

Abstract
Diffuse pulmonary lymphangiomatosis (DPL) is a rare disease caused by uncontrolled lymphatic vessel proliferation resulting in respiratory dysfunction. Lymphatic vessel growth is influenced by vascular endothelial growth factor (VEGF). It has been shown that bevacizumab, a monoclonal antibody to VEGF type A, may be helpful in treating diseases characterized by excessive vessel proliferation. We report the case of a 51‐year‐old man with DPL treated with 1 mg/kg bevacizumab every three weeks for 6 months. A significant improvement in lung infiltrates was seen on post‐treatment computed tomography (CT) chest with a 17.5% improvement in forced expiratory volume in one second (FEV1). The patient reported improved respiratory symptoms, and no significant adverse drug side effects were reported. The authors believe this is the first case of DPL to report lung function improvement [FEV1, forced vital capacity (FVC), and Diffusion Capacity for Carbon Monoxide (DLCO)] following bevacizumab therapy.
Keywords: Bevacizumab, lymphangiomatosis, pulmonary
Introduction
Lymphangiomatosis is a rare disorder characterized by progressive lymphatic vessel proliferation. When predominant in the thorax, it results in diffuse pulmonary lymphangiomatosis (DPL). This condition produces both obstructive and restrictive ventilatory deficits and recurrent pleural effusions and can lead to respiratory failure.
Lymphatic vessel proliferation is influenced by the expression of a signal protein, vascular endothelial growth factor (VEGF), and its subtypes, including VEGF type A (VEGF‐A) 1.
There have been a small number of case reports highlighting the effectiveness of bevacizumab in the treatment of DPL, although without clear radiological or physiological evidence of treatment response 2, 3.
Here, we report on a case of DPL treated with bevacizumab in a 51‐year‐old man. We present objective radiological and physiological assessment before and after treatment.
Case Report
A 51‐year‐old, previously well, man presented to a respiratory physician with a one‐month history of breathlessness on exertion and a dry cough. He was a 10‐pack year ex‐smoker with no other significant past medical history.
Cross‐sectional imaging demonstrated large‐volume mediastinal and hilar lymphadenopathy and asymmetrical multi‐lobar alveolar infiltrates. Respiratory function testing showed a mixed obstructive/restrictive ventilatory deficit with a forced expiratory volume in one second (FEV1) of 2.39 L (69.6% predicted) and a forced vital capacity (FVC) of 3.76 L (88.7% predicted) with FEV1/FVC ratio of 0.64. Total lung capacity (TLC) was 5.31 L (79.7% predicted), and diffusing capacity [Diffusion Capacity for Carbon Monoxide (DLCO) Adj] was 66.2% predicted.
A malignant cause was suspected; however, extensive investigation with bronchoscopy, bronchoalveolar lavage, transbronchial needle aspiration, computed tomography (CT)‐Positron Emission Tomography (PET), bone marrow aspirate, and mediastinal lymph node biopsy failed to demonstrate this or any infective aetiology.
Blood tests demonstrated a markedly elevated IgG4 level of 8 (0.030–2.010), but without histological evidence of IgG4 disease in pulmonary or nodal tissue on surgical biopsy. Concluding these investigations and in the absence of another more likely diagnosis, a decision was made to treat for probable IgG4 disease. Treatment commenced initially with glucocorticosteroids and later methotrexate and propranolol. These treatments were, however, ceased due to poor clinical and radiological response.
In the setting of treatment failure, further diagnostic procedures were undertaken, including surgical wedge biopsy of the right middle lobe and magnetic resonance imaging (MRI) with bilateral inguinal lymph node contrast injection. This demonstrated focal organizing pneumonia, lymphatic dilatation within the thorax, and possible lymphangitis. Additional mediastinal soft tissue was obtained with CT‐guided core biopsy. Immunohistochemical assessment demonstrated positive staining for CD31 antigens, confirming a diagnosis of pulmonary lymphangiomatosis. Due to patient preference, further treatment was postponed for a two‐year period in favour of observation.
At representation, clinical and radiological deterioration was evident. A significant decline in lung function, with FEV1 of 1.6 L (2.39 L at diagnosis) and FVC of 4.18 L (5.31 L at diagnosis), was demonstrated. The patient was referred to our centre, and a decision was made to initiate treatment with bevacizumab at 1 mg/kg given every three weeks for 6 months.
Cross‐sectional imaging and lung function assessment were undertaken before and after treatment. A significant improvement in lung infiltrate following treatment was seen on CT chest (Figs. 1, 2). Lung function assessment demonstrated a 17.5% improvement in FEV1, a 4.6% increase in FVC, a 9.6% increase in TLC, and an 8.9% increase in gas diffusion (Table 1).
Figure 1.
