

Abstract
Phthalocyanines are an important class of industrial dyes with potential commercial applications ranging from photovoltaics to biomedical imaging and therapeutics. We previously demonstrated the versatility of the commercially available zinc(II) hexadecafluorophthalocyanine (ZnF16Pc) as a platform for rapidly developing functional materials for these applications and more. Because this core-platform approach to dye development is increasingly common, it is important to understand the photophysical and structural consequences of the substitution chemistry involved. We present a fundamental study of a series of ZnF16Pc derivatives in which the aromatic fluorine atoms are progressively substituted with thioalkanes. Clear spectroscopic trends are observed as the substituents change from electron-withdrawing to electron-releasing groups. Additionally, there is evidence for significant structural distortion of the normally planar heterocycle, with important ramifications for the photophysics. These results are also correlated to DFT calculations, which show that the orbital energies and symmetries are both important factors for explaining the excited-state dynamics.
Graphical Abstract
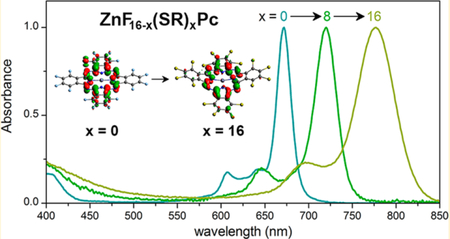
I. INTRODUCTION
Phthalocyanines (Pc) have been an important class of industrial dyes since their serendipitous discovery in the early 20th century.1,2 In addition to accounting for a large percentage of organic dyes currently in production,3 Pcs are also routinely investigated for applications ranging from solar energy conversion4–9 to thin film electronics10–15 to photodynamic therapy.16–21 The chemical and thermal stability of these compounds are augmented by diverse photonic properties, including remarkably high molar extinction coefficients and fluorescence efficiencies. It has been known since their inception that functionalizing the Pc core allows for increased solubility and ease of processing, while simultaneously allowing the aforementioned photonic properties to be systematically tuned because the substituents are directly appended to the macrocycle.
Many research groups are interested in 1,2,3,4,8,9,10,11,−15,16,17,18,22,23,24,25-hexadecafluorophthalocyanine (F16Pc), and its metal complexes (MF16Pc).4,6,10–13,18–25 The electron-withdrawing fluorine substituents slightly perturb the molecular orbitals causing a small bathochromic shift in the absorbance and fluorescence peaks, as well as an enhanced tendency toward aggregation.26 Moreover, the increased dipole moment of the carbon—fluorine bond and the electron-deficient nature of the fused benzenes make this molecule an ideal substrate for attack via nucleophilic aromatic substitution (SNAr).4,6,22,24,25,27,28 This synthetic strategy has proven extremely versatile, allowing the creation of a large variety of substituted Pcs that might not be otherwise accessible via traditional synthetic methods.29 Thiol, amine, and alcohol nucleophiles bearing a range of functional groups can be employed, and the extent of substitution can be carefully controlled by adjusting the reaction conditions.
We demonstrated4 an improved photovoltaic efficiency in bulk heterojunction solar cells fabricated from blends of ZnPcs appended with varying numbers of thioalkane chains. It was shown that successive substitution of the F atoms with these—SR groups on the ZnF16Pc platform causes a red shift in the absorbance Q-band, allowing a mixture of different dyes to cover a much broader range of the solar spectrum. Solar cells utilizing Pc dye blends were compared with cells made from only one dye component and were found to have a power conversion efficiency (PCE) greater than that predicted from summing the corresponding single-component device efficiencies. Surprisingly, this synergistic enhancement was achieved without the need to engineer the dyes into an energetically stepwise hierarchical structure at the molecular level. Using grazing incidence small-angle X-ray scattering, Jurow et al.6 found that the presence of the exocyclic thioalkane chains on the ZnPcs induced liquid crystal like behavior, resulting in an undesirable homogeneous alignment of aggregates parallel to the electrode surface. Further, they showed that incorporating zinc tetra-tert-butylphthalocyanine (Zn(tBu)4Pc) into the active layer increased the cell efficiencies by frustrating this unfavorable packing. It is possible that dye blends with widely varying numbers of thioalkyl substituents could produce the same effect without the need for Zn(tBu)4Pc.
Although molecular structure and packing order are clearly important in determining the PCE, there are many other factors such as photophysical properties to consider in engineering Pcs for solar energy conversion and other photonic applications. Herein we assess the photophysical properties through a rigorous spectroscopic study of the ZnF16−x(SR)xPc series of dyes (where x = 0–16). The photophysical properties of the molecules, including absorbance, fluorescence emission, and excited-state lifetime, are measured as a function of the number of thioalkyl substituents, wherein the specific substitution pattern is discussed below. We use density function theory (DFT) and time-dependent density function theory (TD-DFT) calculations to help support and explain the conclusions regarding the excited state and structural dynamics. A single chain length was used because the number of substituents is the relevant variable for these studies, but the trends observed will map onto other derivatives based on the ZnF16Pc platform under the same conditions. Using n-octylthio groups imparts the desired degree of solubility and processability without resulting in undesirable liquid crystalline behavior. For the theoretical calculations we have modeled the compounds with shorter n-butylthio chains to significantly reduce the computational time required, while still obtaining useful information regarding the electronic and molecular structure. Neither previous reports6,30 nor our own investigations show any difference in the inductive effects of substituents with different carbon chain lengths.
There are diverse applications for materials that harness the excited-state energy of efficient light absorbers such as Pcs, so a detailed understanding of the excited-state dynamics will be beneficial for areas such as solar energy conversion, photo-catalysis, biomedical imaging, photoacoustic spectroscopy, and photodynamic and photothermal therapies.21 The results presented are thus broadly applicable to the development of Pc systems wherein the substituent is appended to the conjugated macrocycle. For ZnF16Pc, the absorbance, fluorescence quantum yields, and excited-state lifetimes can all be systematically tuned by serial substitution of the F atoms on the macrocycle.4 We demonstrate for the first time that the molecular structure of a preformed Pc core can be significantly distorted by simple substitution chemistry. Disrupting the normally planar aromatic system induces drastic changes in the photophysics, with important consequences for potential applications. To date, syntheses of structurally distorted Pcs have only been accomplished through the cyclotetramerization of phthalonitriles or similar precursors already bearing bulky groups in the α position,31–37 thereby limiting the scope of this approach. In this context, exploiting the synthetic versatility of the ZnF16Pc platform is an important part of the rapid design and testing of Pc compounds for commercially viable applications.
II. EXPERIMENTAL SECTION
IIA. Materials, Instruments, and Methods.
[1, 2, 3, 4, 8, 9, 10, 11, 15, 16, 17, 18, 22, 23, 24, 25-Hexadecafluorophthalocyaninato]zinc(II) (ZnF16 Pc), [2,9,16,23-tetra(tert-butyl)phthalocyaninato]zinc(II) (Zn-(tBu)4Pc), tetrahydrofuran (THF), dichloromethane (DCM), acetone, ethyl acetate, petroleum ether, 1-octanethiol, potassium carbonate (K2CO3), sodium hydride (NaH), and anhydrous sodium sulfate (Na2SO4) were purchased from Sigma-Aldrich, Fisher Scientific or Acros Organics. Most reagents were used without further purification. 1-Octanethiol was dried over basic alumina immediately prior to use. THF was freshly distilled over sodium and benzophenone before use to eliminate water, peroxides, and butylated hydroxytoluene (BHT), a commercial stabilizer that has a pronounced fluorescence in the ultraviolet. Analytical thin-layer chromatography (TLC) was performed on polyester-backed TLC plates 254 (precoated, 200 μm, Sorbent Technologies). Preparative scale TLC was performed on glass-backed silica gel TLC plates (precoated, 1000 μm, Analtech). Silica gel 60 (70–230 mesh, Merck) was used for column chromatography.
1H NMR and 19F NMR spectra were recorded at the Hunter College NMR facility on a 500 MHz Bruker Avance and 400 MHz Bruker Avance III spectrometers, respectively. The 19F nucleus resonates at 376.5 MHz on the latter instrument. Proton chemical shifts are expressed in ppm relative to the residual peak of the solvent used, either CDCl3 (7.26 ppm, 1H) or (CD3)2CO (2.05 ppm, 1H). Fluorine chemical shifts are reported relative to trichlorofluoromethane (CCl3F, 0.00 ppm, 19F). Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry was performed at the Shared Instrument Facility at New York University using a Bruker UltrafleXtreme MALDI-TOF acquired through the support of the National Science Foundation under Award Number CHE-0958457. The matrix used was a 20 mg·mL−1 solution of 2,5-dihydroxybenzoic acid (DHB, Sigma) in a 30:70 v/v ratio mixture of THF to 0.1% trifluoroacetic acid (TFA) in water. For each sample, this matrix solution was used to make a submillimolar Pc solution and was subsequently spotted onto a ground steel target plate and allowed to air-dry. The instrument was operated in reflectance mode with positive ion detection. For the starting material of our compounds, ZnF16Pc, the intact molecular ion was observed and used as an external single-point calibration reference for all subsequent samples.
UV—visible absorbance spectra were obtained with a Lambda 35 UV—vis spectrophotometer from PerkinElmer. Calibration curves were generated from dilute solutions with absorbance less than approximately 0.1 at the λmax, as detailed in the Supporting Information. Steady-state fluorescence spectra were acquired on a HORIBA Scientific FluoroLog-3 fluorescence spectrometer. Quantum yields were also calculated from series of dilute solutions using the relative gradient method described in the Supporting Information, with Zn(tBu)4Pc in deaerated toluene as a standard. Fluorescence lifetimes were recorded using the FluoroHub Tau-3 time-correlated single photon counting (TCSPC) module for the same instrument. Excitation for TCSPC was provided by a pulsed diode laser source with a peak wavelength of either 653 nm (NanoLED-650L, HORIBA) or 667 nm (NanoLED-670L, HORIBA), each with a pulse duration of less than 200 ps. The data collected in this manner were subjected to standard multiexponential fitting using the Decay Analysis Software package (v. 6.4) bundled with the instrument.
DFT and TD-DFT calculations were performed with the Gaussian 09 software package using the resources of the City University of New York High Performance Computing Center based at the College of Staten Island and supported under National Science Foundation Grants CNS-0958379, CNS-0855217, and ACI-1126113. Avogadro 1.1.1 was used to construct the models, generate the input files, and process and visualize the orbital surfaces. All DFT and TD-DFT calculations were performed using the B3LYP hybrid exchange—correlation functional and the 6–31G(d,p) basis set. The THF solvent contribution was simulated using the polarizable continuum model (PCM). Ground-state optimizations were performed with equilibrium PCM solvation and accompanied by frequency calculations to ensure the absence of imaginary frequency modes which would indicate a transition state. Vertical excitation (i.e., absorption) energies were obtained by performing single-point TD-DFT calculations of the excited electronic states at the ground-state optimized nuclear geometry under nonequilibrium, state-specific PCM solvation conditions.
IIB. Synthesis.
The synthesis for most of the compounds in the series largely follows the procedure previously reported.4 In short, solid ZnF16Pc was added to dry THF under N2 along with either K2CO3 or NaH as a base. Then, 1-octanethiol was injected into the reaction vessel and brought to temperature for the specified time. The extent of substitution was controlled through a combination of stoichiometry, heat, and reaction duration, with higher thiol:Pc ratios, higher temperatures, and longer times producing more substituted products. For the most highly substituted products, sodium metal was used instead of base to generate the thiolate nucleophile in situ, as described below.
