

Abstract
We observed increased expression of ADAMDEC1 RNA in monocytes from patients with systemic lupus erythematosus. The precise role of ADAMDEC1 is uncertain and uniquely among metalloproteinases it utilizes a zinc-coordinating aspartic acid residue which allows it to escape inhibition by tissue inhibitor of metalloprotease-3 (TIMP-3). A closely related gene encodes the protein ADAM28, which is not up-regulated in lupus. We leveraged the ability to look at both gene’s promoters and enhancers simultaneously. ADAMDEC1 was up-regulated by LPS while ADAM28 was not upregulated in the short term. We identified MAP kinases and NFκB as critical cell pathways regulating the expression of ADAMDEC1. These same pathways were implicated in driving the expression of the ADAMDEC1 upstream enhancer RNAs. We demonstrated that binding of the enhancer RNAs produced from the upstream enhancer were critically important and that p300 bound to both the RNA from the enhancer and the DNA at the enhancer. P300 binding to the enhancer was dependent on NFκB. These data define the critical pathways regulating the expression of ADAMDEC1 and extend our knowledge of the roles of enhancer RNAs and mechanistically links p300 and enhancer RNAs.
Keywords: SLE, lupus, complement, ADAM28, epigenetic, eRNA, decysin
Graphical Abstract
1.0. Introduction
The family of proteins referred to as ADAMs (A Disintegration And Metalloproteinase) have emerged as major regulatory effectors for biological processes. Cellular processes related to fertility, cell fate determination, and cell development have all been implicated are regulated by this family of proteins (Alfandari et al., 2001; Blobel, 2005; Chen and Sampson, 1999; Maretzky et al., 2005; Reiss and Saftig, 2009; Seals and Courtneidge, 2003). Knockout mice for various ADAM family members have had diverse phenotypes (Reiss and Saftig, 2009). One of the main functions of ADAM family proteins is proteolytic cleavage and ectodomain shedding. ADAM family members induce shedding of many cell surface proteins including growth factors, cytokines, cell adhesion molecules, and various receptors (Black et al., 1997; Moss and Lambert, 2002; Yan et al., 2002). ADAM-mediated shedding is both constitutive and inducible and is largely dependent on G-protein coupled receptors and protein kinase C (Ohtsu et al., 2006; Schafer et al., 2004).The role of ADAM proteins in remodeling extracellular matrix has led to intense investigation from the perspective of regulating metastases but there is little information regarding expression in inflammatory states (Han et al., 2001; Zhu et al., 2003).
We identified overexpression of ADAMDEC1 in systemic lupus erythematosus (SLE) (Shi et al., 2014) and studies have found significant upregulation of ADAMs in colitis with ADAMDEC1 knockout mice more susceptible to colitis (O’Shea et al., 2016). ADAMDEC1, also known as decysin, is primarily expressed in myeloid lineage cells. It is upregulated by a variety of stimuli and inflammatory states (Galamb et al., 2008). These data suggest that ADAMDEC1 may be important in inflammation, however, the regulation of expression of ADAMDEC1 has not been investigated in detail.
ADAM28 is encoded upstream of ADAMDEC1 and appears to have overlapping functions. The role of ADAM28 in metastatic spread is well-characterized and ADAM28 is thought to regulate migration (Mitsui et al., 2006; Mochizuki et al., 2012; Ohtsuka et al., 2006). One substrate of ADAM28 is connective tissue growth factor and cleavage by ADAM28 releases vascular endothelial growth factor 165 which drives angiogenesis, a key step in invasion by malignant cells, thus providing another mechanism related to metastasis (Mochizuki et al., 2010). ADAM28 expression has been detected in lymphocytes where it regulates adhesion (McGinn et al., 2011) and it may contribute to atherosclerosis by modulating monocyte attachment to endothelium (Worley et al., 2007). Recently, an interaction with the complement protein C1q has been described (Miyamae et al., 2016). ADAM28 provides protection from C1q-mediated cell death, a critical pathologic process in inflammation. Complement-mediated tissue injury is a key facet of end-organ damage in SLE.
We therefore set out to understand the expression of ADAM28 and ADAMDEC1, two closely related proteins transcribed from adjacent genes and relevant in SLE. We found that ADAMDEC1 is regulated by a cascade of events centered on NFκB regulation of p300 binding of enhancer RNAs (eRNAs).
2.0. Materials and Methods
2.1. Cells and inhibitors:
Primary monocytes were obtained from a campus core facility under an IRB-approved protocol. Subjects gave informed consent. The SLE samples reported were obtained as part of a previously reported study under a separate IRB-approved protocol for which all subjects gave informed consent (Zhang et al., 2018a). The SLE patient RNA samples have been previously reported (Shi et al., 2015; Shi et al., 2014), although these analyses are new. MonoMac6 cells were used as a monocyte cell line and maintained in RPMI with 10% cosmic calf serum with OPI supplement (SigmaAldrich, St. Louis, MO). MonoMac6 cells were obtained from the German source and thawed from primary vials every 2–3 months. The inhibitors utilized included C646 (Santa Cruz Biotechnology, Dallas, Texas) which was used at 20μM. It is an inhibitor of CBP/p300 induced histone acetylation (Bowers et al., 2010). The inhibitor iBET151 (Life science Research, Billerica, Massachusetts) was used at 20μM as a specific inhibitor of BRD3/4, important for transcriptional activation and elongation at paused genes (Yang et al., 2005). DRB (Sigma-Aldrich, Allentown, PA ) was used at 40μM as an inhibitor of elongation regulating eviction of NELF via P-TEFb (Yankulov et al., 1995). JSH-23 (Santa Cruz Biotechnology) was used at 20μM as a specific inhibitor of NFκB translocation (Shin et al., 2004) and SP600125 (Calbiochem, Darmstadt, Germany) was used at 10μM an inhibitor of the JNK MAP kinase. Toxicity analyses confirmed minimal cell death at these concentrations. Transfection of cells was performed by electroporation with the Amaxa Cell Line Lonza Nucleofector Kit (Amaxa Biosystems, Gaithersburg, MD) or Lipofectamine 2000 (ThermoFisher, Waltham, MA). HPLC-purified LPS and phorbol myristate acetate (PMA) were purchased from Sigma-Aldrich (St. Louis, MO). Knock-down phosphorothioate antisense oligonucleotides used were ADAMDEC1–157-ASO1-TTT GGAAGCCAAGTAATAGC and ADAMDEC1–157ASO2-TGTTTATCCAAAGAATTTTC.
Two million cells were transfected with 500nM antisense oligonucleotide using nucleofection. LPS treatment utilized 1μg unless otherwise specified.
2.2. Luciferase reporter assay:
The pGL3-TK vector and the TK-A7 positive control were gifts from the Shiekhattar lab (Orom et al., 2010). The TK-A7 enhancer regulates the UBE2VI gene. The luciferase assay was performed in 96-well white plates using the Dual-Luciferase reporter assay kit (Promega, Madison, WI). 5×104 cells were transfected with 0.2μg of plasmid and 0.01μg pRL renilla reporter vector. Luciferase and renilla signals were measured at 24 hours.
