

Finding “Known Unknown” Chemicals: Suspect Screening in the Environment
High resolution mass spectrometry (HR-MS) has expanded assessment of chemical exposure in the environment well beyond screening for a limited subset of target (“known”) chemicals. “Suspect screening” has evolved into an efficient and popular way to screen for hundreds to thousands of chemicals of interest (the “known unknowns” or “suspects”) in complex samples, based initially only on their molecular formula and the resulting calculated exact mass [1]. However, the exact mass is insufficient evidence for unequivocal identification [2]. Challenges facing comprehensive suspect screening include increasing chemicals of interest (tens of thousands), as well as ever- decreasing detection limits, leading to increased false positives.1 Addressing these challenges requires a foundation of cheminformatics tools and database resources freely available to the community to enable exchange of information between diverse communities and the pooling of resources toward a common good. Thus, open science is poised to play a pivotal role in the evolution of suspect screening.
Chemical Data Curation
Collecting, curating and providing high quality chemical information from multiple data sources is extremely challenging, yet it is essential prior to performing high confidence suspect screening. The variety of data sources supporting chemical identification already makes it difficult to compare scientific results between institutions [1]. Assuming good data quality and correctness is common, yet detrimental to many studies requiring quality data sources, [3] leading to unintentional errors [1]. Challenges for chemical databases include encouraging scientists to submit high-quality data to online platforms and, in return, providing access to curated chemical structures and related data for suspect prioritization such as (1) experimental/ predicted properties; (2) toxicity data; (3) product occurrence/ functional use; (4) production volumes; (5) literature data; (6) previous detections. Accessing substance metadata is critical to the rapid tentative identification of “known unknowns”, [4] even with the most advanced in silico fragmentation methods. The latest Critical Assessment of Small Molecule Identification (CASMI) showed the essential role this metadata, or context, plays in high-throughput (semi-automated) identification, with 70% of 208 challenges ranked Top 1 when including metadata, versus 34% without (www.casmi-contest.org/2016/).
Each chemical data source comes with diverse identifiers (Figure 1), such as systematic, trivial and product names, molecular formula, CAS numbers (including active, alternate and deleted versions), database identifiers and/or structures (e.g. SMILES, InChI Strings, InChIKeys, MOL files). While a molecular formula is sufficient for suspect screening, calculating additional properties requires a structure – especially for in silico fragment confirmation, toxicity prediction or quantitative structure property relationships (QSPRs). Mass spectrometry (MS) cannot detect the commercial forms of many chemical substances (e.g. various salts or polymer mixes). Chemical databases address this limitation via “MS-ready” forms of the structures for lookup during suspect screening, while retaining the link to associated data from the commercial forms, as these “non-MS-ready” forms often contain more information (Figure 1). Once MS-ready forms are available, the calculation of a consistent set of physicochemical properties (e.g., physicochemical and environmental fate and transport) [3] is achievable for large open resources such as the CompTox Chemistry Dashboard. These open science resources offer a way to overcome many chemical curation challenges and help scientists focus on research questions.
Figure 1:
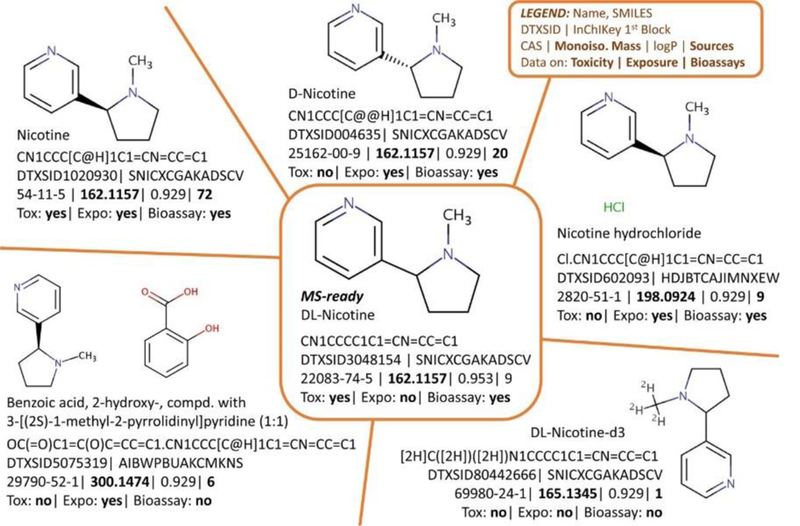
Chemical Curation and “MS-ready” structures demonstrated with Nicotine and selected data from the Chemistry Dashboard. MS will detect, e.g., [M + H]+ 163.1235 (structures top left, top middle, center), not salts or mixtures. Various toxicity, exposure, bioassay, and reference data exist for all forms (bold values).