After the reaction was stopped, the THF was evaporated and the crude mixture was washed with deionized water and extracted into ethyl acetate. The organic layer was dried over Na2SO4 and filtered, and the ethyl acetate was removed under reduced pressure at low temperature to prevent oxidation of the sulfides to sulfoxides or sulfones. This was then passed through a silica column, first with hexanes to remove the unreacted thiol, followed by ethyl acetate to obtain a mixture of substituted phthalocyanines. The individual products were separated from this mixture by silica gel preparative scale TLC using a combination of ethyl acetate and hexanes as the eluent. The separated TLC bands were then scraped off and filtered through hydrophobic 0.20 μm PTFE syringe filter tips (Millipore) using ethyl acetate. See Supporting Information for spectroscopic and mass spectrometry characterization.
III. RESULTS AND DISCUSSION
IIIA. Synthesis and Characterization.
This series of Pcs was synthesized via nucleophilic substitution on the ZnF16Pc platform, as shown in Scheme 1. Considering only the number of substituents on the macrocycle, there are 16 possible products from the reaction described. For brevity, we refer to these as ZnF16−x(SR)xPc where x = 0–16, indicating the number of appended thioalkyl chains. This synthetic route is remarkably robust to variations in the reaction conditions, with virtually any base and any polar aprotic solvent being suitable. NaH greatly accelerated the reaction, reducing both the time and heat required for extensive substitution. The reaction is easily monitored by UV—vis absorbance spectroscopy, because there is a red shift in the absorbance peak of about 6 nm as each F atom is exchanged for a thioalkane group.
Scheme 1.
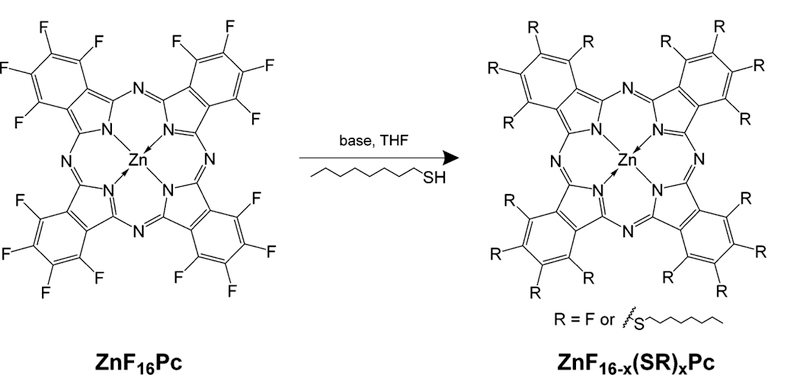
Control of Reaction Conditions and Stoichiometry of the Nucleophilic Aromatic Substitution of the Fluorine Groups on ZnF16Pc by Thiols Results in a Number of Different Substituted Products and Isomers
The starting ZnF16Pc material is a turquoise blue powder that becomes a brilliant green, sticky solid upon substitution. Eventually, after about 10 thiols have been added, the green color fades to a yellowish brown and the products become a thick, viscous oil. The addition of the long chain hydrocarbons at random positions frustrates the crystal packing of the material, which also prevents aggregation in solution and greatly enhances the solubility. The most highly substituted products are fully soluble even in nonpolar hexanes. However, extensive substitution is accompanied by its own set of issues, including more difficult separation and a decreased stability toward oxidation.38
IIIB. Statistical Analysis.
Given the nature of the reaction, discussion of the combinatorial statistics is warranted. The problem of analyzing the isomeric products of porphyrin and phthalocyanine condensation or substitution has been addressed.39–48 One approach to drug development is to generate large libraries of porphyrinoid products which are then tested and optimized for some desired application. For materials, however, mixtures of these dyes may have superior properties. Lindsey et al. presented a detailed discussion of this problem accompanied by a custom program (PorphyrinVi-LiGe) designed to generate a list of the potential products based on a statistical analysis.39–42 Although capable of handling complex reactions involving multiple substituents or precursors, Lindsey’s program has some limitations that prevent a direct analysis of the situation discussed here. Specifically, the simulated reaction types only allow for the derivatization of at most eight equivalent positions of the macrocycle, corresponding to either all β or all α substitution in the case of a Pc. The ZnF16Pc platform utilized may be substituted at any of the 16 fluorine positions, and to fully characterize the series of products, it is important to understand the distribution of isomers among them.
It is trivial to calculate the number of permutations of a given number of substituents around the 16 possible positions of the ZnF16Pc macrocycle. Given n substituents, there are 16Cn = 16!/[n!(16 − n)!] such combinations. However, not all of these arrangements correspond to physically distinct molecules. Many of them are merely rotations of one another under the proper symmetry subgroup of the D4h point group of the parent molecule. Though Pólya’s enumeration theorem may be used to count the true number of unique isomers under this symmetry group, it does not provide a detailed listing of the individual microstructures. To that end, we have written a program that takes the number of substitutions as an input and steps through every possible permutation according to a previously published algorithm.52 After this enumeration, the rotational transformations of the proper symmetry subgroup are applied to each permutation to compare it to the others, eliminate the duplicates, and count the remaining isomers.
The program code is given in the Supporting Information along with a more detailed description and plots (Figure S1) of the number of permutations and distinct isomers as a function of the number of substituents. The two graphs (A and B) shown in Figure S1 correspond to two different assumptions regarding the nature of the reaction. For the unrestricted plot, we assume that all 16 positions have an equal probability of being substituted during the reaction, and we therefore see a maximum number of 1,654 isomers for ZnF8(SR)8Pc. In the restricted plot, we assume that the more kinetically available β positions must all react first, with equal probability, before any α position is substituted. This condition, which is more representative of the actual reaction products, means that ZnF8(SR)8Pc will be nearly isomerically pure, whereas ZnF12(SR)4Pc and ZnF4(SR)12Pc have the largest number of possible isomers (13 each). It is impractical to separate all of the positional isomers, and for materials this is not a priori necessary. Thus, the macroscopic materials properties and spectroscopic data of any given Pc in this family actually represent a weighted statistical average of the ensemble of isomers.
IIIC. UV—Vis Spectroscopy.
The photophysical data are given in Table 1, and the UV—vis absorbance spectra are shown in Figure 1A. Calibration curves of absorbance vs molar concentration (Figures S3 and S4) demonstrate excellent linearity within the observed concentration range for all samples, confirming the applicability of the Beer—Lambert law and the absence of significant aggregation effects.
Table 1.
Photophysical Parameters for All Standards and Compounds Studied, in Dilute THF Solution
| compound | abs λmax (nm) | log ε | fluor λmax (nm) | ϕf | Stokes shift (nm) | τ (ns) | kf (ns−1) | knr (ns−1) |
|---|---|---|---|---|---|---|---|---|
| ZnPc | 666 | 5.48a | 671 | 0.100b | 5 | 3.38 | 0.030 | 0.266 |
| Zn(tBu)4Pc | 672 | 5.43c | 676 | 0.081d | 4 | 3.28 | 0.025 | 0.280 |
| ZnF16Pc | 672 | 5.24e | 678 | 0.057 | 6 | 2.75 | 0.021 | 0.343 |
| ZnF15(SR)Pc | 679 | 5.32 | 685 | 0.061 | 6 | 2.64 | 0.023 | 0.356 |
| ZnF14(SR)2Pc | 686 | 693 | 0.046 | 7 | 2.73 | 0.017 | 0.349 | |
| ZnF13(SR)3Pc | 692 | 5.08 | 701 | 0.037 | 9 | 2.52 | 0.015 | 0.382 |
| ZnF12(SR)4Pc | 698 | 5.44 | 707 | 0.042 | 9 | 2.53 | 0.017 | 0.379 |
| ZnF11(SR)5Pc | 704 | 5.36 | 713 | 0.033 | 9 | 2.27 | 0.015 | 0.426 |
| ZnF10(SR)6Pc | 708 | 5.29 | 718 | 0.034 | 10 | 2.20 | 0.015 | 0.439 |
| ZnF9(SR)7Pc | 716 | 5.46 | 724 | 0.030 | 8 | 2.15 | 0.014 | 0.451 |
| ZnF8(SR)8Pc | 720 | 5.47 | 729 | 0.025 | 9 | 2.08 | 0.012 | 0.469 |
| ZnF7(SR)9Pc | 727 | 743 | 16 | |||||
| ZnF6(SR)10Pc | 731 | 748 | 0.006 | 17 | 1.67f | 0.004 | 0.595 | |
| ZnF5(SR)11Pc | 733 | 4.97 | 753 | 0.009 | 20 | 1.59 | 0.006 | 0.623 |
| ZnF4(SR)12Pc | 740 | 4.89 | 761 | 0.005 | 21 | 1.33 | 0.004 | 0.748 |
| ZnF3(SR)13Pc | 749 | 5.12 | 771 | 0.005 | 22 | 1.29 | 0.004 | 0.771 |
| ZnF2(SR)14Pc | 755 | 778 | 0.004 | 23 | 1.30f | 0.003 | 0.766 | |
| ZnF(SR)15Pc | 768 | 796 | 28 | |||||
| Zn(SR)16Pc | 777 | 5.16 | 802 | 0.003 | 25 | 1.38 | 0.002 | 0.722 |
The extinction coefficient (log ε) of ZnPc in THF taken from ref 53.
Quantum yield of ZnPc in toluene calculated as the average of the values in refs 54 and 53.
The extinction coefficient (log ε) obtained for Zn(tBu)4Pc in THF matches ref 53 to within less than 0.2%.
Quantum yield of Zn(tBu)4Pc standard in toluene, taken directly from ref 53.
The extinction coefficient (log ε) obtained for ZnF16Pc in THF matches ref 16 to within less than 1.3%.
The decay required fitting with a second lifetime, close to the IRF width (<300 ps), as a minor component (<20%).
Figure 1.
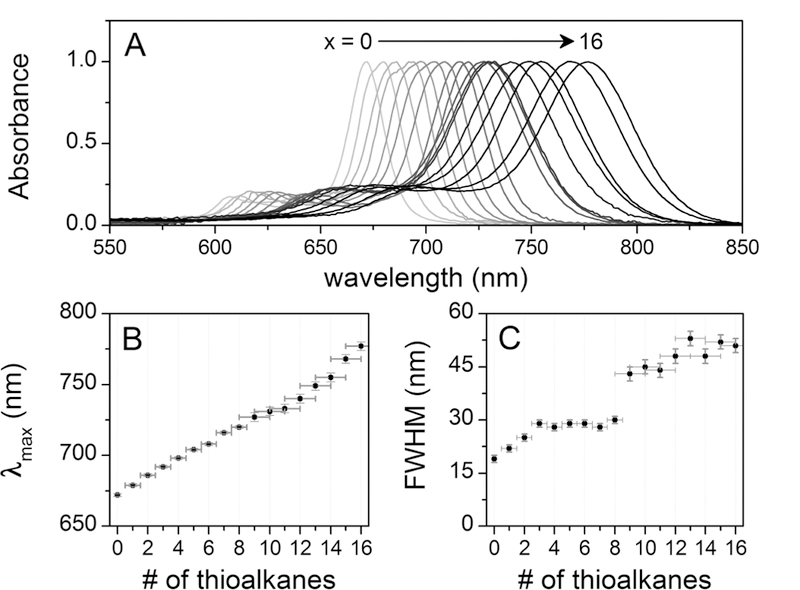
(A) Normalized absorbance spectra of compounds ZnF16−x(SR)xPc (x = 0–16). (B) Wavelength of maximum absorbance (λmax) as a function of the number of thioalkane substituents. (C) Peak widths as a function of the number of thioalkane substituents, as measured by the full-width at half-maximum (fwhm).
The intense color of Pcs is due mainly to the strong absorbance peak in the red region, the Q-band, and its higher energy vibronic satellite. As seen in Figure 1B, these bands shift approximately 6 nm to the red for each appended thioalkane chain. Pcs also have a higher energy B band, which can be seen in the expanded UV—visible plots given in Figure S2 in the Supporting Information. The B band falls mainly in the ultraviolet for the unsubstituted starting material but grows slightly in the visible as more thioalkane groups are added to the macrocycle.
