

Summary
Despite significant progress in hepatitis C virus protease inhibitor (PI) drug design, resistance remains a problem causing treatment failure. Double substitution variants, notably Y56H/D168A have emerged in patients who fail therapy with a PI-containing regimen. The resistance conferred by Asp168 substitutions has been well characterized and avoided in newer inhibitors. However, an additional mutation at Tyr56 confers resistance to even the most robust inhibitors. Here, we elucidate the molecular mechanisms of resistance for the Y56H/D168A variant against grazoprevir (and 4 analogs), paritaprevir, and danoprevir through inhibition assays, co-crystal structures and molecular dynamics simulations. The PIs susceptibility to Y56H/D168A vary, with those stacking on the catalytic His57 losing the most potency. For such inhibitors, the Y56H substitution disrupts favorable stacking interactions with the neighboring catalytic His57. This indirect mechanism of resistance threatens to cause multi-PI failure as all HCV PIs in clinical development rely on interactions with the catalytic triad.
eTOC Blurb
The molecular mechanism of resistance for HCV NS3/4A protease inhibitors (PIs) against the clinically emerging Y56H/D168A variant is described by determining crystal structures of this double mutant and Y56H variant. Substitution at the neighboring Y56 disrupted critical stacking interactions with the catalytic His57, revealing a structural mechanism that threatens cross-resistance.
Graphical Abstract
Introduction
Hepatitis C virus (HCV) is a global threat infecting 75 million people worldwide (Blach et al., 2017). Over 80% of individuals infected with HCV develop chronic liver disease, which often progresses to cirrhosis or becomes malignantly transformed to hepatocellular carcinoma. Prior to the introduction of direct-acting antivirals (DAAs), the standard of care for HCV infection consisted of pegylated interferon alpha and ribavirin (Ghany et al., 2011). This treatment resulted in a sustained virological response (SVR; the standard indication of cure from infection), of less than 50% against genotype 1 (GT1) and had low tolerability due to severe side effects (Fried et al., 2002). Fortunately, in the last several years the advent of DAAs against essential viral proteins NS3/4A, NS5A and NS5B has significantly improved therapeutic options and treatment outcomes for patients infected with HCV (Asselah et al., 2016; Falade-Nwulia et al., 2017).
HCV is a highly diverse virus with seven known genotypes (GT1–7) and multiple subtypes (Gower et al., 2014; Messina et al., 2015; Smith et al., 2014). Patients infected with HCV develop a heterogeneous population of viral species known as quasispecies due to low fidelity of the RNA-dependent RNA polymerase (Rong et al., 2010; Sarrazin and Zeuzem, 2010). The genetic diversity both among genotypes and within a viral population presents a challenge in the treatment of HCV infections. The six U.S. FDA approved all-oral combination therapies have varied effectiveness, and especially the earlier therapies can fail against certain genotypes (Falade-Nwulia et al., 2017). Newer generation inhibitors and various combinations have improved SVR rates across all genotypes to greater than 83% (Kwo et al., 2017). While combinations without a PI are also widely used, the two most recent combination therapies approved in 2017 by the FDA, Vosevi and Mavyret, contain a PI and have pan-genotypic activity, a major milestone in HCV treatment (Bourliere et al., 2017; Poordad et al., 2017). While the treatment options for HCV infection have significantly improved, one major threat to the clinical effectiveness of all anti-HCV drug classes is the emergence of resistance-associated substitutions (RASs) in target proteins (Ng et al., 2017; Pawlotsky, 2016; Sulkowski et al., 2015; Zeuzem et al., 2014).
RASs often weaken inhibitor binding resulting in reduced activity against the target enzyme. The HCV NS3/4A protease inhibitors (PIs) are highly effective drugs with the ability to rapidly reduce the HCV viral titer in infected patients but are susceptible to RASs around the protease active site (Lawitz et al., 2017; Lawitz et al., 2015). There are currently five FDA-approved protease inhibitors: simeprevir (TMC-435) (Rosenquist et al., 2014), paritaprevir (ABT-450) (Pilot-Matias et al., 2015), grazoprevir (MK-5172) (Harper et al., 2012), glecaprevir (ABT-493) (Ng et al., 2017) and voxilaprevir (GS-9857) (Rodriguez-Torres et al., 2016). All of these PIs incorporate large heterocyclic moieties at the P2 position to achieve high potency. However, this large P2 moiety often renders PIs susceptible to RASs, particularly at residues Arg155, Ala156, and Asp168. We have shown that the resistance profile of PIs largely depends on how the PIs protrude beyond the substrate envelope (Romano et al., 2012; Romano et al., 2010), which is largely determined by the identity of their P2 moiety and macrocycle location (Ali et al., 2013). Substitutions typically occur at residues that interact with PIs beyond the substrate envelope, preserving substrate recognition and turnover while disrupting inhibitor binding. Two such residues are Arg155 and Asp168 located in the S2 subsite, which form a critical electronic network that provides a surface essential for inhibitor binding but not for substrate recognition. Our previous crystal structures revealed that disruption of this electrostatic network as a result of substitutions at either Arg155 or Asp168 underlies the mechanism of resistance for earlier generation NS3/4A PIs (O’Meara et al., 2013; Romano et al., 2012).