2.3. Transcriptional analysis:
Cells were harvested and total RNA was extracted by TRI Reagent® (Sigma-Aldrich, St. Louis, MO, USA) and the Direct-zol™ RNA MiniPrep Kit (Zymo Research Irvine, CA). The Clontech Advantage RT for PCR kit (Clontech, Mountain View, CA) was used to make cDNA. RNA levels were detected by real-time PCR using the TaqMan 7900. Transcript levels were normalized to the 18S signal. ncRNAs were detected with custom SYBR green primers. Primer sequences are listed below.
122F: CCC ACT GGAAAA TTG AAA ACA,122R: AAA TCC TCCTCA AAG TGT TGG T
134F: CAA CCC CTG CTT TCT ACA ACA,134R: TTC TCC AGC TCA AAT CTC TGG
142F: CAG CCC AGA ATT TTC CCT TCT TTC,142R: GGG GCT CAG TAG GAG TCA TCC TTT,157F:
AGCCAACTAGAGCCGAGATA,157R: GAACACAGAGGGAAGGAATCAA
158F: AAGAAGCCTGTCTCAAGGATATG,158R: AGGGATTCCTGAGCATCAAAG
146F: GCTGACTTGTACTCCCAGTATC,146R: CTGGTCCTCAGCGTAATTTCTA
2.4. ChIP assays:
ChIP experiments were carried out as previously described (Garrett et al., 2008; J. Y. Lee et al., 2003). Briefly, 5–10 X 106 cells in each condition were treated with 1 % formaldehyde for 10 min at room temperature to crosslink. Glycine was used to quench the cross-linking. Lysed cells were sonicated and immunoprecipitated overnight at 4°C. Immune complexes were collected with protein A (Invitrogen, Carlsbad, CA), washed extensively and eluted. DNA was extracted by phenol-chloroform and proteinase K (200 μg/ml, Roche) digestion. DNA was RNase treated (40 μg/ml, Roche) for 30 min at 37°C. Antibodies utilized included those to: CDK9 (a component of P-TEFb), NELF-A, c-JUN, NFκB p65, and p300 (all from Santa Cruz Biotechnology, Santa Cruz, CA); H3K4me3 (Active Motif, Carlsbad, CA) and H3K27ac (Active Motif). The GST antibody (Invitrogen, Camarillo, CA) was used as a negative control for all ChIP assays. Cells were harvested at 30 minutes after LPS stimulation. Signals are reported as units normalized to 10% input according to the formula 2^(10%input CT-Sample CT). The primers for the ChIP assays are listed below:
Enhancer-1 F-CCATCTGTTCCCTCCATTTCT
Enhancer-1 probe-ACACAGGAGGAAGTGGTTGGAACC
Enhancer-1 R-TGATCATGACTCACTTGGTAATCT
Enhancer-2 F-GGGAGTCTGAGAGAGAGAAAGA
Enhancer-2probe-ATGCATTGTTGTGCTTCTCTGGCC
Enhancer-2R-ACTAGGACTAGTTCCAGCTTCT
ADAM28-Pro F-TGTGCTGTATGAAATTGTCCCT
ADAM28-Pro Probe-TGGTTCAAGACCTCTTGTTGCCTCT
ADAM28-Pro F-GGCTGCGAGTGATACAAAGA
ADAMDEC1-Pro F-GGAGACCACAACTTCATGCT
ADAMDEC1-Pro Probe-TTGGGTCCTGCTGCCTGTACTTT
ADAMDEC1-Pro F-CGTACCTTGAGTTTGAACAATGAG
GAPDH F-CGG TGC GTG CCC AGT T
GAPDH R-CCC TAC TTT CTC CCC GCT TT
GAPDH Probe-ACC AGG CGG CTG CGG AAA AAA
2.5. RNA immunoprecipitation assays:
The RNA immunoprecipitation (RNA-IP) assay was modified as described by Niranjanakumari (Niranjanakumari et al., 2002). Briefly, 20–40 million cells were crosslinked using 1% formaldehyde for 10 min and quenched with glycine. The fixed cells were lysed in RIPA buffer (1%NP40, 0.5% sodium deoxycholate, 0.05%SDS, 1mM EDTA, 150mM NaCl) and sonicated. The precleared lysates were incubated with antibodies and protein A agarose at 4°C. Immune complexes were washed and reverse cross-linked. Direct-zol™ RNA MiniPrep Kit was used to extract the immunoprecipitated RNA.
2.6. Statistical analyses
The results represent a minimum of three separate biological experiments. Each figure legend describes the number of individual experiments. Comparisons across treatments were performed using Student’s t test.
3.0. Theory
The interaction of CBP with eRNAs is a newly described aspect of transcriptional regulation (Bose et al., 2017). CBP binding of eRNAs was defined using ChIP-seq on a genome-wide basis and CBP binding of eRNAs was found to activate acetyltransferase activity. P300, a closely related histone acetyltransferase, was not examined in the same manner but is known to have the same RNA-binding motif. This study analyzed the role of p300 as a nexus of eRNA activation of transcription with gene-level detail in a system relevant for human disease.
4.0. Results
4.1. MAP kinase and NFκB regulation of ADAMDEC1
We noted on RNA-seq of peripheral blood monocytes that ADAMDEC1 was overexpressed in SLE while the adjacent ADAM28 was not (Table 1) (Shi et al., 2014). The expression of ADAM28 has been primarily investigated in the setting of malignancy while ADAMDEC1 has been investigated in inflammatory disease states (Fritsche et al., 2003; Galamb et al., 2008). To understand the pathways regulating ADAMDEC1 expression, we contrasted the expression of these two directly adjacent ADAM family member genes in a carefully controlled setting using qRT-PCR. We stimulated primary monocytes with LPS, a classic pro-inflammatory stimulus with a well-understood signaling cascade involving MAP kinases and NFκB. The expression of ADAMDEC1 was upregulated after 90 minutes of LPS stimulation while ADAM28 remained unchanged at this time-point in primary monocytes (Figure 1). To investigate the key elements responsible for the regulation of ADAMDEC1, we utilized a panel of inhibitors designed to probe the role of each component of the signaling cascade for LPS. U0126 was used as an inhibitor of MAP kinase ERK, SB203580 was used as an inhibitor of the MAP kinase p38, SP600125 was used as an inhibitor of the MAP kinase JNK, and JSH-23 was used as an inhibitor of NFκB. The p38 inhibitor, SB203580, had the greatest inhibitory effect on ADAMDEC1 expression while the ERK inhibitor, U0126, had no effect. The inhibitory effects of the agents used were confirmed by analyzing the expression of the classic inflammatory cytokines, TNF and IL1A after LPS. We concluded that ADAMDEC1 is regulated through the classical signaling pathway for LPS with contributions from p38, JNK, and NFκB. With this foundation, we set out to understand the interplay of chromatin dynamics and eRNA expression at this key locus.