In addition to the approximately linear increase in the absorbance λmax, there is also an increase in the full width at half-max (fwhm, Figure 1C). The fwhm increases slightly at first, and then discontinuously jumps by about 15 nm after eight thioalkyl substituents have been added to the core, followed by only slight increases thereafter. There are a few possible explanations for this broadening. First, the chromatographic separation becomes more difficult with increased substitution. As more thioalkanes are added, the preparative TLC bands become very close with even the most nonpolar eluents. Though this does lead to traces of other compounds with x ± 1 for the more substituted compounds, this does not cause the observed broadening and cannot account for the discontinuity. The broadening could also be the result of contributions from different positional isomers with slightly different absorption properties arising from small differences in the HOMO—LUMO gaps. However, if isomers were the primary cause of the Q-band broadening, the trends in the FWHW would be expected to generally correlate with the calculated distributions of isomers (Figure S1). For the case in which the β positions react first, ZnF12(SR)4Pc and ZnF4(SR)12Pc would have the broadest peaks, whereas ZnF8(SR)8Pc would have the broadest spectrum if the reaction were completely random. Because the observed changes in the peak widths do not correlate to either of these cases, the mixture of isomers is unlikely to be a dominant cause of the broadening.
A third potential explanation is that the broadening is due to fundamental changes in the electronic structure of the molecules as substituents are added. Breaking the symmetry of the degenerate LUMO orbitals, for instance, could cause the Q-band transitions to become nondegenerate.55 An extreme case of this is seen in free-base Pcs, in which the D4h symmetry has been completely broken to D2h, resulting in the Q-band splitting into two distinct peaks. An intermediate case might produce some of the broadening observed, as well as the asymmetry seen in some of the absorbance bands. It was reported that the asymmetry found in absorbance bands due to splitting is not mirrored in the emission band shape.55 This is an example of Kasha’s rule, which states that the initially formed excited state internally converts to the lower excited state before fluorescing. Several of the less substituted Pcs exhibit an asymmetry in their absorbance bands whereas all of the emission bands are nearly symmetrical. However, the emission does retain the broadness of the absorption bands (vide infra), indicating that Kasha’s rule is not relevant. Orbital splitting is therefore unlikely to cause all of the observed band broadening.
Because the width of the absorbance spectra is related to the vibrational structure of the ground and excited electronic states, the sharp increase in the fwhm after eight substitutions is best explained by a change in the conformational dynamics of the molecule. Substitution of the β positions is kinetically favored, and thus the first eight substitutions do not greatly alter the structure of the planar macrocycle. Upon substitution of the remaining α positions, however, steric interactions begin to either lower the energy barrier to nonplanar vibrations or force nonplanar distortions (vide infra).30–37,56–60
IIID. Steady-State Fluorescence Spectroscopy.
Fluorescence emission spectra of all of the compounds, including ZnF16Pc, are shown in Figure 2A. The peaks of the spectra are scaled to the experimental quantum yield values to highlight the quenching that occurs upon substitution. Like the absorbance spectra, the fluorescence bands also broaden as they shift toward the red. The quantum yields and Stokes shifts are plotted vs the number of substituents in Figure 2B,C, respectively.
Figure 2.
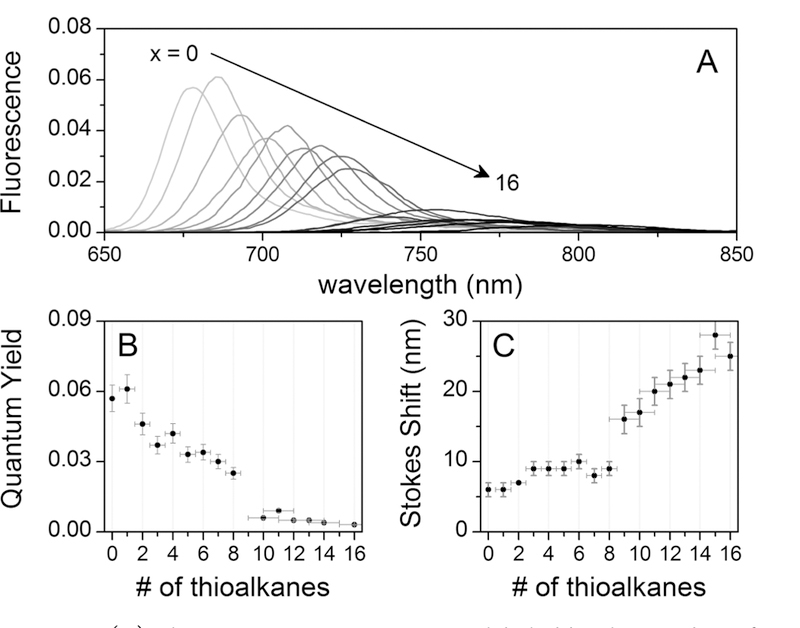
(A) Fluorescence emission spectra labeled by the number of thioalkane substituents, ZnF16−x(SR)xPc (x = 0–16). The peak heights are scaled to match the quantum yield of the compound. (B) Quantum yields as a function of the number of thioalkane substituents.(C) Stokes shifts as a function of the number of thioalkane substituents.
Accurate values for fluorescence quantum yields can be difficult to obtain experimentally, so there is a large range of literature values for any ostensible standard. It is often impossible to ascertain from the detail given whether these discrepancies arise from aggregation effects, the presence of molecular oxygen, inner filter effects, or other unknown causes. Pcs are especially vexing in this regard due to their well-documented tendency to aggregate. For instance, addition of 1% pyridine to coordinate the zinc and frustrate π-stacking apparently increases the solubility and quantum yields.61 However, we have preferred pure solvents for comparative purposes. As one example of the spread of literature values, Nyokong gives a ϕf value of 0.07 for ZnPc in deaerated toluene54 but then reports a value of 0.13 for the same compound relative to the same standard (chlorophyll a in ether) in a subsequent paper.53 Therefore, Zn(tBu)4Pc in toluene, with ϕf = 0.081,53 was chosen as a more reliable standard to generate an internally consistent set of quantum yields for this series.
Although the quantum yield of ZnF16Pc in THF can also be found in the literature (ϕf = 0.15),26 the value was calculated from a solution with relatively high absorbance near 630 nm, indicating significant formation of dimers and aggregates that do not contribute to fluorescence. We calculated ϕf = 0.057 for ZnF16Pc (Table 1), which is almost 3 times less than theirs (Supporting Information). The decrease in fluorescence emission between ZnPc and ZnF16Pc is best explained by the heavy atom effect62–66 and the electron-withdrawing effects caused by replacing hydrogens with fluorines.
The steady-state fluorescence data mirror the absorbance data, showing a systematic red shift in the emission λmax with increasing number of thioalkane substituents (Figure 2A). The fwhm of the fluorescence peaks slightly increases for the first eight substitutions, followed by a discontinuous jump of about 20 nm. Although there is no obvious relationship between the extent of substitution and the extinction coefficients or oscillator strengths, there is a clear correlation with the fluorescence quantum yield. Figure 2B shows how ϕf decreases with each additional octylthio group until it is almost completely quenched by about the eighth or ninth substitution. This is consistent with the work of Kobayashi et al.,67 who showed that the fluorescence is quenched for compounds with an emission λmax greater than about 740 nm. The authors explain this relationship in terms of the Energy Gap Law,68 which predicts an exponential increase in the rate of radiationless transitions between two electronic states as the energetic separation between them decreases.
The Stokes shift also correlates to the number of thioalkanes (Figure 2C), with a slight increase observed over the first 8 substitutions, followed by a jump around the 11th substitution and a steeper increase thereafter. The Stokes shift is a measure of the difference between the excitation and emission energies, which arises from differences in the nuclear configurations of the ground and excited states. The range of values for Pcs is generally around 5–10 nm.37 Increased Stokes shifts and broadened, asymmetric peak shapes are characteristic of nonplanar porphyrins and Pcs.37,69 The Stokes shifts for the most substituted compounds in this study are almost 30 nm. These are among some of the highest reported for Pc compounds in the literature, indicating that substantial conformational changes are induced upon substitution at the α position.
IIIE. Lifetimes and Rate Constants.
The fluorescence lifetimes are also given in Table 1 and plotted vs the number of appended thioalkyl chains in Figure 3. A general decrease in the lifetime with substitution is observed, tracking the decrease in quantum yield. The lifetime and the quantum yield allow determination of the fluorescent radiative rate constant using eq 1.
| (1) |
Here, kf is the natural fluorescence rate constant, and τ is the observed fluorescence lifetime. In the absence of other nonradiative deactivation pathways, the observed lifetime would approach the natural fluorescent lifetime, 1/kf, and the quantum yield would approach unity. The knr term accounts for all of the nonradiative excited-state energy and can be calculated through the complement to the fluorescence quantum yield using eq 2.
Figure 3.
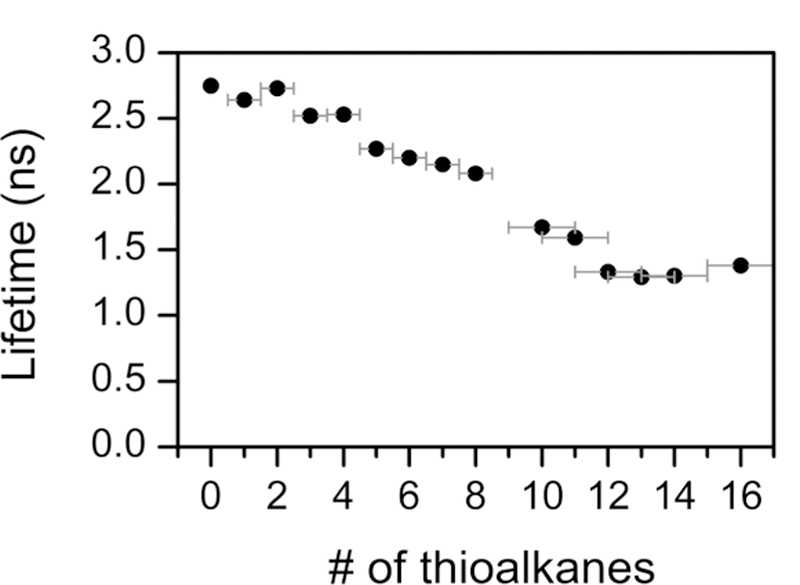
Fluorescence lifetimes as a function of the number of thioalkane substituents.
| (2) |
These photophysical parameters are plotted as a function of substitution number in Figure S6. In the absence of quenching, excited-state reactions, or other competing pathways, knr only includes contributions from intersystem crossing to the triplet state and internal conversion. The fluorescence rate constant kf decreases with the quantum yield and lifetime, reaching an apparent minimum by the 10th substitution. The nonradiative rate constant knr steadily increases and then jumps up at the 10th substitution. Both trends are consistent with the observations regarding the fwhm and the Stokes shift.
Intersystem crossing to the triplet state generally takes place through spin—orbit coupling and is enhanced by the presence of atoms with greater atomic numbers. This heavy atom effect is known to be promoted by sulfur atoms.70–73 At the same time, thermal relaxation can be enhanced through the addition of thioalkyl groups. First, the addition of long chain alkanes will introduce new vibrational modes that help to dissipate heat.74 The contribution of this “loose-bolt effect”75 depends on the coupling between the substituents and the core macrocycle. Another mechanism is the aforementioned Energy Gap Law.68 When there is a small energy difference between the ground and excited states, and correspondingly similar molecular geometries, there will be a large overlap in the Franck—Condon factors determining vibronic coupling between them. This leads to an approximately exponential increase in the strength of radiationless transitions from S1 to S0 as the energy gap decreases upon substitution. In addition to these internal conversion processes, there are also external conversion mechanisms that transfer excess energy as heat to another particle, such as a solvent molecule. Attaching thioalkyl chains to the Pc core changes the size, weight, aggregation, and polarity of the molecule, all of which will affect how it interacts with solvent molecules to dissipate energy.
