc.e15-01-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mackay DR, Makise M, Ullman KS. Defects in nuclear pore assembly lead to activation of an Aurora B-mediated abscission checkpoint. J Cell Biol. 2010;191:923–931. doi: 10.1083/jcb.201007124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mackay DR, Ullman KS. Coordinating postmitotic nuclear pore complex assembly with abscission timing. Nucleus. 2011;2:283–288. doi: 10.4161/nucl.2.4.16189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Courjol F, et al. Characterization of a nuclear pore protein sheds light on the roles and composition of the Toxoplasma gondii nuclear pore complex. Cell Mol Life Sci. 2017;74:2107–2125. doi: 10.1007/s00018-017-2459-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shannon KB, Salmon ED. Chromosome dynamics: new light on Aurora B kinase function. Curr Biol. 2002;12:R458–R460. doi: 10.1016/S0960-9822(02)00945-4. [DOI] [PubMed] [Google Scholar]
- 75.Song J, Salek-Ardakani S, So T, Croft M. The kinases aurora B and mTOR regulate the G1-S cell cycle progression of T lymphocytes. Nat Immunol. 2007;8:64–73. doi: 10.1038/ni1413. [DOI] [PubMed] [Google Scholar]
- 76.Mackay DR, Ullman KS. ATR and a Chk1-Aurora B pathway coordinate postmitotic genome surveillance with cytokinetic abscission. Mol Biol Cell. 2015;26:2217–2226. doi: 10.1091/mbc.e14-11-1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mathieu J, et al. Aurora B and cyclin B have opposite effects on the timing of cytokinesis abscission in Drosophila germ cells and in vertebrate somatic cells. Dev Cell. 2013;26:250–265. doi: 10.1016/j.devcel.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Thoresen SB, Campsteijn C, Vietri M, Schink KO, Liestol K, Andersen JS, Raiborg C, Stenmark H. ANCHR mediates Aurora-B-dependent abscission checkpoint control through retention of VPS4. Nat Cell Biol. 2014;16:550–560. doi: 10.1038/ncb2959. [DOI] [PubMed] [Google Scholar]
- 79.Dhara A, de Paula Baptista R, Kissinger JC, Snow EC, Sinai AP. Ablation of an ovarian tumor family deubiquitinase exposes the underlying regulation governing the plasticity of cell cycle progression in Toxoplasma gondii. MBio. 2017;8:e01846-17. doi: 10.1128/mBio.01846-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.van der Horst A, Vromans MJ, Bouwman K, van der Waal MS, Hadders MA, Lens SM. Inter-domain cooperation in INCENP promotes Aurora B relocation from centromeres to microtubules. Cell Rep. 2015;12:380–387. doi: 10.1016/j.celrep.2015.06.038. [DOI] [PubMed] [Google Scholar]
- 81.de Groot CO, et al. A cell biologist’s field guide to aurora kinase inhibitors. Front Oncol. 2015;5:285. doi: 10.3389/fonc.2015.00285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Falchook GS, Bastida CC, Kurzrock R. Aurora kinase inhibitors in oncology clinical trials: current state of the progress. Semin Oncol. 2015;42:832–848. doi: 10.1053/j.seminoncol.2015.09.022. [DOI] [PubMed] [Google Scholar]
- 83.Lok W, Klein RQ, Saif MW. Aurora kinase inhibitors as anti-cancer therapy. Anticancer Drugs. 2010;21:339–350. doi: 10.1097/CAD.0b013e3283350dd1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.