Table 1.
RNA levels from RNA-seq data on SLE monocytes
| Gene | Fold Change | P Value | FDR |
|---|---|---|---|
| ADAM28 | 1.046 | 8.2E-01 | 0.998 |
| STC1 | 1.057 | 7.2E-01 | 0.973 |
| ADAMDEC1 | 2.402 | 2.5E-04 | 0.033 |
Figure 1. LPS induction of ADAMDEC1 but not ADAM28.

Primary monocytes were stimulated by 1μg/ml LPS for 90 minutes after pretreatment with 20μM SB203580, 20μM U0126, 20μM SP600125 or 20μM JSH-23 (JSH). ADAM28 mRNA and ADAMDEC1 mRNA were quantitated by qRT-PCR. SB203580 inhibits the p38 MAP kinase pathway. U0126 inhibits the ERK pathway. SP600125 inhibits the JNK pathway. JSH is an NFκB inhibitor. (n=4, error bars in panels denote SD). P values are indicated for each comparison.
4.2. Enhancer regulation of ADAMDEC1
The distinct expression pattern of two highly related genes that are transcribed from adjacent locations provided an opportunity to directly compare features to elucidate the mechanisms of regulation. Using ChIP-seq tracks for regulatory regions, we noted two prominent enhancers as defined by H3K27ac peaks with low H3K4me3 (Figure 2A). Approximately 20kb upstream of ADAM28 there is a region with chromatin features of an enhancer (Enhancer 1). Downstream of ADAM28, lying between ADAM28 and ADAMDEC1 is a second region with chromatin marks of an enhancer (Enhancer 2). To ensure that we had identified active enhancers based on chromatin characteristics, we examined our RNA-seq data for potential eRNAs, observed as short non-coding RNAs (Zhang et al., 2018b). Enhancer 1, upstream of ADAM28 and Enhancer 2, upstream of ADAMDEC1, both exhibited short transcripts consistent with eRNAs (shown schematically in Figure 2A above the histone tracks and as RNA-seq tracks indicated as the eRNA track). To further validate that these transcripts likely represented regulatory eRNAs, we analyzed their cellular compartment localization by separating cytoplasm, nucleoplasm and chromatin fractions and performing qRT-PCR (Figure 2B). For this analysis and subsequent analyses where large numbers of cells were required, we utilized MonoMac6 cells as a human monocyte cell line representative of mature monocytes. XIST was used as a control non-coding RNA that is known to regulate X chromosome inactivation and remains associated with chromatin. The eRNAs were strongly associated with the chromatin fraction, similar to XIST, and distinct from the cytoplasm-associated ACTB RNA that encodes cellular actin. These data support a regulatory role of the putative eRNAs.
Figure 2. The characterization of identified enhancer RNAs.

(A) The UCSC genome browser view shows the position of ADAM family genes and enhancers. The detailed annotation of the eRNAs was identified by the Tophat-Cufflinks pipeline in human SLE monocytes (Shi et al., 2014). The eRNAs are shown schematically a the top of the figure and the eRNA tracks from our RNA-seq dataset are displayed in the eRNA track with the mRNAs removed due to the magnitude of their expression compared to the eRNAs. Histone marks in human CD14+ monocytes from the ENCODE data indicated that these eRNAs are located within active enhancer regions, termed Enhancer 1 and Enhancer 2. (B) The eRNAs were localized primarily to the chromatin fraction. RNA was extracted from MonoMac6 cytoplasm, nucleoplasm and chromatin fractions with or without 1μg/ml LPS treatment. qRT-PCR was performed. Actin B and XIST are shown as positive controls for cytoplasmic and chromatin fractions, respectively (n=3, error bars in panels denote SD). Symbols indicate statistical significance: * indicates p<0.05, ** p<0.01.
As an additional strategy to confirm that these were functional enhancers, we subcloned the DNA corresponding the eRNA 134, which is centered on the H3K27ac peak upstream of ADAM28 (Enhancer 1), into a minimal thymidine kinase (TK) promoter construct. This construct was tested in a luciferase assay for enhancer function (Figure 3A). The 134 DNA construct was able to transactivate the TK promoter, as was the positive control, A7 (Orom et al., 2010), while a negative control, 172 that represents a non-enhancer-associated non-coding RNA, had no effect. Based on characteristic chromatin features and eRNA production, we conclude that Enhancers 1 and 2 represent active enhancers in monocytes.
Figure 3. DNA from Enhancer 1 activates luciferase transcription.

(A) The enhancer DNA corresponding to eRNA 134 was subcloned downstream of luciferase under the control of the minimal thymidine kinase promoter (TK). The firefly luciferase vectors were co-transfected with a constitutive Renilla control, pRL-TK, into HEK293 cells. PGL3-TK-A7 is a positive control from near the UBE2VI gene. Construct 172 is a negative control, which encodes a non-coding RNA but is not an eRNA and is not located in the ADAM locus. Inducible firefly luciferase was normalized to the Renilla signal. DNA from Enhancer 1 was able to transactivate TK expression in this system. (B) The Enhancer 1 and 2 eRNAs were coordinately regulated with ADAMDEC1 in primary monocytes. The cells were pretreated by SB203580, JSH-23, SP60025 or U0126 for 30 minutes and then stimulated with LPS for 90 minutes (n=3, error bars in panels denote SD). Symbols indicate statistical significance: * indicates p<0.05, ** p<0.01, and *** p<0.001.
Enhancer RNAs are produced in a roughly linear fashion with the mRNAs that are regulated by the enhancer. To determine whether these eRNAs were coordinately regulated with ADAM28 or ADAMDEC1, we stimulated with LPS and used the same inhibitors that we used to define the regulatory pathways impacting the mRNAs in Figure 1 (Figure 3B). The effects of the inhibitors on eRNA expression were similar but somewhat distinct. Enhancer 1 exhibited suppression of all three tested eRNAs with the P38 inhibitor, SB203580 while the effects of JNK and NFκB inhibition were limited to 134 and 142. Notably, eRNA 122 is the farthest away from the peak of the enhancer. Both sets of eRNAs (Enhancer 1 and Enhancer 2) were induced by LPS and the effect was inhibited by the p38 inhibitor (SB203580) and the JNK inhibitor (SP60025), which was also true for ADAMDEC1 expression. In contrast to the pattern for ADAMDEC1 where LPS induction of expression was inhibited by the NFκB inhibitor, JSH-23, the eRNAs located at Enhancer 1 were not affected by JSH-23 and the eRNAs at Enhancer 2 were minimally affected. Thus, both enhancers had an expression pattern that displayed significant but imperfect overlap with that of ADAMDEC1. Enhancer 2 eRNAs exhibited a pattern more consistent with that of ADAMDEC1 than Enhancer 1 eRNAs.