In addition to these factors, there are literature reports asserting that distortion of the macrocycle also enhances nonradiative relaxation. The photophysical consequences of nonplanar distortions are reported for porphyrins,76–84 but are much less well understood in Pcs.57,60,69 Distorted or nonplanar Pcs have been reported, but the accompanying photophysical studies focus on the electronic and emission spectra.30–36,58,59 Conversely, detailed photophysical studies of Pcs often neglect considering the possibility of structural distortions.67 In porphyrins, distortion is known to cause broadened, red-shifted absorption and emission bands, increased Stokes shifts, lower fluorescence quantum yields and lifetimes, and even higher rates of intersystem crossing.85–87 All of these characteristics were unexpectedly observed in our series of Pcs, lending credibility to the hypothesis that the more highly substituted members adopt nonplanar conformations. Our assumption that the β position reacts first is also consistent with the trends in the photophysical data, because the distortion is induced by steric crowding of the α substituents after the eighth addition, with this effect being further exacerbated after the 11th substitution.
It is important to note that the nuclear potential energy surfaces will be more complex for a distorted Pc than a planar one. The former will have multiple local minima separated by barriers to interconversion, and these barriers may be lower in the excited states. Upon light absorption, the initially formed excited state will internally convert to the lowest excited state S1, imparting a significant amount of energy into the macrocycle. This energy can be enough to overcome the barriers in the potential energy surface and open up new conformational dynamics.88
IIIF. DFT and TD-DFT.
Many authors explain the photophysical properties in terms of the electronic effects of electron-withdrawing and -releasing groups, with mixed success. It is generally agreed67,89–92 that the HOMO is more sensitive to substituent effects than the LUMO, and that α substitution has a larger effect on the size of the HOMO—LUMO gap than β substitution. In turn, these observations are justified by noting the relative size of the orbital coefficients at the α and β positions of the HOMO. However, octa-α substituted Pcs can be as much as 3 times more red-shifted than their tetra-α substituted counterparts, and the red shift induced by octa-α substitution is nearly 20 times larger than that caused by octa-β substitution, despite there being only small differences in the α and β orbital coefficients.67 Thus, inductive electronic effects cannot account for all of the photophysics. To further clarify these electronic and structural factors, we performed quantum chemical calculations on a series of model compounds.
Representative compounds from this series were analyzed using density functional theory (DFT) and time-dependent DFT (TD-DFT). Given computational limitations, it was necessary to choose a handful of compounds spanning the series of products. To fully elucidate both structural and electronic inductive effects, this analysis was undertaken in three parts. First, a series of compounds in which the β positions have been substituted first were studied. The particular isomers are depicted in the first row of Figure 4. These structures were optimized to find the minimum energy conformations, ground-state energy levels, and molecular orbital surfaces by DFT. Subsequent to this, TD-DFT was used to find all of the excited states with energies below about 3.5 eV, including the transition energies, oscillator strengths, and the configuration interaction (CI) expansions of the excited-state wave functions.
Figure 4.
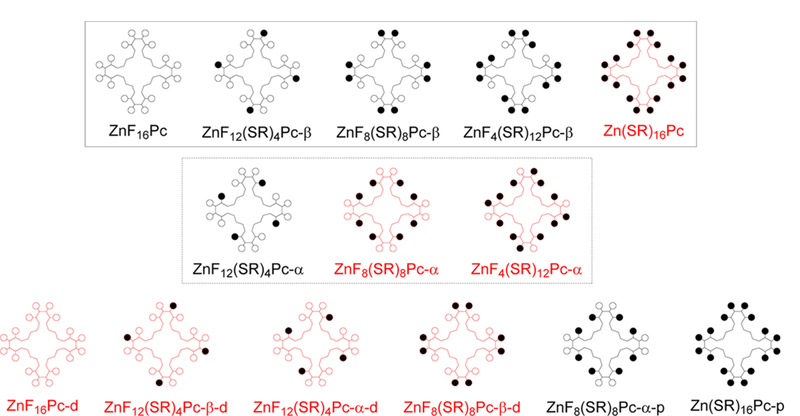
Schematic diagram of the specific compounds studied by DFT. Open circles (○) represent fluorine atoms and closed circles (●) represent n-butylthio substituents. Structures drawn in black are nominally planar, and those drawn in red are distorted. The first row, enclosed in a solid rectangle, includes isomers in which the β positions have been substituted first. The second row, enclosed in a dashed rectangle, includes isomers in which the α positions have been substituted first. The last row includes some of the same positional isomers, but with the core macrocycle artificially “frozen” into either a distorted or planar geometry, indicated by the suffix “-d” or “-p”, respectively. For this row, geometry optimization was only performed on the substituent chains, and then a single-point energy calculation was done to find the orbital energies.
A second series of compounds was also generated in which the α positions were substituted first. This series is seen in the second row of Figure 4. Although these α isomers are probably only a very minor component of the overall ensembles to which they belong, they are nevertheless physically realistic. They can be taken to provide a rough estimate of the range of HOMO and LUMO energies expected within the isomeric ensembles and provide some insight into how substitution can distort the Pc core. As with the first series, the structures were optimized by DFT, and the orbital energies and surfaces were calculated.
For the final series, seen in the last row of Figure 4, we wished to investigate the effects of structural distortions on the core macrocycle. Several compounds were constructed and analyzed with fixed, nonequilibrium geometries to compare them to their optimized counterparts. For ZnF16Pc-d, ZnF12(SR)4Pc-β-d, ZnF12(SR)4Pc-α-d, and ZnF8(SR)8Pc-β-d, the molecules were constructed with a distorted conformation by taking the optimized Zn(SR)16Pc result from the first series and modifying the substituents appropriately. On the contrary, the core macrocycles for ZnF8(SR)8Pc-α-p and Zn(SR)16Pc-p were frozen into a planar structure taken from the optimized ZnF16Pc result from the first series. The structures for this series were not optimized, so as to preserve the artificially imposed conformations chosen. Instead, single-point energy calculations were performed by DFT to find the MO levels and surfaces.
For the compounds in the first series, isosurfaces of the two eg LUMOs and the a1u HOMO are depicted in Figure 5, along with an edge-on perspective of the molecular structure to help visualize deviations from planarity. It should be noted that whereas ZnF4(SR)12Pc-β is designated as nominally planar in Figure 4, there is some distortion despite it being one of the least sterically hindered 12-substituted isomer possible. It is clear from the figure that the overall shapes of the relevant frontier orbitals are not dramatically changed by the addition of the thioalkyls. Interestingly, this is even true for the most highly substituted compounds, despite significantly distorted geometries. The underlying symmetry of the parent molecule is largely preserved, with only small perturbations depending on the substitution pattern.
Figure 5.
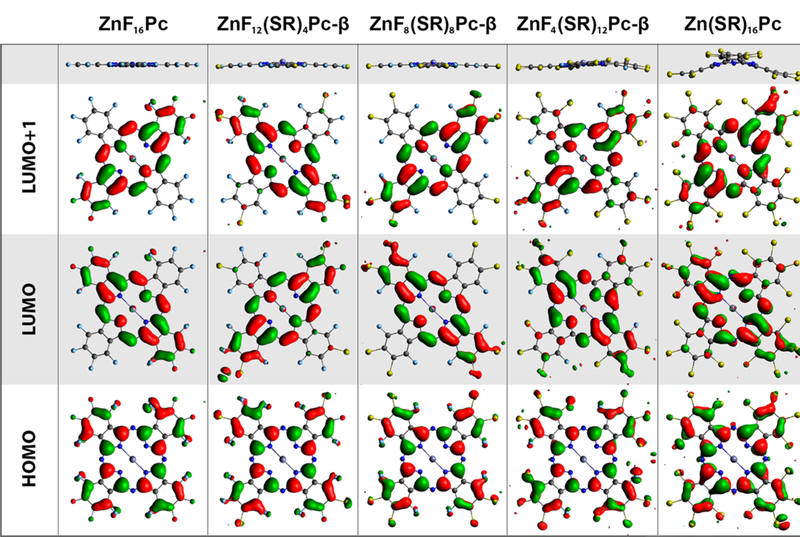
Frontier molecular orbitals (bottom rows) and edge-on views (top row) of the optimized molecular structures for representative (alkylthio)—Zn(II)Pcs. Gray atoms are carbon, dark blue are nitrogen, light blue are fluorine, and yellow are sulfur. Structures were optimized with n-butylthio groups in place to reduce computation time. For visual clarity, the hydrocarbon chains have been deleted from the images, along with some small regions of electron density associated with them.
In Gouterman’s four-orbital model for metalloporphyrins,93,94 the two LUMOs are a set of degenerate eg orbitals, and the two HOMOs are “accidentally” degenerate, with a1u and a2u symmetry designations. Configuration interaction between these four states results in a pair of pseudoparity forbidden, low energy transitions (the Q-bands) and a pair of fully allowed, high energy transitions (the B or Soret bands). In Pcs, the near-degeneracy of the HOMO levels is broken by the presence of the aza-bridge nitrogens and the fused benzene rings.89,90,95 The a2u orbital, having significant electron density on the more electronegative aza bridges, is lowered in energy by several electronvolts relative to the a1u orbital. This electronic structure is essentially the same in ZnF16Pc, despite the overall energies being slightly lowered by the presence of the fluorine atoms.96 From the DFT results we have obtained for ZnF16Pc, it is actually the HOMO—9 level (i.e., the 10th highest MO, Figure 6) that corresponds to the a2u HOMO found in metalloporphyrins. With the a1u/a2u degeneracy removed, the low energy transition no longer cancels, and the Q-band becomes fully allowed. The eg LUMOs retain their degeneracy but are slightly stabilized, red-shifting the Pc peak relative to porphyrin.
Figure 6.
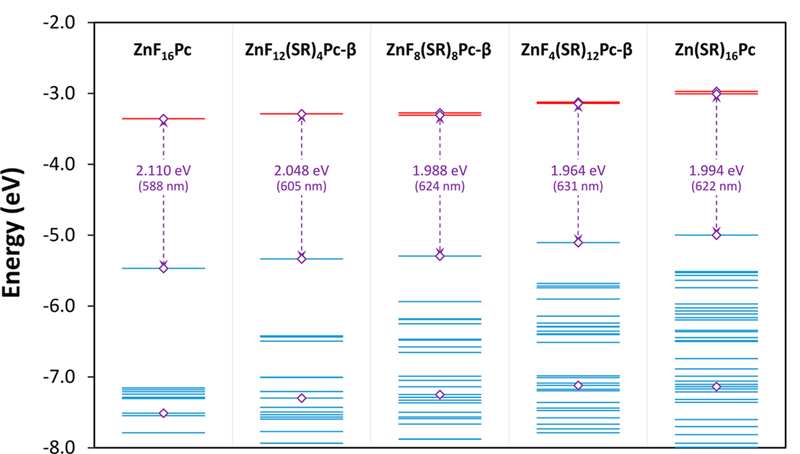
Energies for the frontier molecular orbitals of ZnF16Pc, ZnF12(SR)4Pc-β, ZnF8(SR)8Pc-β, ZnF4(SR)12Pc-β, and Zn(SR)16Pc between −2.0 and −8.0 eV, as calculated by DFT using the B3LYP hybrid functional and the 6–31G(d,p) basis set. The highest occupied orbitals are shown as blue lines, and the lowest unoccupied virtual orbitals are shown as red lines. There are two LUMOs shown for every compound, though they appear as a single line when the energies are degenerate or nearly so. Purple diamonds (◇) mark the orbitals associated with Gouterman’s original model. The HOMO—LUMO energy gaps are also shown in both electron volts and nanometers.