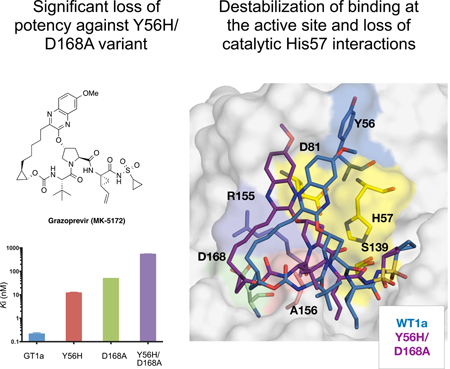
Grazoprevir is a highly potent P2-P4 macrocyclic inhibitor with cross-genotypic activity but reduced potency against GT3, a difficult to treat HCV variant (Summa et al., 2012). Grazoprevir was the first inhibitor with a unique binding mode whereby the P2 quinoxaline moiety, which still protrudes beyond the substrate envelope, stacks on the residues of the invariant catalytic triad. The catalytic residues cannot mutate without compromising substrate recognition and turnover, avoiding resistance. Robustness of grazoprevir against resistance prompted this binding mode to be exploited by all newer generation inhibitors including glecaprevir and voxilaprevir, which share a similar scaffold to grazoprevir. This binding mode also minimizes interactions with S2 subsite residues that typically mutate to confer resistance (Figure 1) (Romano et al., 2012) and thus reduces grazoprevir’s susceptibility to substitutions at residue Arg155. However, grazoprevir is still moderately susceptible to substitutions at Asp168 due to the packing of the P2-P4 macrocycle.
Figure 1: The binding mode and chemical structure of NS3/4A protease inhibitors.
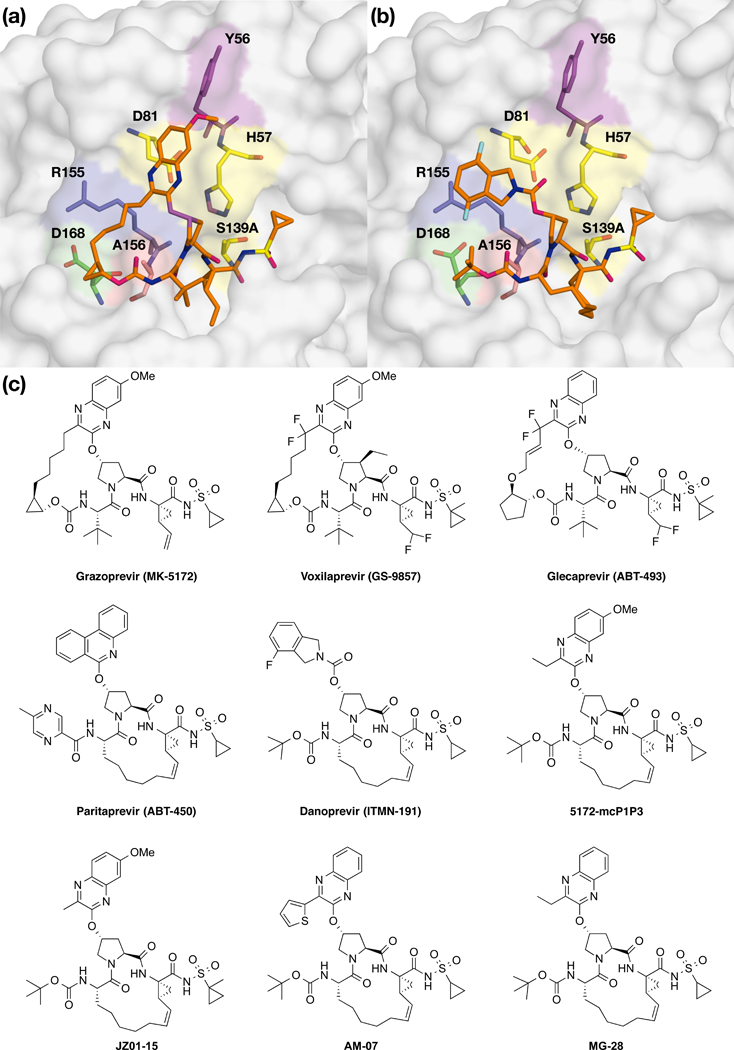
Surface view of NS3/4A protease inhibitors (a) grazoprevir and (b) danoprevir bound to the active site. Danoprevir’s P2 isoindoline moiety occupies two conformations in the protease active site. The catalytic triad is shown in yellow and drug resistance residues Tyr56, Argl55, Ala156, and Asp 168 are shown in magenta, blue, red and green, respectively. Residues Tyr56 and Asp 168 are located almost 15 Å apart in the protease active site, (c) Grazoprevir, voxilaprevir, glecaprevir and paritaprevir are approved by the FDA. Danoprevir was in clinical development. All other inhibitors were synthesized in house as P1-P3 macrocyclic analogs of grazoprevir.