To demonstrate the functional role of the eRNAs, we transfected 20nt phosphorothioate antisense oligonucleotides (ASOs) directed at eRNAs (Figure 4A). The eRNA 157 from Enhancer 2 was knocked down using two separate ASOs (right panel) and we noted no difference in ADAM28 expression (left panel) compared to the LACZ control siRNA while ADAMDEC1 expression was reduced compared to the LACZ siRNA control after LPS treatment (middle panel). The ASOs did not affect basal expression of ADAMDEC1, as expected, since eRNAs do not regulate basal expression. These data confirm the role of eRNA 157 in the regulation of ADAMDEC1. We saw no effect of knocking down the eRNAs from Enhancer 1 on either ADAM28 or ADAMDEC1 expression (data not shown). We noted in a careful kinetic analysis that the eRNAs associated with Enhancer 1 (122, 134, 142) had a slightly later response to LPS compared to the eRNAs associated with Enhancer 2 (157, 158) (Figure 4B). In these MonoMac 6 cells, ADAM28 was modestly up-regulated with later kinetics than ADAMDEC1. We hypothesize that the later kinetics for Enhancer 1 eRNAs may be responsible for the late activation of ADAM28 or that the effect may be indirect. Based on the ASO knockdowns and the expression pattern of the eRNAs, we assigned Enhancer 2 as regulating ADAMDEC1 expression while Enhancer 1 may be involved in the regulation of ADAM28 at later time points or indirectly involved in the regulation of ADAMDEC1. With this clear functional association attributable to Enhancer 2 eRNAs regulating ADAMDEC1, we wished to understand the chromatin dynamics at the promoter and enhancer related to ADAMDEC1.
Figure 4. The eRNAs regulate transcription.

(A) Knockdown of eRNA 157 from Enhancer 2 decreased the LPS-induced ADAMDEC1 mRNA but not ADAM28 mRNA. MonoMac6 cells were transfected with the indicated 20-bp phosphorothioate antisense oligonucleotides (ASO) designed to knock down Enhancer 2 eRNA157 (two different ASO) or LACZ for 3 hours and stimulated with or without 1μg/ml LPS for 60 minutes. The eRNAs and mRNAs were measured by qRT-PCR. The ASO effectively knocked down eRNA 157. ADAMDEC1 expression after LPS was diminished although basal expression was not affected. ADAM28 expression was unchanged. (n =3, * p<0.05). (B) LPS-induced mRNA and eRNA kinetic analysis in the primary monocytes. The eRNA and mRNA expression levels were measured by qRT-PCR at various time points and normalized to 18S (mRNAs) or ACTB (eRNAs) (n=3, error bars indicate SD).
4.3. Chromatin characteristics
Acute stimulation of expression is often accompanied by changes in the local chromatin at either the promoter, enhancer or both. We hypothesized that differential expression of ADAM28 and ADAMDEC1 might be regulated at the level of chromatin and could be reflected in eRNA effects. We therefore determined the changes in histone modifications at the ADAM28 and ADAMDEC1 promoters, Enhancer 1, and Enhancer 2 after LPS stimulation (Figure 5). H3K4me3 and H3K27ac at the ADAM28 promoter did not exhibit any changes with LPS stimulation, consistent with its lack of regulation by that stimulus in this early timeframe. The ADAMDEC1 promoter and Enhancer 2 exhibited increased H3K27ac after LPS with Enhancer 2 also having slightly increased H3K4me3. These findings support the important role for Enhancer 2 in the regulation of ADAMDEC1. NFκB p65 and c-JUN, as a downstream effector of the MAP kinase pathway, were also examined. We observed that c-JUN was inducibly loaded onto Enhancer 1 and the ADAM28 promoter after LPS. Thus, the differences in expression are not due to a lack of transcription factor binding at ADAM28. We did not identify NFκB or c-Jun binding on the ADAMDEC1 promoter, possibly because our primers amplified only the proximal promoter. Enhancer 2 exhibited increased NFκB p65 after LPS. The inhibitor data supports a role for c-JUN in the regulation of ADAMDEC1, in spite of not observing acute loading at the proximal promoter or Enhancer 2. These data collectively demonstrate critical chromatin changes at the ADAMDEC1 promoter and enhancer related to LPS stimulation. Increased H3K27ac is associated with activation of enhancers and is therefore biologically relevant.
Figure 5. ChIP-seq of the enhancer regions.

MonoMac6 cells were treated with 1μg/ml LPS for 90 minutes. ChIP assays were performed with antibodies H3K4me3, H3K27ac, c-JUN and P65. In resting cells, H3K4me3 was highest at Enhancer 1. LPS stimulation let to increased c-JUN at Enhancer 1, ADAM28 promoter, H3K27ac at the ADAMDEC1 promoter and Enhancer 2 as well as binding of p65 to the ADAM28 promoter and Enhancer 2. (n=4, error bars represent SD, * indicates p<0.05, ** p<0.01).
4.4. Regulation of eRNA expression
We hypothesized that these histone modifications were central to the transcriptional activation of ADAMDEC1 and not simply epiphenomenon. To gain mechanistic insights, we examined the effect of chromatin modification inhibitors on the expression of the eRNAs and the mRNAs (Figure 6). C646 was used as an inhibitor of p300, iBET 151 was used as an inhibitor of bromodomains such as BRD3 and BRD4, regulating elongation, and DRB was used as an inhibitor of P-TEFb, critical for regulating release from pausing. Most inducible genes are regulated at the level of pause-release (Adelman and Lis, 2012). Expression of TNF was used as a pause-release control gene for the inhibitors and LPS induction of expression. GAPDH was used as a non-inducible control. ADAM28 was not induced by LPS and the inhibitors had no effect on expression levels. ADAMDEC1 expression was markedly inhibited by iBET151, DRB and C646, consistent with enhancer regulation of pause-release. The eRNAs at Enhancer 2 (157, 158) were also strongly inhibited by DRB and iBET with some inhibition by C646. Only eRNA 142 from Enhancer 1 had a similar pattern. These data again suggest that Enhancer 2 is more tightly tied to the ADAMDEC1 promoter than Enhancer 1 although it does appear that Enhancer 1 is expressed and may contribute to gene regulation tangentially. One of the key functions of eRNAs is to activate the acetylase function of CBP and p300, pivotal enhancer-binding proteins (Bose et al., 2017). We hypothesized that acetylation of histones by CBP or p300 could be responsible for the increased H3K27ac observed at ADAMDEC1 after stimulation.
Figure 6. The effect of chromatin inhibitors on transcript levels.

Primary monocytes from healthy controls were pretreated with 20μM C646, 40μM DRB or 10 μM iBET for 30 minutes before adding 1μg/ml LPS. Total RNA was isolated and qRT-PCR was used to measure the enhancer RNA or mRNA level. DRB and iBET uniformly led to diminished RNA levels. The effect of C646 was more variable (n=4, error bars represent SD, * indicates p<0.05, ** p<0.01). The y-axis represents fold change over LPS-treated cells.