The energies of these frontier orbitals are shown in Figure 6, along with some other lower lying occupied MOs that are involved in transitions in the UV—visible region. From the diagram it is clear that the orbitals are generally destabilized as electron-releasing thioalkane groups are introduced. This effect is greater for the HOMO, which has significant probability density around all 16 reactive positions, than for the LUMOs, which have density on only eight of these positions each. This leads to the red shift observed in the Q-band and is also consistent with cyclic voltammetry studies carried out by Varotto et al., which show the HOMO is raised relative to the LUMO.4 Furthermore, the addition of various thioalkyl chains introduces many new orbitals at energies between the original Gouterman a1u and a2u HOMOs. These new orbitals are likely to play some part in the band broadening and could also have an effect on the peak position as they begin to mix with the HOMO—LUMO transition.
The DFT results shown in Figure 6 predict that the additive electronic effect of the substituents will only be approximately linear through the first eight substitutions, after which it becomes smaller and even reverses by the time all 16 fluorines have been replaced. It appears that at some point after the first eight thioalkanes have been added, further destabilization of the HOMO is offset by a corresponding destabilization of the LUMOs. In fact, these data suggest that the fully substituted Zn(SR)16Pc should actually be blue-shifted relative to ZnF8(SR)8Pc, a prediction that is reinforced by the TD-DFT transitions calculated for these compounds.
The TD-DFT calculated oscillator strengths of these transitions are plotted vs wavelength in Figure 7. These data are also presented in Table 2, along with the weights and characters of the singly excited configurations contributing to each transition. The decomposition of the excited-state wave functions shows that, in all cases, the observed Q-bands arise almost exclusively from HOMO → LUMO transitions. This also yields a double-band structure due to the LUMO and LUMO+1 levels, which are degenerate or nearly degenerate. The degeneracy is broken by the addition of the thioalkyl chains, which leads to a splitting of the transitions associated with the Q-bands. Again, the splitting is not resolved in the experimental UV—visible spectra, but it is likely to be at least partially responsible for the broadening of the bands observed in more substituted compounds.
Figure 7.
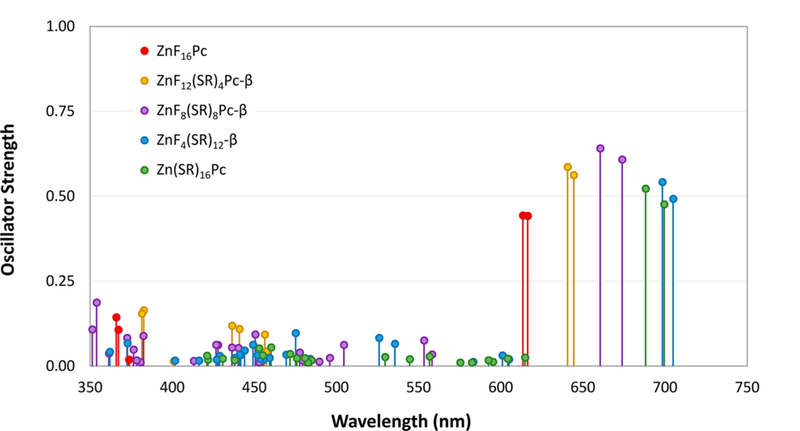
Electronic transitions between 350 and 750 nm for compounds ZnF16Pc, ZnF12(SR)4Pc-β, ZnF8(SR)8Pc-β, ZnF4(SR)12Pc-β, and Zn(SR)16Pc. Only transitions with oscillator strengths above 0.01 are included.
Table 2.
Electronic Transitions for ZnF16Pc, ZnF12(SR)4Pc-β, ZnF8(SR)8Pc-β, ZnF4(SR)12Pc-β, and Zn(SR)16Pc, Calculated by TD-DFT Using the B3LYP Hybrid Exchange—Correlation Functional and the 6–31G(d,p) Basis Seta
| compound | λb | f c | wave functiond |
|---|---|---|---|
| ZnF16Pc | 616 | 0.442 | 95% (H → L) + … |
| 613 | 0.444 | 95% (H→ L+1) + … | |
| 367 | 0.107 | 5% (H−9 → L+1) + 30% (H−5 → L+1) + 63% (H−3 → L) + … | |
| 366 | 0.144 | 6% (H−9 → L) + 21% (H−5 → L) + 67% (H−3 → L+1) + … | |
| ZnF12(SR)4Pc−β | 644 | 0.562 | 96% (H → L) + … |
| 641 | 0.586 | 96% (H → L+1) + … | |
| 441 | 0.110 | 88% (H−4 → L) + 7% (H−2 → L) + … | |
| 436 | 0.119 | 94% (H−4 → L+1) + … | |
| 383 | 0.164 | 93% (H−6 → L) + … | |
| 382 | 0.155 | 93% (H−6 → L+1) + … | |
| ZnF8(SR)8Pc−β | 674 | 0.608 | 96% (H → L) + … |
| 661 | 0.641 | 95% (H → L+1) + … | |
| 354 | 0.187 | 6% (H−18 → L) + 36% (H−14 → L) + 25% (H−13→L) + 14% (H−13 → L+1) + 6% (H−12 → L+1) + … | |
| 351 | 0.108 | 10% (H−13 → L) + 31% (H−13 → L+1) + 13% (H−12 → L) + 29% (H−12 → L+1) + … | |
| ZnF4(SR)12Pc−β | 705 | 0.492 | 95% (H → L) + … |
| 698 | 0.542 | 95% (H → L+1) + … | |
| Zn(SR)16Pc | 700 | 0.476 | 93% (H → L) + … |
| 688 | 0.522 | 92% (H → L+1) + … |
A more complete table of transitions is given in the Supporting Information (Table S2).
Transition wavelengths in nanometers. Only transitions with energies below ∼3.5 eV (>350 nm) are given.
Calculated oscillator strengths. Only transitions with oscillator strengths greater than 0.10 are given.
Excited-state wave function, in terms of the contributions of single excitations of the ground-state Slater determinant. Only single excitations with contributions greater than 5% are given. The HOMO is designated “H”, the second HOMO is “H−1″, etc. The LUMO is designated “L”, the second LUMO is “L+1”, etc. Excitations involving only orbitals corresponding to Gouterman’s four-orbital model are shown in italics.
The simulated TD-DFT spectra also predicts the appearance of a very broad, shallow absorbance in the region between the Q and B bands as the Pc is progressively substituted. This feature is, in fact, observed in the experimentally observed UV—visible spectra shown in Figure S2. The decomposition of the wave functions for these transitions show that they arise due to the manifold of new energy levels introduced between the two original Gouterman HOMOs, marked with purple diamonds in the chart. Inspection of the orbital surfaces for these new levels shows that they are primarily associated with the sulfur atoms introduced around the periphery. Many of them also appear to be hybridized with the core macrocycle orbitals, breaking the symmetry enough to relax the Laporte selection rules.
It is perhaps more interesting to note the predictive failures of these calculations. In particular, the reversal of the peak wavelength on going from ZnF8(SR)8Pc-β to ZnF4(SR)12Pc-β to Zn(SR)16Pc is not observed in the UV—visible data for the series. Although the absolute energies of DFT calculations for porphyrinoids are known to be overestimated by several tenths of an electronvolt,97–99 we would expect the relative energies to be more consistent with observations.
Considering the possibility that this deviation is an artifact of using single isomers to represent large ensembles, we next calculated energy levels for the α substituted compounds shown in the second row of Figure 4. Though the α and β isomers may not strictly bound the range of energies of all possible isomers for a given Pc, they will give a general idea of the magnitude of that range. The difference in the effects of α and β substitution can be seen in Figure 8A. It is clear from the trends in the energy gaps that the β series more accurately predicts the red shift up until at least ZnF8(SR)8Pc-β, after which both the α and β models begin to deviate significantly from observation.
Figure 8.
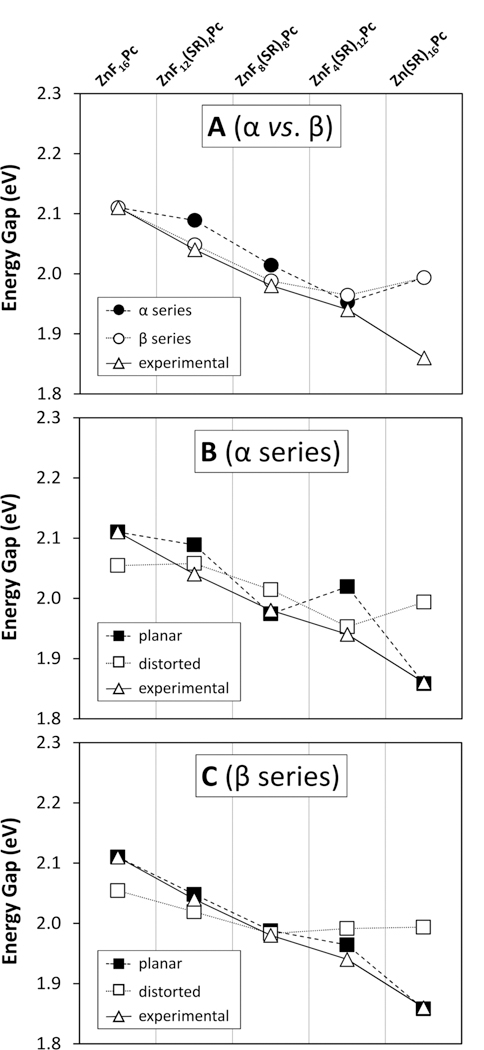
Calculated and experimental HOMO—LUMO energy gaps of various model Pc compounds. In all three panels, the open triangles (Δ) indicate the experimentally observed energy gaps calculated by converting the peak absorbance wavelengths to eV and adding a constant factor of 0.260 eV. This factor is an ad hoc correction which represents the difference between the calculated and observed energy gaps for ZnF16Pc. The top panel (A) shows a comparison of gap energies for α (filled circles: ●) and β (open circles: ○) substituted Pcs. The middle and bottom panels show comparisons of gap energies for planar (filled squares: ■) and distorted (open squares: □) geometries for α (middle panel, B) and β (bottom panel, C) substituted compounds.
Because conformational dynamics are likely the next most important factors in determining the energy levels, we went on to study the artificially manipulated structures shown in the last line of Figure 4. Figure 8B depicts the HOMO—LUMO gaps for planar and nonplanar geometries of the α Pcs, and Figure 8C shows the same for the β Pcs. It is again clear from these two panels that the β series is in better agreement with the experimental values. It is also interesting to note that, for both α and β Pcs, the planar geometries display a much steeper slope and more variation than the distorted geometries, indicating a greater red shift over the series. This is expected because the distorted geometry will tend to disrupt the conjugation of the macrocycle and thus reduce the electronic influence of the substituents. However, it is surprising for two reasons. First, it predicts that distorted Pcs will sometimes be blue-shifted relative to their planar counterparts, by as much as 40 nm or more. In fact, these calculations predict that a distorted Zn(SR)16Pc should have approximately the same energy gap as a ZnF8(SR)8Pc in any conformation, with any substitution pattern. This is, of course, in sharp contrast to the observed UV—vis spectra and therefore requires some explanation. Second, it is the series of Pcs with planar geometries that most accurately matches the observed red shifts, even for the most highly substituted compounds, which we expect to be severely distorted.