In fact, most PIs are susceptible to substitutions at Asp168, which are often present in patients who fail therapy (Pawlotsky, 2016). Notably, the polymorphism Gln168 at this position underlies reduced efficacy of PIs against GT3 (Soumana et al., 2016a). Glecaprevir and voxilaprevir have improved resistance profiles against D168A and are active against GT3, but like grazoprevir are highly susceptible to substitutions at Ala156 (Ng et al., 2017) due to van der Waals clashes with their P2-P4 macrocycles (Ali et al., 2013; Soumana et al., 2016b). Unfortunately, even with the newest combinations some patients still fail therapy, with more than one RAS detected in the infecting viral population (Lawitz et al., 2014). The emergence of such double and sometimes triple-site RAS variants in clinic is threatening the effectiveness of current anti-HCV therapies (Gane et al., 2016).
While the molecular basis of drug resistance caused by single-site RASs has been well characterized (Romano et al., 2012; Romano et al., 2010; Soumana et al., 2014; Soumana et al., 2016b), the impact of clinically relevant NS3/4A protease double substitutions on inhibitor binding and the mechanisms of drug resistance remain largely unexplored. One key clinically relevant protease variant is Y56H/D168A, often present in patients who fail therapy with the newer generation PIs. Grazoprevir and paritaprevir are highly susceptible to this signature variant and exhibit over 500-fold loss in potency (Guo et al., 2017; Lontok et al., 2015; Zeuzem et al., 2014). Substitutions at Tyr56 rarely occur alone, but are becoming common in combination with substitutions at Asp168. These residues are not in physical contact, with Tyr56 located next to the catalytic His57, approximately 15 Å away from residue Asp168. Nevertheless, co-evolution of these two sites results in a detrimental loss of potency for all PIs. The molecular mechanism underlying high-level resistance of PIs against the Y56H/D168A double substitution variant is unknown.
To elucidate the molecular mechanism of resistance for PIs against the Y56H/D168A NS3/4A protease variant, we used a multi-disciplinary approach involving enzyme inhibition and antiviral assays, co-crystal structures, and molecular dynamics simulations. A panel of 7 NS3/4A PIs (grazoprevir and 4 analogs, paritaprevir, and danoprevir) with varying macrocycle locations and P2 binding modes were tested for enzyme inhibition and antiviral potency. To tease out the impact of individual substitutions, the double substitution variant was compared to both single substitutions and wild-type GT1a NS3/4A protease. While all inhibitors were 3–10 orders of magnitude less active against the Y56H/D168A NS3/4A protease variant, the potency loss was exacerbated for PIs that stack on the catalytic triad, including grazoprevir. Crystal structures and dynamic analysis of grazoprevir bound to protease variants revealed that this resistance is largely due to the Y56H substitution disrupting the favorable stacking interactions with the neighboring catalytic residue His57. Thus, in addition to the loss of the ionic network due to D168A substitution (O’Meara et al., 2013; Romano et al., 2012), decreased direct interactions with catalytic His57 underlie resistance against this double substitution variant. To prevent such mechanisms of clinically relevant resistance, inhibitor design limiting interactions with Tyr56 while still maintaining stacking against the catalytic residues is warranted.