4.5. P300 binds eRNAs
CBP and p300 are highly homologous proteins that drive the stimulus-inducible histone acetylation at enhancers (Bose et al., 2017). Additionally, we previously reported the binding of eRNAs to NELF and PTEF-b in the setting of pause-release transcriptional activation of SERPINB2 (Shi et al., 2017). We therefore examined the binding of the eRNAs to CDK9 (a component of PTEF-b), NELF-A, CBP, and p300 by RNA -immunoprecipitation (RNA-IP). With very rapid kinetics after stimulation, we saw binding of Enhancer 1 (122, 142) and Enhancer 2 (157) eRNAs to p300. (Figure 7). The effect was transient and had returned to baseline at 30 minutes. CDK9 binding was only observed for Enhancer 1 eRNAs (122, 142). NELF-A binding to 157 and 122 also exhibited rapid kinetics after LPS. A control eRNA that is not inducible by LPS (477) did not change with stimulation. None of the eRNAs exhibited binding by CBP. These studies suggested that p300 was the acetyltransferase responsible for binding to eRNAs and writing the H3K27ac mark at the enhancer and promoter.
Figure 7. LPS induces NELF and P300 binding to eRNAs.

MonoMac6 cells were treated with 1μg/ml LPS for 0,10 and 30 minutes. RNA-IPs were performed using NELF-A, CDK9 (a component of P-TEFb), P300 or CBP antibodies. Bound RNAs were detected by qRT-PCR. 477 is an enhancer RNA located on chromosome 1 but not induced by LPS. Results are the means plus standard deviation (n=4). Symbols indicate statistical significance: * indicates p<0.05, ** p<0.01.
To confirm the role of p300, we performed a ChIP assay for p300, reasoning that if the p300 bound to eRNAs was active, we should observe it on the enhancer DNA after LPS. Indeed, LPS stimulation increased p300 on both Enhancer 1 and Enhancer 2. In cell differentiation, p300 marks super-enhancers required for cell fate decisions (Witte et al., 2015). Recruitment in that setting is often dependent on NFκB (Holmqvist et al., 2012; Ong and Corces, 2011; Rikitake and Moran, 1992; Visel et al., 2009). We investigated whether NFκB was critical for p300 recruitment in response to LPS in our model system by using JSH-23, a specific NFκB inhibitor (Figure 8). JSH-23 was already demonstrated to diminish mRNA and eRNA transcript levels for ADAMDEC1 In this p300 ChIP analysis, JSH-23 led to diminished p300 recruitment after LPS at Enhancer 1 and Enhancer 2. These data support a role for NFκB in the early stages of enhancer activation at the ADAM28 and ADAMDEC1 locus through the regulation of p300 recruitment to enhancers after stimulation.
Figure 8. NFκB regulating p300 recruitment after LPS induction.

MonoMac6 cells were pretreated with NFκB inhibitor JSH for 30 minutes and then stimulated with 1μg/ml LPS for 15 minutes. ChIP assays were performed using P300 or GST antibodies. Bound promoter and enhancer were detected by qPCR. NFκB inhibition led to diminished recruitment of P300 to Enhancer 1 and Enhancer 2. Results are the means plus standard deviation (n=4, * p<0.05).
5.0. Discussion
This analysis took advantage of two highly homologous ADAM family genes with discordant regulation. ADAMDEC1, upregulated in monocytes in SLE, and ADAM28 which has been functionally implicated in SLE pathogenesis but which had normal expression in SLE monocytes, were examined in detail in this study. In a rigorous kinetic analysis, Enhancer 1, closest to ADAM28, had slower upregulation in response to LPS than the eRNAs from Enhancer 2, closest to ADAMDEC1. We did not perform detailed time course studies to determine if late events after LPS could be responsible for Enhancer 1 regulating ADAM28 since the time frame suggested indirect regulation. We also observed that eRNA 157 from Enhancer 2 was functionally important for the up-regulation of ADAMDEC1 after LPS. The kinetics and ASO effects suggest that Enhancer 2 is primarily responsible for ADAMDEC1 regulation, however, we did not rigorously exclude a late role for Enhancer 1. H3K27ac was strongly upregulated after LPS at Enhancer 2, whereas that of Enhancer 1 was not changed and Enhancer 1 had much higher basal H3K4me3, a mark found in lesser amounts at active enhancers (Djebali et al., 2012). In other ways, the behavior of the two enhancers was comparable. Both enhancers had inducible binding of p300 that in turn bound the eRNAs. Both enhancers exhibited p300 binding to chromatin that was dependent on NFκB. These data provide important gene-level support for a model of enhancer activation that has been largely proposed based on genome-wide level ChIP-seq and other next generation sequencing approaches.
The model for activation of transcription in the setting of inducible expression at pause-release promoters involves early events setting the stage for eRNA production. These include establishing H3K4me1and recruitment of BRD4 (Dorighi et al., 2017; J. E. Lee et al., 2017). These early stages are known to be important for recruitment of RNA polymerase II and the Mediator complex (Jeronimo et al., 2016). Our study did not analyze those early steps because the permissive landscape is often established in development. For pause-release genes such as most of those responding to LPS, RNA polymerase II is loaded onto the promoter and short 60–80nt transcripts are produced with RNA polymerase blocked from elongation by NELF and DSIF (Patel et al., 2013). Release from pausing requires P-TEFb which we analyzed by looking for CDK9, the RNA-binding subunit of P-TEFb. We found that Enhancer 1 eRNA bound to CDK9 after stimulation but not eRNAs from Enhancer 2. Whether this reflects a non-pause-release mechanism for Enhancer 2 regulation of ADAMDEC1 is not known. We also found NFκB-dependent recruitment of p300 to the enhancers extending our understanding of the mechanisms regulating p300 recruitment (Ong and Corces, 2011). CBP and p300 have been examined as scaffolding proteins supporting NFκB-regulated promoters (Kamitani et al., 2011; Li et al., 2018; Mukherjee et al., 2013). Thus, interactions between NFκB and p300 are well described at promoters. Less well understood are their interactions at enhancers. H3K27ac and p300 are often used to define enhancers bioinformatically (Creyghton et al., 2010). When an inducible system was used, activation of transcription was associated with acquisition of p300 at enhancers (Ramos et al., 2010). P300 has been found to mark enhancers that are co-bound by NFκB and in our studies, inhibition of NFκB led to diminished recruitment of p300 to the enhancers after LPS stimulation, a novel finding. Our studies confirm the prevailing model for enhancer activation in inducible systems and extend the data to a single locus where the details were elucidated mechanistically and further define the stepwise acquisition of transcriptional activation through the NFκB-mediated recruitment of p300 at some enhancers.