There are a few possible reasons for the above discrepancies between calculated and observed spectra. First, it may be that the DFT calculations are simply inaccurate, most likely due to limitations in the 6–31G(d,p) basis set used. This basis in particular gives acceptable accuracy for planar chromophores but is known to break down for more polarizable, nonplanar systems that require better descriptions of the diffuse states. A second possible issue with the calculations themselves could be that the energy levels are accurate for the structures found, but that the optimization procedure has generated incorrect structures. For instance, the planar structures could be the true minimum conformations, despite the distortion ostensibly found by DFT optimization. However, this would contradict a great deal of the literature, especially concerning α-thioalkyl substituted Pcs. Unfortunately, we were unable to obtain crystals of these more substituted compounds to conclusively determine their structure by X-ray diffraction.
It may also be that there is no significant deficiency in the computation at all, but rather that the model compounds we have chosen do not faithfully represent the compounds actually present in solution. Nonplanar porphyrinoids are known to twist or “flap” from one conformation to another,60 most likely through a planar or nearly planar transition state. It is unclear how the transient appearance of these planar states would affect the observed spectra, if at all, but it may serve to shift the absorbance of a molecule with a distorted minimum closer to that expected for a planar structure. Lastly, there has been some debate100–106 in the literature over the nature of distorted porphyrins and the photophysical implications thereof. Although the controversy has not explicitly involved Pcs, there is no reason that the arguments presented on either side would not apply equally well to them. In short, some groups have attributed the observed red shift in distorted porphyrins to the accompanying in-plane nuclear reorganizations (IPNRs) rather than to the out-of-plane distortion per se. These IPNRs consist of changes in the nuclear coordinates, bond lengths, bond angles, etc. Our method, which freezes the nuclear positions of the artificially flattened or distorted Pcs during the calculations, would not be able to account for these changes and thus could be introducing substantial error. Detailed photophysical and dynamic NMR studies versus temperature and solvent may shed light on these issues for Pcs.
IV. CONCLUSIONS
We have presented a comprehensive study of a family of substituted Pcs, including their photophysical and structural characteristics. As electron-releasing thioalkanes are appended to the core macrocycle, the absorbance band is red-shifted due to the inductive effect. However, although the literature reports typically show a larger red shift for α substituents than for β, our experimental spectroscopy indicates that the change remains mostly linear. This may be a consequence of the fluorine atoms around the periphery strongly counterbalancing the electron-donating thioalkanes. We have also shown that the red shift is accompanied by some strongly nonlinear effects, including broadening of the absorbance and emission bands, increase in the Stokes shift, and decreases in the fluorescence quantum yields and lifetimes. All of these results point toward distortion of the planar macrocycle caused by extensive substitution. DFT calculations further support this interpretation, although there still remain some unresolved discrepancies between the theory and experiment.
One of the most important outcomes of this study is the development of a facile, flexible method for rapidly generating Pcs with tunable properties, using only inexpensive, commodity reagents. Investigating the structure—function relationships in Pcs and other photoactive macrocycles is an active area of research,21 and our results offer a new perspective on the competing mechanisms at play. In particular, we have highlighted a distinct approach to easily manipulating these properties through distortion of the planar Pc structure. These kinds of nonplanar chromophores have found use in a variety of applications such as organic photovoltaics, photodynamic therapy, nonlinear optics, and photoacoustic spectroscopy, all of which underscores the importance of further exploring this pathway. For instance, correlating molecular properties (e.g., solubility, cell permeability, tumor uptake, phototoxicity, singlet oxygen yield, etc.) to spectroscopic signatures for aromatic distortion (e.g., fwhm or Stokes shift) could lead to new insights for the development of the next generation of phototherapeutics.
Supplementary Material
ACKNOWLEDGMENTS
We thank Naxhije Berisha for help with preparing the manuscript and for taking some of the spectra. This work was supported by the National Science Foundation, United States (NSF), through CHE-1213962 and IGERT-0965983 to C.M.D. Hunter College science infrastructure is supported by the NSF, the City University of New York, and the National Institute on Minority Health and Health Disparities of the National Institutes of Health under award number 8G12MD007599. This work is dedicated to Dr. Barry B. Corden, honoring 35 years of physical inorganic chemistry.
Footnotes
ASSOCIATED CONTENT
Supporting Information
PHP program for enumerating statistical isomers; plot of the number of isomers vs substitution number; discussion of oxidative decomposition; discussion of UV—vis spectroscopy and expanded UV—vis spectra for all compounds; calibration curves for determining extinction coefficients of all compounds; discussion of fluorescence spectroscopy and plots of fluorescence lifetimes vs. concentration; plots of radiative and nonradiative rate constants vs. the number of substituents; detailed description of methods and analytical data; discussion of MALDI-TOF mass spectrometry and MALDI-TOF mass spectra; full table of TD-DFT transitions (PDF)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Braun A; Tcherniac J Über die Produkte der Einwirkung von Acetanhydrid auf Phthalamid. Ber. Dtsch. Chem. Ges 1907, 40, 2709–2714. [Google Scholar]
- (2).de Diesbach H; von der Weid E Quelques sels complexes des o-dinitriles avec le cuivre et la pyridine. Helv. Chim. Acta 1927, 10, 886–888. [Google Scholar]
- (3).Löbbert G Phthalocyanines. Ullmann’s Encyclopedia of Industrial Chemistry, Electronic Release; Wiley-VCH: Weinheim, 2000; Vol. 27. [Google Scholar]
- (4).Varotto A; Nam CY; Radivojevic I; Tome JP; Cavaleiro JA; Black CT; Drain CM Phthalocyanine Blends Improve Bulk Heterojunction Solar Cells. J. Am. Chem. Soc 2010, 132, 2552–2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Walter MG; Rudine AB; Wamser CC Porphyrins and Phthalocyanines in Solar Photovoltaic Cells. J. Porphyrins Phthalocyanines 2010, 14, 759–792. [Google Scholar]
- (6).Jurow MJ; Hageman BA; Dimasi E; Nam CY; Pabon C; Black CT; Drain CM Controlling Morphology and Molecular Packing of Alkane Substituted Phthalocyanine Blend Bulk Hetero-junction Solar Cells. J. Mater. Chem. A 2013, 1, 1557–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Ragoussi M-E; Ince M; Torres T Recent Advances in Phthalocyanine-Based Sensitizers for Dye-Sensitized Solar Cells. Eur. J. Org. Chem 2013, 2013, 6475–6489. [Google Scholar]
- (8).Ince M; Yum J-H; Kim Y; Mathew S; Graẗzel M; Torres T; Nazeeruddin MK Molecular Engineering of Phthalocyanine Sensitizers for Dye-Sensitized Solar Cells. J. Phys. Chem. C 2014, 118, 17166–17170. [Google Scholar]
- (9).Martín-Gomis L; Fernańdez-Laźaro F; Sastre-Santos Á Advances in phthalocyanine-sensitized solar cells (PcSSCs). J. Mater. Chem. A 2014, 2, 15672–15682. [Google Scholar]
- (10).Debnath AK; Kumar A; Samanta S; Prasad R; Singh A; Chauhan AK; Veerender P; Singh S; Basu S; Aswal DK; et al. Fluorinated Copper-Phthalocyanine/Cobalt-Phthalocyaine Organic Heterojunctions: Charge Transport and Kelvin Probe Studies. Appl. Phys. Lett 2012, 100, 142104. [Google Scholar]
- (11).Wang H; Liu Z; Lo MF; Ng TW; Yan D; Lee C-S Electron Depletion And Accumulation Regions in N-type Copper-Hexadecafluoro-Phthalocyanine and their Effects on Electronic Properties. Appl. Phys. Lett 2012, 100, 103302. [Google Scholar]
- (12).Komolov AS; Lazneva EF; Komolov SA; Repin PS; Gavrikov AA Potential Barrier and Photovoltage at Interfaces of Hexadecafluoro-Copper-Phthalocyanine and Copper Phthalocyanine Films on the Surface of Tin Dioxide. Semiconductors 2012, 46, 988–992. [Google Scholar]
- (13).Gao YL; Ding HJ; Wang HB; Yan DH Electronic Structure of Interfaces Between Copper-Hexadecafluoro-Phthalocyanine and 2,5-Bis(4-biphenylyl) Bithiophene. Appl. Phys. Lett 2007, 91, 142112. [Google Scholar]
- (14).Jiang H; Ye J; Hu P; Wei F; Du K; Wang N; Ba T; Feng S; Kloc C Fluorination of Metal Phthalocyanines: Single-Crystal Growth, Efficient N-Channel Organic Field-Effect Transistors, and Structure-Property Relationships. Sci. Rep 2014, 4, 7573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Jurow MJ; Schuckman AE; Batteas JD; Drain CM Porphyrins as Molecular Electronic Components of Functional Devices. Coord. Chem. Rev 2010, 254, 2297–2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Decreau R; Richard M-J; Julliard M Photodynamic Therapy Against Achromic M6Melanocytes: Phototoxicity Of Lipophilic Axially Substituted Aluminum Phthalocyanines And Hexadecahalogenated Zinc Phthalocyanines. J. Porphyrins Phthalocyanines 2001, 05, 390–396. [Google Scholar]
- (17).Stanley CF Photophysical Evaluation of Substituted Zinc Phthalocyanines as Sensitisers for Photodynamic Therapy. Ph.D. Thesis; Durham University: Durham, England, 1997. [Google Scholar]
- (18).Boyle RW; Rousseau J; Kudrevich SV; Obochi MOK; van Lier JE Hexadecafluorinated Zinc Phthalocyanine: Photo-dynamic Properties Against the EMT-6 Tumour In Mice and Pharmacokinetics Using 65Zn As A Radiotracer. Br. J. Cancer 1996, 73, 49–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Alleḿann E; Rousseau J; Brasseur N; Kudrevich SV; Lewis K; van Lier JE Photodynamic Therapy of Tumours with Hexadecafluoro Zinc Phthalocyanine Formulated in PEG-Coated Poly(Lactic Acid) Nanoparticles. Int. J. Cancer 1996, 66, 821–824. [DOI] [PubMed] [Google Scholar]
- (20).Allémann E; Brasseur N; Kudrevich SV; La Madeleine C; van Lier JE Photodynamic Activities and Biodistribution Of Fluorinated Zinc Phthalocyanine Derivatives in the Murine EMT-6 Tumour Model. Int. J. Cancer 1997, 72, 289–294. [DOI] [PubMed] [Google Scholar]
- (21).Sekkat N; van den Bergh H; Nyokong T; Lange N Like a Bolt from the Blue: Phthalocyanines in Biomedical Optics. Molecules 2012, 17, 98–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Leznoff CC; Hiebert A; Ok S Titration Syntheses of Polyaminosubstituted Phthalocyanines via Nucleophilic Aromatic Substitutions on Zinc(II) 1,2,3,4,8,9,10,11,15,16,17,18,22,23,24,25-Hexadecafluorophthalocyanine. J. Porphyrins Phthalocyanines 2007, 11, 537–546. [Google Scholar]
- (23).Alonso MI; Garriga M; Osso JO; Schreiber F; Barrena E; Dosch H Strong Optical Anisotropies of F[sub 16]CuPc Thin Films Studied by Spectroscopic Ellipsometry. J. Chem. Phys 2003, 119, 6335–6340. [Google Scholar]
- (24).Leznoff CC; Sosa-Sanchez JL Polysubstituted Phthalocyanines by Nucleophilic Substitution Reactions on Hexadecafluorophthalocyanines. Chem. Commun 2004, 338–339. [DOI] [PubMed]
- (25).Jurow MJ; Varotto A; Manichev V; Travlou NA; Giannakoudakis DA; Drain CM Self-organized Nanostructured Materials of Alkylated Phthalocyanines and Underivatized C60 on ITO. RSC Adv 2013, 3, 21360–21364. [Google Scholar]
- (26).Garcia AM; Alarcon E; Munoz M; Scaiano JC; Edwards AM; Lissi E Photophysical Behaviour and Photodynamic Activity of Zinc Phthalocyanines Associated to Liposomes. Photochem. Photobiol. Sci 2011, 10, 507–514. [DOI] [PubMed] [Google Scholar]
- (27).Bartoli G; Ciminale F; Todesco PE Electronic and Steric Effects in Nucleophilic Aromatic Substitution. Reaction by Phenoxides as Nucleophiles in Dimethyl Sulfoxide. J. Org. Chem 1975, 40, 872–874. [Google Scholar]
- (28).Wang J; Khanamiryan AK; Leznoff CC Multisubstituted Phthalonitriles for Phthalocyanine Synthesis. J. Porphyrins Phthalocyanines 2004, 08, 1293–1299. [Google Scholar]
- (29).Bhupathiraju NV; Rizvi W; Batteas JD; Drain CM Fluorinated Porphyrinoids as Efficient Platforms for New Photonic Materials, Sensors, and Therapeutics. Org. Biomol. Chem 2016, 14, 389–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Chambrier I; Cook MJ; Wood PT Conformationally Stressed Phthalocyanines: The Non-Planarity of the 1,4,8,11,15,18,22,25-Octaisopentyl Derivative. Chem. Commun 2000, 2133–2134.