Results
Inhibitors are highly susceptible to the Y56H/D168A NS3/4A variant
Enzyme inhibition and replicon potency of HCV NS3/4A PIs correlate closely with efficacy in the clinic, as does loss of potency due to site-specific substitutions. As Y56H and D168A substitutions have been selected in clinic together under the selective pressure of PIs, these two substitutions may have interdependent effects in conferring resistance. Thus to determine the impact of Y56H/D168A and potential interdependency of these substitutions, enzyme inhibition and replicon assays were performed against WT (GT-1a), Y56H, D168A and the double substitution variant for a panel of diverse NS3/4A PIs (Figure 2 and Tables S1, S2). This panel included FDA-approved inhibitors (grazoprevir, paritaprevir), P1-P3 macrocyclic analogs of grazoprevir (5172-mcP1P3, JZ01–15, AM-07, MG-28) and danoprevir (Figure 1). The inhibitors were selected to test the role of the macrocycle (P1-P3 versus P2-P4) and the packing of the P2 moiety on susceptibility to these substitutions.
Figure 2: In vitro resistance profile of NS3/4A protease inhibitors.
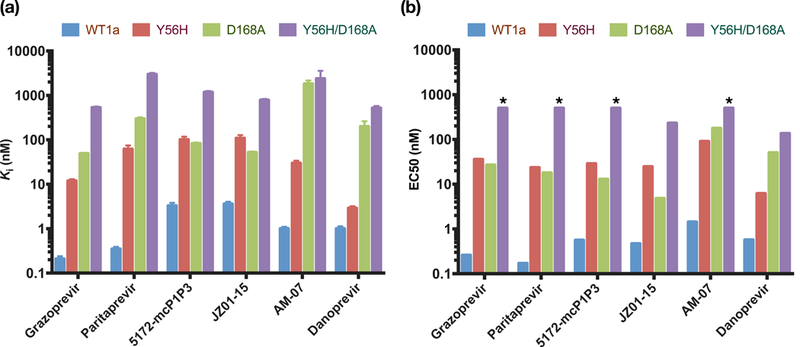
a) Enzyme inhibition constants (against protease domain) and b) replicon-based half maximal effective concentrations for WT HCV NS3/4A and drug-resistant variants Y56H, D168A and Y56H/D168A. All inhibitors lost activity against the Y56H/D168A protease variant. * Indicates EC50 value greater than 500. See also Tables S1 and S2.
All seven inhibitors were potent against WT with inhibition constants (Ki) ranging from 0.2 to 7.2 nM, (Figure 2A and Table S1), in agreement with previous reports (Matthew et al., 2017). Similarly, in replicon assays, all inhibitors exhibited sub-nanomolar to 1.4 nM potency against WT HCV (Figure 2B and Table S2). Relative to WT protease, all PIs lost potency against the D168A variant, as has been observed for NS3/4A inhibitors (Soumana et al., 2016a; Summa et al., 2012). However, the potency losses varied significantly from 14- to 1800-fold, with the thiophene substituted P1-P3 macrocyclic analog AM-07 losing the most potency and danoprevir exhibiting a 200-fold reduction in potency in enzyme inhibition assays. As we previously reported (Matthew et al., 2017), grazoprevir exhibited > 200-fold lower potency against the D168A variant in enzyme inhibition assays but was still one of the most potent inhibitors against this drug resistant protease variant (Ki =49.1 nM). The P1-P3 macrocyclic inhibitors JZ01–15 and 5172-mcP1P3 (with a methyl and ethyl substituted quinoxaline, respectively) exhibited lower potency against WT protease compared to grazoprevir but were less susceptible to the D168A variant, resulting in an inhibition constant similar to grazoprevir (Ki = 52 and 82.4 nM, respectively). Similar trends were observed in replicon assays with inhibitors losing 10- to 124-fold in potency compared to WT. Grazoprevir was over 100-fold less potent against the D168A variant (EC50 = 26.8 nM). Interestingly, JZ01–15 exhibited the best activity against the D168A variant in replicon assays with EC50 below 5 nM (EC50 = 4.8 nM). Thus the P1-P3 macrocyclic inhibitor with a small methyl substitution on the P2 quinoxaline showed a much flatter resistance profile and was less susceptible to D168A substitution.