The finding of CDK9 binding eRNAs at Enhancer 1 suggested that the enhancer role might be primarily in pause-release. This is not surprising since most LPS-inducible genes are regulated by pause-release. Only Enhancer 2, however, exhibited binding to NELF, a feature we previously identified as part of pause-release at the SERPINB2 enhancer (Shi et al., 2017). It is possible that the enhancers collaborate to regulate expression with high fidelity. While it remains difficult to extrapolate these findings to precise mechanistic differences between the regulation of ADAM28 and ADAMDEC1, it is clear that they have basal and inducible chromatin features that are distinct. Inducible H3K27ac was seen only at the ADAMDEC1 promoter and Enhancer 2, a process thought to be secondary to eRNA binding to p300 or CBP at the site. Yet both Enhancer 1 and Enhancer 2 eRNAs bound to p300 and p300 was correctly positioned on DNA at both enhancers. We cannot exclude that H3K27ac appears later at the ADAM28 and Enhancer 1 sites.
Although the distinction between ADAM28 and ADAMDEC1 regulation remains less than fully elucidated, these studies are extremely important in demonstrating directly the pivotal role for eRNAs in the transcriptional regulation of ADAMDEC1. There are still very few examples where eRNA function has been directly demonstrated. We showed that knockdown of Enhancer 2 eRNA 157 blunted the response to LPS. Additionally, p300 at Enhancer 2 bound eRNA157 and both the ADAMDEC1 promoter and Enhancer 2 exhibited increased H3K27ac after stimulation, consistent with activation of the acetylase function of p300 by the eRNAs (Bose et al., 2017). Chemical inhibition of p300 by C646 also led to a blunted transcriptional response of ADAMDEC1 to LPS, supporting a role specifically for the acetylase function being required for transcriptional activation. Additionally, p300 recruitment to the enhancer was diminished by NFκB inhibitor treatment.
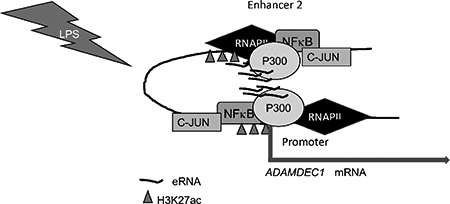
In our model, the paused state at the ADAMDEC1 promoter is maintained by NELF and DSIF. After LPS, NFκB is rapidly recruited to Enhancer 2, leading to assembly of a p300-NFκB complex on Enhancer 2. After eRNA production, p300 activation occurs, leading to increased H3K27ac at both the enhancer and promoter. The eRNA binds to CDK9 and NELF-A on the promoter, serving to activate the super-elongation complex and relieve pausing. Collectively, these data represent key insights into the regulation of a gene commonly affected by inflammatory diseases. The production of eRNAs represents a rapid-on-rapid-off kick-start for transcriptional activation. Recognition of these mechanisms is of critical importance as therapeutics directed at bromodomains and methyltransferases are being piloted for humans (Copeland et al., 2009; Liu et al., 2017; Perez-Salvia and Esteller, 2017; Zhao et al., 2017). Off-target effects on responses to inflammatory stimuli may occur and a better understanding of the transcriptional regulation allows for prediction of effects on the immune system.
6.0. Conclusions
P300 represents the pivotal histone acetyltransferase for the ADAMDEC1 enhancer. Given the role of ADAMDEC1 in inflammatory diseases, a complete understanding of its regulation is imperative. These data demonstrate a complex interplay between eRNAs and p300 with NFκB linked to p300 recruitment leading to transcriptional activation.
Highlights.
LPS induction of ADAMDEC1 expression is regulated by enhancer RNAs
Enhancer RNAs interact with p300
Histone acetylation is upregulated by LPS at the ADAMDEC1 enhancer
Acknowledgement
The authors would like to thank the patients.
Formatted funding sources
R01AR058547, R01AR043727 and R01AR069572.
Funding details: This work was supported by the Wallace Chair of Pediatrics and NIH grant R01AR058547. The Hopkins site was supported by NIH R01AR043727 and R01AR069572.
7.0. Glossary
- Pause-release:
RNA polymerase II arrests 60–80bp downstream of the transcriptional start site until a signal leads to P-TEFb releasing DSIF and NELF from pausing RNA polymerase
- Enhancer RNA:
Transcription of enhancers is central to enhancer function, however, the mechanism by which it elicits its effect is not understood. A key aspect is activation of P300 and CBP histone acetyltransferase activity
- Chromatin immunoprecipitation:
This technique captures protein with attendant DNA within a population of cells
Abbreviations
- ADAM
A Disintegration And Metalloproteinase
- CDK9
Cyclin dependent kinase 9
- DSIF
DRB sensitivity inducing factor
- LPS
Lipopolysaccharide
- NELF
Negative elongation factor
- RNAPII
RNA polymerase II
- SLE
Systemic lupus erythematosus
Footnotes
Conflict of interest
The authors have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adelman K, Lis JT, 2012. Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat Rev Genet 13, 720–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfandari D, Cousin H, Gaultier A, Smith K, White JM, Darribere T, DeSimone DW, 2001. Xenopus ADAM 13 is a metalloprotease required for cranial neural crest-cell migration. Curr. Biol 11, 918–930. [DOI] [PubMed] [Google Scholar]
- Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, Nelson N, Boiani N, Schooley KA, Gerhart M, Davis R, Fitzner JN, Johnson RS, Paxton RJ, March CJ, Cerretti DP, 1997. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 385, 729–733. [DOI] [PubMed] [Google Scholar]
- Blobel CP, 2005. ADAMs: key components in EGFR signalling and development. Nat. Rev. Mol. Cell Biol 6, 32–43. [DOI] [PubMed] [Google Scholar]
- Bose DA, Donahue G, Reinberg D, Shiekhattar R, Bonasio R, Berger SL, 2017. RNA Binding to CBP Stimulates Histone Acetylation and Transcription. Cell 168, 135–149 e122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers EM, Yan G, Mukherjee C, Orry A, Wang L, Holbert MA, Crump NT, Hazzalin CA, Liszczak G, Yuan H, Larocca C, Saldanha SA, Abagyan R, Sun Y, Meyers DJ, Marmorstein R, Mahadevan LC, Alani RM, Cole PA, 2010. Virtual ligand screening of the p300/CBP histone acetyltransferase: identification of a selective small molecule inhibitor. Chem. Biol 17, 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Sampson NS, 1999. Mediation of sperm-egg fusion: evidence that mouse egg alpha6beta1 integrin is the receptor for sperm fertilinbeta. Chem. Biol 6, 1–10. [DOI] [PubMed] [Google Scholar]