- (31).Zorlu Y; Kumru U; Isci U; Divrik B; Jeanneau E; Albrieux F; Dede Y; Ahsen V; Dumoulin F 1,4,8,11,15,18,22,25-Alkylsulfanyl Phthalocyanines: Effect of Macrocycle Distortion on Spectroscopic and Packing Properties. Chem. Commun 2015, 51, 6580–6583. [DOI] [PubMed] [Google Scholar]
- (32).Honda T; Kojima T; Kobayashi N; Fukuzumi S Crystal Structures and Electronic Properties of Saddle-Distorted and Protonated Phthalocyanines. Angew. Chem., Int. Ed 2011, 50, 2725–2728. [DOI] [PubMed] [Google Scholar]
- (33).Furuyama T; Satoh K; Kushiya T; Kobayashi N Design, Synthesis, and Properties of Phthalocyanine Complexes with Main-Group Elements Showing Main Absorption and Fluorescence Beyond 1000 nm. J. Am. Chem. Soc 2014, 136, 765–776. [DOI] [PubMed] [Google Scholar]
- (34).Kobayashi N; Fukuda T; Ueno K; Ogino H Extremely Non-Planar Phthalocyanines with Saddle or Helical Conformation: Synthesis and Structural Characterizations. J. Am. Chem. Soc 2001, 123, 10740–10741. [DOI] [PubMed] [Google Scholar]
- (35).Cammidge AN; Tseng C-H; Chambrier I; Hughes DL; Cook MJ Phthalocyanines Bearing Bulky Cycloalkylmethyl Substituents on Non-Peripheral Sites. Tetrahedron Lett 2009, 50, 5254–5256. [Google Scholar]
- (36).Kobayashi N; Furuyama T; Satoh K Rationally Designed Phthalocyanines Having their Main Absorption Band Beyond 1000 nm. J. Am. Chem. Soc 2011, 133, 19642–19645. [DOI] [PubMed] [Google Scholar]
- (37).Isago H Optical Emission Spectra of Phthalocyanines. Optical Spectra of Phthalocyanines and Related Compounds; Springer: New York, 2015; pp 107–131. [Google Scholar]
- (38).Golchoubian H; Hosseinpoor F Effective Oxidation of Sulfides to Sulfoxides with Hydrogen Peroxide under Transition-Metal-Free Conditions. Molecules 2007, 12, 304–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Taniguchi M; Du H; Lindsey JS Virtual Libraries of Tetrapyrrole Macrocycles. Combinatorics, Isomers, Product Distributions, and Data Mining. J. Chem. Inf. Model 2011, 51, 2233–2247. [DOI] [PubMed] [Google Scholar]
- (40).Taniguchi M; Soares ARM; Chandrashaker V; Lindsey JS A Tandem Combinatorial Model for the Prebiogenesis of Diverse Tetrapyrrole Macrocycles. New J. Chem 2012, 36, 1057–1069. [Google Scholar]
- (41).Taniguchi M; Lindsey JS Diversity, Isomer Composition, and Design of Combinatorial Libraries of Tetrapyrrole Macrocycles. J. Porphyrins Phthalocyanines 2012, 16, 1–13. [Google Scholar]
- (42).Taniguchi M; Lindsey JS Enumeration of Isomers of Substituted Tetrapyrrole Macrocycles: From Classical Problems in Biology to Modern Combinatorial Libraries. Handbook of Porphyrin Science; World Scientific: Singapore, 2012; Vol. 23, pp 1–80. [Google Scholar]
- (43).Drain CM; Singh S Combinatorial Libraries of Porphyrins: Chemistry and Applications. Handbook of Porphyrin Science; Kadish KM, Smith KM, Guilard R, Eds.; World Scientific: Singapore, 2011; Vol. 3, pp 485–530. [Google Scholar]
- (44).Berlin K; Jain RK; Tetzlaff C; Steinbeck C; Richert C Spectrometrically Monitored Selection Experiments: Quantitative Laser Desorption Mass Spectrometry of Small Chemical Libraries. Chem. Biol 1997, 4, 63–77. [DOI] [PubMed] [Google Scholar]
- (45).Stulz E; Scott SM; Bond AD; Teat SJ; Sanders JK Selection and Amplification of Mixed-Metal Porphyrin Cages From Dynamic Combinatorial Libraries. Chem. -Eur. J 2003, 9, 6039–6048. [DOI] [PubMed] [Google Scholar]
- (46).Ding S; Gray NS; Wu X; Ding Q; Schultz PG A Combinatorial Scaffold Approach toward Kinase-Directed Heterocycle Libraries. J. Am. Chem. Soc 2002, 124, 1594–1596. [DOI] [PubMed] [Google Scholar]
- (47).Drain CM; Gong X; Ruta V; Soll CE; Chicoineau PF Combinatorial Synthesis and Modification of Functional Porphyrin Libraries: Identification of New, Amphipathic Motifs for Biomolecule Binding. J. Comb. Chem 1999, 1, 286–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Samaroo D; Vinodu M; Chen X; Drain CM meso-Tetra(pentafluorophenyl)porphyrin as an Efficient Platform for Combinatorial Synthesis and the Selection of new Photodynamic Therapeutics using a Cancer Cell Line. J. Comb. Chem 2007, 9, 998–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Read RC Pólya’s Theorem and Its Progeny. Math. Mag 1987, 60, 275–282. [Google Scholar]
- (50).Pólya G Kombinatorische Anzahlbestimmungen für Gruppen, Graphen und chemische Verbindungen. Acta Math 1937, 68, 145–254. [Google Scholar]
- (51).Redfield JH The Theory of Group-Reduced Distributions. Am. J. Math 1927, 49, 433–455. [Google Scholar]
- (52).Williams A Loopless Generation of Multiset Permutations using a Constant Number of Variables by Prefix Shifts. Proceedings of the Twentieth Annual ACM-SIAM Symposium on Discrete Algorithms 2009, 987–996.
- (53).Chidawanyika W; Nyokong T The Synthesis And Photo-physicochemical Properties of Low-Symmetry Zinc Phthalocyanine Analogues. J. Photochem. Photobiol., A 2009, 206, 169–176. [Google Scholar]
- (54).Ogunsipe A; Maree D; Nyokong T Solvent Effects on the Photochemical and Fluorescence Properties of Zinc Phthalocyanine Derivatives. J. Mol. Struct 2003, 650, 131–140. [Google Scholar]
- (55).Iagatti A; Doria S; Marcelli A; Angelini N; Notarantonio S; Paoletti AM; Pennesi G; Rossi G; Zanotti G; Calogero G; et al. Photophysical Processes Occurring in a Zn-phthalocyanine in Ethanol Solution and on TiO2 Nanostructures. J. Phys. Chem. C 2015, 119, 20256–20264. [Google Scholar]
- (56).Dick S; Peisert H; Dini D; Hanack M; Cook MJ; Chambrier I; Chasse T Influence of the Alkyl-Chains Length on the Electronic Structure and Interface Properties of 1,4-Octasubstituted Zinc Phthalocyanines on Gold. J. Appl. Phys 2005, 97, 073715. [Google Scholar]
- (57).Mack J; Kobayashi N; Stillman MJ Re-examination of the Emission Properties of Alkoxy-and Thioalkyl-Substituted Phthalocyanines. J. Inorg. Biochem 2010, 104, 310–317. [DOI] [PubMed] [Google Scholar]
- (58).Gorun SM; Rathke JW; Chen MJ Long-range Solid-state Ordering and High Geometric Distortions Induced in Phthalocyanines by Small Fluoroalkyl Groups. Dalton Trans 2009, 1095–1097. [DOI] [PubMed]
- (59).Gao Y; Chen Y; Li R; Bian Y; Li X; Jiang J Nonperipherally Octa(butyloxy)-substituted Phthalocyanine Derivatives with Good Crystallinity: Effects of Metal-Ligand Coordination on the Molecular Structure, Internal Structure, and Dimensions of Self-Assembled Nanostructures. Chem. -Eur. J 2009, 15, 13241–13252. [DOI] [PubMed] [Google Scholar]
- (60).Gunaratne TC; Gusev AV; Peng X; Rosa A; Ricciardi G; Baerends EJ; Rizzoli C; Kenney ME; Rodgers MA Photophysics of Octabutoxy Phthalocyaninato-Ni(II) in Toluene: Ultrafast Experiments and DFT/TDDFT Studies. J. Phys. Chem. A 2005, 109, 2078–2089. [DOI] [PubMed] [Google Scholar]
- (61).Bishop SM; Beeby A; Parker AW; Foley MSC; Phillips D The Preparation and Photophysical Measurements of Perdeutero Zinc Phthalocyanine. J. Photochem. Photobiol., A 1995, 90, 39–44. [Google Scholar]
- (62).Alberto ME; De Simone BC; Mazzone G; Sicilia E; Russo N The Heavy Atom Effect on Zn(II) Phthalocyanine Derivatives: A Theoretical Exploration of the Photophysical Properties. Phys. Chem. Chem. Phys 2015, 17, 23595–23601. [DOI] [PubMed] [Google Scholar]
- (63).Azenha E. l. G.; Serra AC; Pineiro M; Pereira MM; Seixas de Melo J; Arnaut LG; Formosinho SJ; Rocha Gonsalves A. M. d. A. Heavy-atom Effects on Metalloporphyrins and Polyhalogenated Porphyrins. Chem. Phys 2002, 280, 177–190. [Google Scholar]
- (64).Bonnett R; Harriman A; Kozyrev AN Photophysics of Halogenated Porphyrins. J. Chem. Soc., Faraday Trans 1992, 88, 763–769. [Google Scholar]
- (65).Makhseed S; Ghazal B; Abdelmoniem AM; Novakova V; Zimcik P Photophysical and Theoretical Studies of Peripherally Halogenated Octaphenoxyphthalocyanines. RSC Adv 2015, 5, 58854–58864. [Google Scholar]
- (66).Lo P-C; Wang S; Zeug A; Meyer M; Röder B; Ng DKP Preparation and Photophysical Properties of Halogenated Silicon(IV) Phthalocyanines Substituted Axially with Poly(Ethylene Glycol) Chains. Tetrahedron Lett 2003, 44, 1967–1970. [Google Scholar]
- (67).Kobayashi N; Ogata H; Nonaka N; Luk’yanets EA Effect of Peripheral Substitution on the Electronic Absorption and Fluorescence Spectra of Metal-free and Zinc Phthalocyanines. Chem.-Eur. J 2003, 9, 5123–5134. [DOI] [PubMed] [Google Scholar]
- (68).Bixon M; Jortner J; Cortes J; Heitele H; Michel-Beyerle MD Energy Gap Law for Nonradiative and Radiative Charge Transfer in Isolated and in Solvated Supermolecules. J. Phys. Chem 1994, 98, 7289–7299. [Google Scholar]
- (69).Gutierrez-Meza E; Noria R; Granados G; Gomez-Vidales V; Ramirez JZ; Beltran HI; Peon J Photophysics of a Cis Axially Disubstituted Macrocycle: Rapid Intersystem Crossing in A Tin(IV) Phthalocyanine with A Half-Domed Geometry. J. Phys. Chem. B 2012, 116, 14107–14114. [DOI] [PubMed] [Google Scholar]