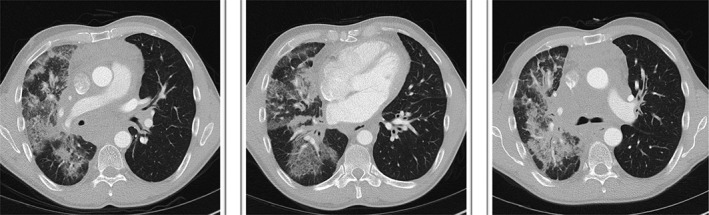
Computed tomography chest prior to treatment.
Figure 2.
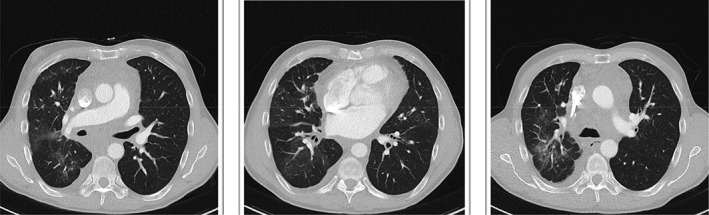
Computed tomography chest after 6 months bevacizumab treatment.
Table 1.
Lung function assessment prior to, during, and after treatment.
| Spirometry | Pre‐treatment | 3 months’ treatment | 6 months’ treatment |
|---|---|---|---|
| FEV1 (L) | 1.6 (48% predicted) | 1.47 (44% predicted) | 1.88 (57% predicted) |
| FVC (L) | 3.07 (73% predicted) | 3.04 (72% predicted) | 3.21 (75% predicted) |
| FEV1/FVC | 0.52 | 0.49 | 0.59 |
| Lung volume | |||
| TLC (L) | 4.18 (64% predicted) | 4.28 (66% predicted) | 4.58 (70% predicted) |
| Diffusing capacity | |||
| DLCO Adj (mL/mmHg/min/L) | 16.8 (60% predicted) | 17.1 (61% predicted) | 18.3 (66% predicted) |
At clinical review, the patient reported an improvement in breathless and no significant adverse reactions to the treatment.
Discussion
This case describes the diagnostic work‐up and treatment of the rare condition DPL. The case objectively documents treatment response with physiological and imaging assessments before and after treatment. Lung function, especially FEV1 and cross‐sectional imaging, demonstrated a clear positive response to bevacizumab.
This case demonstrates the diagnostic challenge DPL may pose. An extensive diagnostic work‐up was required yielding many negative results, including CT‐PET, which did not demonstrate Fluorodeoxyglucose (FDG) avidity in the abnormal tissues within the thorax. This would imply a non‐malignant, non‐infectious, and non‐inflammatory aetiology with a limited number of differentials. DPL should be considered a cause for large‐volume, widespread thoracic disease when a CT‐PET does not demonstrate substantial avidity. Lymphatic scintigraphy is an investigation that can aid in diagnosing DPL, although this was not conducted. In addition, Podoplanin is a further, more specific marker for lymphatic channels, and therefore, testing for this could be considered during investigation 4.
Of note, lung function in our patient did not improve significantly until six months of Bevacizumab had been administered. Aman et al. 3 documented radiological response in a patient with DPL following 10 weeks of bevacizumab therapy and stable CT findings at 7 months post‐treatment. Presently, it is unknown how long treatment should continue and whether continued improvement in lung function could be expected with further bevacizumab treatment in our patient. Moreover, it is unclear whether VEGF‐A levels may be helpful in monitoring disease activity, and unfortunately, this test was not available at the patient’s initial presentation.
The authors believe this is the first case of DPL to report lung function improvement (FEV1, FVC, and DLCO) following bevacizumab therapy. In rare conditions such as DPL, case reports are often the only literature available to help guide treatment, and therefore, we hope to have provided important data on the possible management of this condition.
Disclosure Statement
Appropriate written informed consent was obtained for publication of this case report and accompanying images.
Onyeforo, E , Barnett, A , Zagami, D , Deller, D , Feather, I . (2018) Diffuse pulmonary lymphangiomatosis treated with bevacizumab. Respirology Case Reports, 7(1), e00384. 10.1002/rcr2.384
Associate Editor: Arata Azuma
References
- 1. Tammela T, and Alitalo K. 2010. Lymphangiogenesis: molecular mechanisms and future promise. Cell 140:460–476. [DOI] [PubMed] [Google Scholar]
- 2. Grunewald T, Damke L, Maschan M, et al. 2010. First report of effective and feasible treatment of multifocal lymphangiomatosis (Gorham‐Stout) with bevacizumab in a child. Ann. Oncol. 21:1733–1734. [DOI] [PubMed] [Google Scholar]
- 3. Aman J, Thunnissen E, Paul M, et al. 2012. Successful treatment of diffuse pulmonary lymphangiomatosis with bevacizumab. Ann. Intern. Med. 156:839–840. [DOI] [PubMed] [Google Scholar]
- 4. Satria M, Pacheco‐Rodriguez G, and Moss J. 2011. Pulmonary lymphangiomatosis. Lymphat. Res. Biol. 9:191–193. [DOI] [PMC free article] [PubMed] [Google Scholar]