- Copeland RA, Solomon ME, Richon VM, 2009. Protein methyltransferases as a target class for drug discovery. Nat Rev Drug Discov 8, 724–732. [DOI] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, Boyer LA, Young RA, Jaenisch R, 2010. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. U. S. A 107, 21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, Xue C, Marinov GK, Khatun J, Williams BA, Zaleski C, Rozowsky J, Roder M, Kokocinski F, Abdelhamid RF, Alioto T, Antoshechkin I, Baer MT, Bar NS, Batut P, Bell K, Bell I, Chakrabortty S, Chen X, Chrast J, Curado J, Derrien T, Drenkow J, Dumais E, Dumais J, Duttagupta R, Falconnet E, Fastuca M, Fejes-Toth K, Ferreira P, Foissac S, Fullwood MJ, Gao H, Gonzalez D, Gordon A, Gunawardena H, Howald C, Jha S, Johnson R, Kapranov P, King B, Kingswood C, Luo OJ, Park E, Persaud K, Preall JB, Ribeca P, Risk B, Robyr D, Sammeth M, Schaffer L, See LH, Shahab A, Skancke J, Suzuki AM, Takahashi H, Tilgner H, Trout D, Walters N, Wang H, Wrobel J, Yu Y, Ruan X, Hayashizaki Y, Harrow J, Gerstein M, Hubbard T, Reymond A, Antonarakis SE, Hannon G, Giddings MC, Ruan Y, Wold B, Carninci P, Guigo R, Gingeras TR, 2012. Landscape of transcription in human cells. Nature 489, 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorighi KM, Swigut T, Henriques T, Bhanu NV, Scruggs BS, Nady N, Still CD 2nd, Garcia BA, Adelman K, Wysocka J, 2017. Mll3 and Mll4 Facilitate Enhancer RNA Synthesis and Transcription from Promoters Independently of H3K4 Monomethylation. Mol. Cell 66, 568–576 e564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsche J, Muller A, Hausmann M, Rogler G, Andreesen R, Kreutz M, 2003. Inverse regulation of the ADAM-family members, decysin and MADDAM/ADAM19 during monocyte differentiation. Immunology 110, 450–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galamb O, Gyorffy B, Sipos F, Spisak S, Nemeth AM, Miheller P, Tulassay Z, Dinya E, Molnar B, 2008. Inflammation, adenoma and cancer: objective classification of colon biopsy specimens with gene expression signature. Dis. Markers 25, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett S, Dietzmann-Maurer K, Song L, Sullivan KE, 2008. Polarization of primary human monocytes by IFN-gamma induces chromatin changes and recruits RNA Pol II to the TNF-alpha promoter. J. Immunol 180, 5257–5266. [DOI] [PubMed] [Google Scholar]
- Han DC, Rodriguez LG, Guan JL, 2001. Identification of a novel interaction between integrin beta1 and 14–3-3beta. Oncogene 20, 346–357. [DOI] [PubMed] [Google Scholar]
- Holmqvist PH, Boija A, Philip P, Crona F, Stenberg P, Mannervik M, 2012. Preferential genome targeting of the CBP co-activator by Rel and Smad proteins in early Drosophila melanogaster embryos. PLoS Genet 8, e1002769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeronimo C, Langelier MF, Bataille AR, Pascal JM, Pugh BF, Robert F, 2016. Tail and Kinase Modules Differently Regulate Core Mediator Recruitment and Function In Vivo. Mol. Cell 64, 455–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamitani S, Togi S, Ikeda O, Nakasuji M, Sakauchi A, Sekine Y, Muromoto R, Oritani K, Matsuda T, 2011. Kruppel-associated box-associated protein 1 negatively regulates TNFalpha-induced NF-kappaB transcriptional activity by influencing the interactions among STAT3, p300, and NF-kappaB/p65. J. Immunol 187, 2476–2483. [DOI] [PubMed] [Google Scholar]
- Lee JE, Park YK, Park S, Jang Y, Waring N, Dey A, Ozato K, Lai B, Peng W, Ge K, 2017. Brd4 binds to active enhancers to control cell identity gene induction in adipogenesis and myogenesis. Nat Commun 8, 2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Kim NA, Sanford A, Sullivan KE, 2003. Histone acetylation and chromatin conformation are regulated separately at the TNF alpha promoter in monocytes and macrophages. J. Leukoc. Biol 73, 862–871. [DOI] [PubMed] [Google Scholar]
- Li Y, Li X, He K, Li B, Liu K, Qi J, Wang H, Wang Y, Luo W, 2018. C-peptide prevents NF-kappaB from recruiting p300 and binding to the inos promoter in diabetic nephropathy. FASEB J. 32, 2269–2279. [DOI] [PubMed] [Google Scholar]
- Liu Z, Wang P, Chen H, Wold EA, Tian B, Brasier AR, Zhou J, 2017. Drug Discovery Targeting Bromodomain-Containing Protein 4. J. Med. Chem 60, 4533–4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maretzky T, Reiss K, Ludwig A, Buchholz J, Scholz F, Proksch E, de Strooper B, Hartmann D, Saftig P, 2005. ADAM10 mediates E-cadherin shedding and regulates epithelial cell-cell adhesion, migration, and beta-catenin translocation. Proc. Natl. Acad. Sci. U. S. A 102, 9182–9187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinn OJ, English WR, Roberts S, Ager A, Newham P, Murphy G, 2011. Modulation of integrin alpha4beta1 by ADAM28 promotes lymphocyte adhesion and transendothelial migration. Cell Biol. Int 35, 1043–1053. [DOI] [PubMed] [Google Scholar]
- Mitsui Y, Mochizuki S, Kodama T, Shimoda M, Ohtsuka T, Shiomi T, Chijiiwa M, Ikeda T, Kitajima M, Okada Y, 2006. ADAM28 is overexpressed in human breast carcinomas: implications for carcinoma cell proliferation through cleavage of insulin-like growth factor binding protein-3. Cancer Res. 66, 9913–9920. [DOI] [PubMed] [Google Scholar]
- Miyamae Y, Mochizuki S, Shimoda M, Ohara K, Abe H, Yamashita S, Kazuno S, Ohtsuka T, Ochiai H, Kitagawa Y, Okada Y, 2016. ADAM28 is expressed by epithelial cells in human normal tissues and protects from C1q-induced cell death. FEBS J 283, 1574–1594. [DOI] [PubMed] [Google Scholar]
- Mochizuki S, Soejima K, Shimoda M, Abe H, Sasaki A, Okano HJ, Okano H, Okada Y, 2012. Effect of ADAM28 on carcinoma cell metastasis by cleavage of von Willebrand factor. J. Natl. Cancer Inst 104, 906–922. [DOI] [PubMed] [Google Scholar]
- Mochizuki S, Tanaka R, Shimoda M, Onuma J, Fujii Y, Jinno H, Okada Y, 2010. Connective tissue growth factor is a substrate of ADAM28. Biochem. Biophys. Res. Commun 402, 651–657. [DOI] [PubMed] [Google Scholar]
- Moss ML, Lambert MH, 2002. Shedding of membrane proteins by ADAM family proteases. Essays Biochem. 38, 141–153. [DOI] [PubMed] [Google Scholar]
- Mukherjee SP, Behar M, Birnbaum HA, Hoffmann A, Wright PE, Ghosh G, 2013. Analysis of the RelA:CBP/p300 interaction reveals its involvement in NF-kappaB-driven transcription. PLoS Biol. 11, e1001647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niranjanakumari S, Lasda E, Brazas R, Garcia-Blanco MA, 2002. Reversible cross-linking combined with immunoprecipitation to study RNA-protein interactions in vivo. Methods 26, 182–190. [DOI] [PubMed] [Google Scholar]