- (70).Peceli D; Hu H; Fishman DA; Webster S; Przhonska OV; Kurdyukov VV; Slominsky YL; Tolmachev AI; Kachkovski AD; Gerasov AO; Masunov AE; Hagan DJ; Van Stryland EW Enhanced Intersystem Crossing Rate in Polymethine-Like Molecules: Sulfur-Containing Squaraines Versus Oxygen-Containing Analogues. J. Phys. Chem. A 2013, 117, 2333–2346. [DOI] [PubMed] [Google Scholar]
- (71).Orchin M; Macomber RS; Pinhas AR; Wilson RM The Vocabulary and Concepts of Organic Chemistry; second ed.; John Wiley & Sons, Inc.: Hoboken, NJ, 2005. [Google Scholar]
- (72).Zander M The Intra-annular Internal Heavy-atom Effect on the Fluorescence and Phosphorescence Properties of Oxygen, Sulphur or Selenium Containing Heterocyclic Systems Related to Dibenzo [b,n] perylene. Z. Naturforsch., A: Phys. Sci 1989, 44, 1116–1118. [Google Scholar]
- (73).Goldacker W; Schweitzer D; Zimmermann H Electronic Properties of the Triplet State of Fluorene, Carbazole, Dibenzofuran and Dibenzothiophene (X-traps). Chem. Phys 1979, 36, 15–26. [Google Scholar]
- (74).Wróbel D; Graja A Photoinduced Electron Transfer Processes in Fullerene—Organic Chromophore Systems. Coord. Chem. Rev 2011, 255, 2555–2577. [Google Scholar]
- (75).Turro NJ; Ramamurthy V; Scaiano JC Principles of Molecular Photochemistry: An Introduction; University Science Books: Sausalito, CA, 2009. [Google Scholar]
- (76).Ravikanth M; Reddy D; Chandrashekar TK Fluorescence Properties of Distorted Short-Chain Basket Handle Porphyrins. J. Photochem. Photobiol., A 1993, 72, 61–67. [Google Scholar]
- (77).Maiti NC; Ravikanth M Photophysical Properties of Structurally Deformed Basket-Handle Prophyrins. J. Chem. Soc., Faraday Trans 1995, 91, 4369–4373. [Google Scholar]
- (78).Senge MO Highly Substituted Porphyrins. In The Porphyrin Handbook; Kadish KM, Smith KM, Guilard R, Eds.; Academic Press: New York, 2000; Vol. 1, pp 239–348. [Google Scholar]
- (79).Ivashin NV; Shchupak EE; Panarin AY; Sagun EI Photophysical Properties of Porphyrins with Sterically Distorted and Partially Screened Macrocycles. Opt. Spectrosc 2015, 118, 882–892. [Google Scholar]
- (80).Sazanovich IV; Galievsky VA; van Hoek A; Schaafsma TJ; Malinovskii VL; Holten D; Chirvony VS Photophysical and Structural Properties of Saddle-Shaped Free Base Porphyrins: Evidence for an “Orthogonal” Dipole Moment. J. Phys. Chem. B 2001, 105, 7818–7829. [Google Scholar]
- (81).Nifiatis F; Su W; Haley JE; Slagle JE; Cooper TM Comparison of the Photophysical Properties of a Planar, PtOEP, and a Nonplanar, PtOETPP, Porphyrin in Solution and Doped Films. J. Phys. Chem. A 2011, 115, 13764–13772. [DOI] [PubMed] [Google Scholar]
- (82).Gentemann S; Medforth CJ; Forsyth TP; Nurco DJ; Smith KM; Fajer J; Holten D Photophysical Properties of Conformationally Distorted Metal-Free Porphyrins. Investigation into the Deactivation Mechanisms of the Lowest Excited Singlet State. J. Am. Chem. Soc 1994, 116, 7363–7368. [Google Scholar]
- (83).Röder B; Büchner M; Rückmann I; Senge MO Correlation of Photophysical Parameters with Macrocycle Distortion in Porphyrins with Graded Degree of Saddle Distortion. Photochem. Photobiol. Sci 2010, 9, 1152–1158. [DOI] [PubMed] [Google Scholar]
- (84).Lebedev AY; Filatov MA; Cheprakov AV; Vinogradov SA Effects of Structural Deformations on Optical Properties of Tetrabenzoporphyrins: Free-Bases and Pd Complexes. J. Phys. Chem. A 2008, 112, 7723–7733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Drain CM; Kirmaier C; Medforth CJ; Nurco DJ; Smith KM; Holten D Dynamic Photophysical Properties of Conformationally Distorted Nickel Porphyrins. 1. Nickel(II) Dodecaphenylporphyrin. J. Phys. Chem 1996, 100, 11984–11993. [Google Scholar]
- (86).Drain CM; Gentemann S; Roberts JA; Nelson NY; Medforth CJ; Jia S; Simpson MC; Smith KM; Fajer J; Shelnutt JA; et al. Picosecond to Microsecond Photodynamics of a Nonplanar Nickel Porphyrin: Solvent Dielectric and Temperature Effects. J. Am. Chem. Soc 1998, 120, 3781–3791. [Google Scholar]
- (87).Retsek JL; Drain CM; Kirmaier C; Nurco DJ; Medforth CJ; Smith KM; Sazanovich IV; Chirvony VS; Fajer J; Holten C Photoinduced Axial Ligation and Deligation Dynamics of Nonplanar Nickel Dodecaarylporphyrins. J. Am. Chem. Soc 2003, 125, 9787–9800. [DOI] [PubMed] [Google Scholar]
- (88).Rodriguez J; Kirmaier C; Holten D; Time-resolved. and Static Optical Properties of Vibrationally Excited Porphyrins. J. Chem. Phys 1991, 94, 6020–6029. [Google Scholar]
- (89).Mack J; Kobayashi N Low Symmetry Phthalocyanines and their Analogues. Chem. Rev 2011, 111, 281–321. [DOI] [PubMed] [Google Scholar]
- (90).Rio Y; Salome Rodriguez-Morgade M; Torres T Modulating the Electronic Properties of Porphyrinoids: A Voyage from the Violet to the Infrared Regions of the Electromagnetic Spectrum. Org. Biomol. Chem 2008, 6, 1877–1894. [DOI] [PubMed] [Google Scholar]
- (91).Li R; Zhang X; Zhu P; Ng DK; Kobayashi N; Jiang J Electron-donating or -Withdrawing Nature of Substituents Revealed by the Electrochemistry of Metal-Free Phthalocyanines. Inorg. Chem 2006, 45, 2327–2334. [DOI] [PubMed] [Google Scholar]
- (92).Shinohara H; Tsaryova O; Schnurpfeil G; Wöhrle D Differently Substituted Phthalocyanines: Comparison of Calculated Energy Levels, Singlet Oxygen Quantum Yields, Photo-Oxidative Stabilities, Photocatalytic And Catalytic Activities. J. Photochem. Photobiol., A 2006, 184, 50–57. [Google Scholar]
- (93).Gouterman M Spectra of Porphyrins. J. Mol. Spectrosc 1961, 6, 138–163. [Google Scholar]
- (94).Gouterman M; Wagnier̀e GH; Snyder LC Spectra of Porphyrins: Part II. Four Orbital Model. J. Mol. Spectrosc 1963, 11, 108–127. [Google Scholar]
- (95).Chidawanyika W; Mack J; Shimizu S; Kobayashi N; Nyokong T Effect of Peripheral Fused Ring Substitution on the Optical Spectroscopy and Electronic Structure of Metal Phthalocyanine Complexes. J. Porphyrins Phthalocyanines 2009, 13, 1053–1062. [Google Scholar]
- (96).Das SK; Mahler A; Wilson AK; D’Souza F High-Potential Perfluorinated Phthalocyanine-Fullerene Dyads for Generation of High-Energy Charge-Separated States: Formation and Photoinduced Electron-Transfer Studies. ChemPhysChem 2014, 15, 2462–2472. [DOI] [PubMed] [Google Scholar]
- (97).Lee SU Influence of Exchange-Correlation Functional in the Calculations of Vertical Excitation Energies of Halogenated Copper Phthalocyanines using Time-Dependent Density Functional Theory (TD-DFT). Bull. Korean Chem. Soc 2013, 34, 2276–2280. [Google Scholar]
- (98).Furche F; Rappoport D Density Functional Methods for Excited States: Equilibrium Structure and Electronic Spectra. In Computational Photochemistry; Olivucci, M., Ed.; Theoretical and Computational Chemistry; Elsevier: New York, 2005; Vol. 16. [Google Scholar]
- (99).Fukuda R; Ehara M Excited States and Electronic Spectra of Annulated Dinuclear Free-Base Phthalocyanines: A Theoretical Study on Near-Infrared-Absorbing Dyes. J. Chem. Phys 2012, 136, 114304. [DOI] [PubMed] [Google Scholar]
- (100).Zakavi S; Omidyan R; Talebzadeh S Porphine Core Saddling: Effects on the HOMO/LUMO Gap and the Macrocycle Bond Lengths and Bond Angles. Polyhedron 2013, 49, 36–40. [Google Scholar]
- (101).Zhang YH; Zhao W; Jiang P; Zhang LJ; Zhang T; Wang J Structural Parameters and Vibrational Spectra of a Series of Zinc Meso-Phenylporphyrins: A DFT and Experimental Study. Spectrochim. Acta, Part A 2010, 75, 880–890. [DOI] [PubMed] [Google Scholar]
- (102).Haddad RE; Gazeau S; Pecaut J; Marchon JC; Medforth CJ; Shelnutt JA Origin of the Red Shifts in the Optical Absorption Bands of Nonplanar Tetraalkylporphyrins. J. Am. Chem. Soc 2003, 125, 1253–1268. [DOI] [PubMed] [Google Scholar]
- (103).Ryeng H; Ghosh A Do Nonplanar Distortions of Porphyrins Bring about Strongly Red-Shifted Electronic Spectra? Controversy, Consensus, New Developments, and Relevance to Chelatases. J. Am. Chem. Soc 2002, 124, 8099–8103. [DOI] [PubMed] [Google Scholar]
- (104).Wertsching AK; Koch AS; DiMagno SG On the Negligible Impact of Ruffling on the Electronic Spectra of Porphine, Tetramethylporphyrin, and Perfluoroalkylporphyrins. J. Am. Chem. Soc 2001, 123, 3932–3939. [DOI] [PubMed] [Google Scholar]
- (105).Parusel ABJ; Wondimagegn T; Ghosh A Do Nonplanar Porphyrins Have Red-Shifted Electronic Spectra? A DFT/SCI Study and Reinvestigation of a Recent Proposal. J. Am. Chem. Soc 2000, 122, 6371–6374. [Google Scholar]
- (106).DiMagno SG; Wertsching AK; Ross CR Electronic Consequences of Nonplanar Core Conformations in Electron-Deficient Porphyrins: The Structure and Spectroscopic Properties of [5,10,15,20-Tetrakis(heptafluoropropyl)porphinato]cobalt(II). J. Am. Chem. Soc 1995, 117, 8279–8280. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.