Complex Mixtures and UVCBs: The Next Frontier
Even greater challenges exist for chemicals of Unknown or Variable composition, Complex reaction products, and Bio- logical materials, the UVCBs. Examples include chlorinated biphenyls, C10−C12 chloroalkanes, surfactant mixtures such as linear alkylbenzenesulfonates, biopesticides, and even polymer mixes. UVCB chemicals often have valuable data for prioritization (e.g., tonnage, functional use), yet many structures are absent from databases. We (the authors) are working toward saving representative UVCB structures into virtual chemical libraries, to enable the screening of these substances in the environment with HR-MS.
Open Science – Help Your Data Live!
Ideally, scientific data should be deposited into online resources for community access and requirements to do this are increasing at, for example, research institutes and funding agencies. For example, the Global Natural Products Social Molecular Networking (GNPS) system enables easy deposition of any raw data (e.g., MSV000079601), providing continuous monitoring and data interpretation in return.5 Many other repositories exist, with various pros and cons. However, even providing data in published manuscripts in machine-readable formats would be beneficial. As a central collection point, journals can and should support these efforts. Reporting chemical data with an appropriate structure identifier (e.g., one or more of SMILES, InChI String, InChIKey, PubChem ID, ChemSpider ID, or Dashboard DTXSID), not just a name and mass, is essential for unique identification and database upload. CAS numbers are problematic due to many commercial forms of a chemical (versus “MS-ready”, see Figure 1), as well as restricted (fee-based) access to check CAS systems. Including data, when feasible, in an open machine-readable format in Supporting Information files (e.g., tables in spreadsheet form) or repositories also facilitates database generation. The peak lists behind mass spectra−especially of new substances or transformation products elucidated in great detail−are extremely valuable, yet difficult to extract from figures. The upload of tentatively identified spectra (with confidence level tagging2) as Supporting Information via massbank.eu has already enabled cross-annotations of surfactants and trans- formation products in GNPS data sets. Saving data in open resources can benefit the whole community, supporting smarter suspect screening, prioritization, higher confidence identifica- tions (see refs 1 and 2), and even new discoveries with initiatives such as GNPS [5].
Outlook
HR-MS will detect the presence of chemicals in the environment, including those not yet captured in chemical databases such as “unknown unknowns” and those considered confidential business information. Deposition of high quality, curated open data on chemicals and environmental observa- tions will be vital for improving chemical identification with HR-MS, empowering international efforts to protect human and ecological health.
Acknowledgements:
ES is supported by SOLUTIONS (Grant 603437).
Footnotes
Disclaimer: The views expressed in this paper are those of the authors and do not necessarily reflect the views or policies of the US Environmental Protection Agency. Mention of trade names or commercial products does not constitute endorsement or recommendation for use.
References
- [1].Schymanski EL; Singer HP; Slobodnik J; Ipolyi IM; Oswald P; Krauss M; Schulze T; Haglund P; Letzel T; Grosse S; Thomaidis NS; Bletsou A; Zwiener C; Ibáñez M; Portolés T; De Boer R; Reid MJ; Onghena M; Kunkel U; Schulz W; Guillon A; Noyon N; Leroy G; Bados P; Bogialli S; Stipaničev D; Rostkowski P; Hollender J Anal. Bioanal. Chem. 2015, 407 (21), 6237–6255. [DOI] [PubMed] [Google Scholar]
- [2].Schymanski EL; Jeon J; Gulde R; Fenner K; Ruff M; Singer HP; Hollender J Environ. Sci. Technol. 2014, 48 (4) 2097–2098. [DOI] [PubMed] [Google Scholar]
- [3].Mansouri K; Grulke C; Judson R; Williams AJ SAR and QSAR in Environmental Research, 2016, 27:11, 911–937 [DOI] [PubMed] [Google Scholar]
- [4].McEachran AD; Sobus JR; Williams AJ Anal. Bioanal. Chem. 2017, 409 (7), 1729–1735. [DOI] [PubMed] [Google Scholar]
- [5].Wang M; Carver JJ; Phelan VV; Sanchez LM; Garg N; Peng Y; Nguyen DD; Watrous J; Kapono CA; Luzzatto-Knaan TW; et al. Nature Biotechnology, 2016, 34 (8), 828–837. [DOI] [PMC free article] [PubMed] [Google Scholar]