- O’Shea NR, Chew TS, Dunne J, Marnane R, Nedjat-Shokouhi B, Smith PJ, Bloom SL, Smith AM, Segal AW, 2016. Critical Role of the Disintegrin Metalloprotease ADAM-like Decysin-1 [ADAMDEC1] for Intestinal Immunity and Inflammation. J Crohns Colitis 10, 1417–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsu H, Dempsey PJ, Eguchi S, 2006. ADAMs as mediators of EGF receptor transactivation by G protein-coupled receptors. Am. J. Physiol. Cell Physiol 291, C1–10. [DOI] [PubMed] [Google Scholar]
- Ohtsuka T, Shiomi T, Shimoda M, Kodama T, Amour A, Murphy G, Ohuchi E, Kobayashi K, Okada Y, 2006. ADAM28 is overexpressed in human non-small cell lung carcinomas and correlates with cell proliferation and lymph node metastasis. Int. J. Cancer 118, 263–273. [DOI] [PubMed] [Google Scholar]
- Ong CT, Corces VG, 2011. Enhancer function: new insights into the regulation of tissuespecific gene expression. Nat Rev Genet 12, 283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, Lai F, Zytnicki M, Notredame C, Huang Q, Guigo R, Shiekhattar R, 2010. Long noncoding RNAs with enhancer-like function in human cells. Cell 143, 46–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel MC, Debrosse M, Smith M, Dey A, Huynh W, Sarai N, Heightman TD, Tamura T, Ozato K, 2013. BRD4 coordinates recruitment of pause release factor P-TEFb and the pausing complex NELF/DSIF to regulate transcription elongation of interferon-stimulated genes. Mol. Cell. Biol 33, 2497–2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Salvia M, Esteller M, 2017. Bromodomain inhibitors and cancer therapy: From structures to applications. Epigenetics 12, 323–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos YF, Hestand MS, Verlaan M, Krabbendam E, Ariyurek Y, van Galen M, van Dam H, van Ommen GJ, den Dunnen JT, Zantema A, t Hoen PA, 2010. Genome-wide assessment of differential roles for p300 and CBP in transcription regulation. Nucleic Acids Res. 38, 5396–5408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss K, Saftig P, 2009. The “a disintegrin and metalloprotease” (ADAM) family of sheddases: physiological and cellular functions. Semin. Cell Dev. Biol 20, 126–137. [DOI] [PubMed] [Google Scholar]
- Rikitake Y, Moran E, 1992. DNA-binding properties of the E1A-associated 300-kilodalton protein. Mol. Cell. Biol 12, 2826–2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer B, Marg B, Gschwind A, Ullrich A, 2004. Distinct ADAM metalloproteinases regulate G protein-coupled receptor-induced cell proliferation and survival. J. Biol. Chem 279, 47929–47938. [DOI] [PubMed] [Google Scholar]
- Seals DF, Courtneidge SA, 2003. The ADAMs family of metalloproteases: multidomain proteins with multiple functions. Genes Dev. 17, 7–30. [DOI] [PubMed] [Google Scholar]
- Shi L, Song L, Maurer K, Zhang Z, Sullivan K, 2017. SERPINB2 is regulated by dynamic interactions with pause release proteins and enhancer RNAs. Mol. Immunol 88, 20–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Zhang Z, Song L, Leung YT, Petri MA, Sullivan KE, 2015. Monocyte enhancers are highly altered in systemic lupus erythematosus. Epigenomics 7, 921–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Zhang Z, Yu AM, Wang W, Wei Z, Akhter E, Maurer K, Reis PC, Song L, Petri M, Sullivan KE, 2014. The SLE Transcriptome Exhibits Evidence of Chronic Endotoxin Exposure and Has Widespread Dysregulation of Non-Coding and Coding RNAs. PLoS One 9, e93846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin HM, Kim MH, Kim BH, Jung SH, Kim YS, Park HJ, Hong JT, Min KR, Kim Y, 2004. Inhibitory action of novel aromatic diamine compound on lipopolysaccharide-induced nuclear translocation of NF-kappaB without affecting IkappaB degradation. FEBS Lett. 571, 50–54. [DOI] [PubMed] [Google Scholar]
- Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, Afzal V, Ren B, Rubin EM, Pennacchio LA, 2009. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457, 854–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte S, Bradley A, Enright AJ, Muljo SA, 2015. High-density P300 enhancers control cell state transitions. BMC Genomics 16, 903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worley JR, Hughes DA, Dozio N, Gavrilovic J, Sampson MJ, 2007. Low density lipoprotein from patients with Type 2 diabetes increases expression of monocyte matrix metalloproteinase and ADAM metalloproteinase genes. Cardiovasc. Diabetol 6, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y, Shirakabe K, Werb Z, 2002. The metalloprotease Kuzbanian (ADAM10) mediates the transactivation of EGF receptor by G protein-coupled receptors. J. Cell Biol 158, 221–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Yik JH, Chen R, He N, Jang MK, Ozato K, Zhou Q, 2005. Recruitment of PTEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 19, 535–545. [DOI] [PubMed] [Google Scholar]
- Yankulov K, Yamashita K, Roy R, Egly JM, Bentley DL, 1995. The transcriptional elongation inhibitor 5,6-dichloro-1-beta-D-ribofuranosylbenzimidazole inhibits transcription factor IIH-associated protein kinase. J. Biol. Chem 270, 23922–23925. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Shi L, Song L, Maurer K, Petri M, Sullivan K, 2018a. Overall Downregulation of mRNAs and Enrichment of H3K4me3 Change near Genome-Wide Association Study Signals in Systemic Lupus Erythematosus: Cell-Specific Effects. Front. Immunol 9, article 497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Shi L, Song L, Maurer K, Petri MA, Sullivan KE, 2018b. Overall Downregulation of mRNAs and Enrichment of H3K4me3 Change Near Genome-Wide Association Study Signals in Systemic Lupus Erythematosus: Cell-Specific Effects. Front. Immunol 9, 497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Wang Y, Wang Y, Liu Y, Gao S, 2017. Histone methyltransferase TXR1 is required for both H3 and H3.3 lysine 27 methylation in the well-known ciliated protist Tetrahymena thermophila. Sci China Life Sci 60, 264–270. [DOI] [PubMed] [Google Scholar]
- Zhu P, Sun Y, Xu R, Sang Y, Zhao J, Liu G, Cai L, Li C, Zhao S, 2003. The interaction between ADAM 22 and 14–3-3zeta: regulation of cell adhesion and spreading. Biochem. Biophys. Res. Commun 301, 991–999. [DOI] [PubMed] [Google Scholar]