The impact of Y56H on inhibition by NS3/4A PIs has not been well characterized, as this substitution is rarely observed alone. Enzyme inhibition assays on our panel of PIs showed that all inhibitors except danoprevir were susceptible to the Y56H substitution, exhibiting reduced potency ranging from 29–177 fold. The antiviral activities closely correlated with enzyme inhibition assays with inhibitors losing 51–138 fold in potency. In contrast, danoprevir, whose P2 isoindoline moiety does not stack on the catalytic triad (Romano et al., 2012), maintained potency similar to WT (Ki = 1 nM, EC50 = 0.57 nM) against the Y56H variant (Ki = 3 nM, EC50 = 6.2 nM).
The potency loss of Pis against Y56H variant could be due to disruption of Tyr56 residue’s direct interactions with the P2 quinoxaline. To isolate the effect of direct hydrophobic interactions of the methoxy group at the 7-position of the P2 quinoxaline with the Tyr56 residue, we tested MG-28, which lacks this substituent. MG-28 is an analog of 5172-mcPlP3 and differs only at this position. MG-28 was more susceptible to the Y56H substitution compared to 5172- mcPlP3 (Ki = 205 nM compared with 101 nM). While 5172-mcPlP3 was more potent against WT protease, fold-changes against Y56H variant were comparable regardless of the presence of this group (31 and 29 fold), also for a very close analog (JZ01–15; 30-fold). Thus loss of direct hydrophobic interactions with Tyr56 does not underlie susceptibility to the Y56H substitution, suggesting another mechanism of resistance.
All inhibitors were highly susceptible to the Y56H/D168A variant with enzyme inhibition constants in the mid-nanomolar to micromolar range (Ki = 500 nM - 2 μM). In fact, all inhibitors exhibited a greater than 200-fold loss in potency relative to WT protease. Danoprevir was 500- fold less potent against the Y56H/D168A protease (Ki = 520 nM). However, most of this potency loss is due to the D168A substitution (Ki = 199 nM) with only an additional 2.5-fold reduction in potency with Y56H. The same trend was observed for danoprevir in replicón assays (D168A EC50 = 50.6 nM, Y56H/D168A EC50 = 136 nM). In contrast, grazoprevir and paritaprevir were highly susceptible to the Y56H/D168A protease showing 2500-fold and 8500-fold lower potency, respectively compared to WT. The replicón assays correlated with enzyme potency, with grazoprevir and paritaprevir exhibiting 1923- and 2941-fold lower potency against the Y56H/D168A variant. JZ01–15 and 5172-mcPlP3 exhibited a relatively flatter resistance profile, losing 200–400 fold in enzyme inhibition assays. In fact, of the inhibitors with a P2 quinoxaline moiety, JZ01–15 was the only compound with measurable activity against the double substitution variant in replicon assays (EC50 = 231 nM). Thus, although the extent of the loss of potency was dependent on the particular inhibitor, the Y56H/D168A variant was detrimental to potency of all the inhibitors.
Double mutant cycle analysis reveals interdependency of Tyr56 and Aspl68 for most PIs
To delineate whether or not there is an interdependency between the two sites of mutation on the loss of inhibitor potency against the double substitution variant Y56H/D168A, the free energy of binding, ΔG (Table S3), was calculated for each inhibitor from the experimentally measured inhibition constants (Table S1). If the change in binding free energy (ΔΔG) to the double mutant relative to WT is equal to the sum of the free energy changes to each single mutant, then the substitutions are additive. If not, then the substitutions are coupled or interdependent in conferring resistance. This type of analysis is referred to as a double mutant cycle (Horovitz, 1996). The double mutant cycle analysis revealed that the substitutions were not additive but coupled for most of the inhibitors with the exception of danoprevir (difference in ΔΔG only ~0.06 kcal/mol) (Figure 3), which does not stack on the catalytic residues. The interdependency was most pronounced in other inhibitors with differences in ΔΔG for the double mutant compared to sum of the single mutants of 0.45 kcal/mol for 5172-mcPlP3 and JZ01–15, and 1–2 kcals/mol for grazoprevir, paritaprevir and AM-07. Surprisingly, the substitutions were negatively coupled, with the two changes together having less impact on inhibitor binding than what would be predicted by the simple addition of each independently. This negative coupling implies that the two substitutions may have overlapping effects on inhibitor binding, possibly individually destabilizing similar interactions between the inhibitor and protease active site.
Figure 3: Double mutant cycle analysis of NS3/4A protease inhibitors.
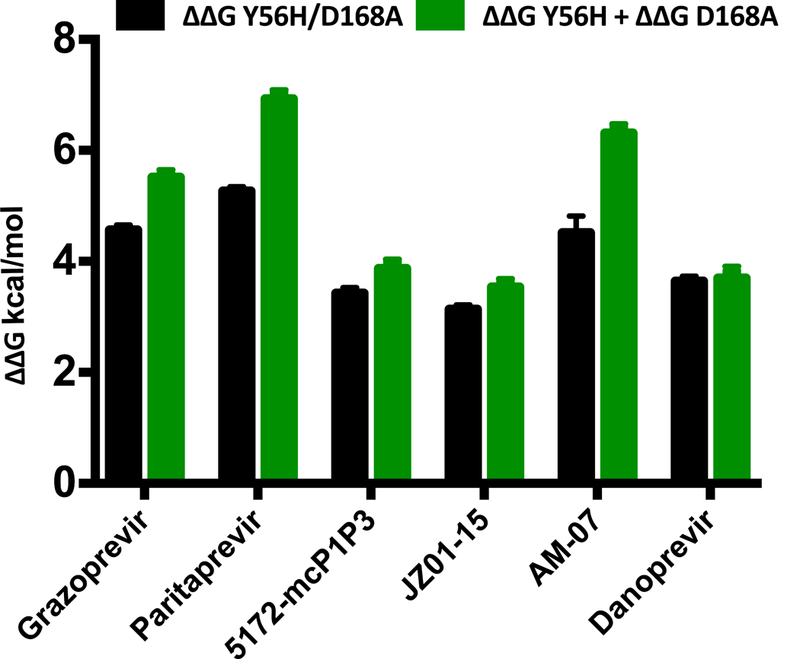
ΔΔG for each inhibitor used in the enzyme inhibition assay against the Y56H/D168A double substitution (black) and the sum of each single substitution (green). The substitutions Y56H and D168A show a coupled effect for grazoprevir, paritaprevir and AM-07. However, these substitutions are additive for danoprevir and to a lesser extent for 5172-mcPlP3 and JZ01–15. See also Table S3.
Crystal structures of protease-inhibitor complexes
As danoprevir and grazoprevir represent the extremes of whether or not the impact of Y56H and D168A are additive or coupled, high-resolution crystal structures were determined of these inhibitors bound to Y56H/D168A and Y56H protease variants. The structures had resolutions ranging from 1.2—1.9 Å, and complemented our previously determined structures of danoprevir (PDB ID: 3M5L and 3SU1 for WT, and D168A respectively) and grazoprevir (3SUD and 3SUF for WT and D168A respectively) (Romano et al., 2012). The grazoprevir-Y56H/D168A complex failed to crystalize despite extensive crystallization efforts, and in total three structures of danoprevir-Y56H, danoprevir-Y56H/D168A and grazoprevir-Y56H were determined (Table 1). These high-resolution structures afforded us atomic level details of protein-inhibitor interactions to elucidate the structural mechanism of resistance for NS3/4A PIs against the Y56H/D168A protease variant.
Table 1.
X-ray data collection and crystallographic refinement statistics.
| Y56H- grazoprevir |
Y56H-danoprevir | Y56H/D168A- danoprevir |
|
|---|---|---|---|
| PDB code | 6C2M | 6C2O | 6C2N |
| Resolution | 1.86 Å | 1.18 Å | 1.80 Å |
| Space group | P21 | P212121 | P212121 |
| Twin Law | h,-k,-h-l | ||
| Twin Fraction | 0.26 | ||
| Molecules in AUa | 4 | 1 | 1 |
| Cell dimensions: | |||
| a (Å) | 56.6 | 55.4 | 60 |
| b (Å) | 103.3 | 59.0 | 55.4 |
| c (Å) | 74.0 | 60.0 | 58.9 |
| β (°) | 90 | 90 | 90 |
| Completeness (%) | 96.0 | 99.8 | 96.4 |
| Total reflections | 208132 | 528153 | 139059 |
| Unique reflections | 63506 | 65383 | 18030 |
| Average I/σ | 11.4 | 18.5 | 17.8 |
| Redundancy | 3.3 | 8.1 | 7.7 |
| Rsym (%)b | 6.6 (28.1) | 6.9 (41.1) | 9.8 (29.9) |
| RMSDc in: | |||
| Bond lengths (Å) | 0.005 | 0.015 | 0.009 |
| Bond angles (°) | 0.9 | 1.9 | 1.3 |
| Rfacoor (%)d | 18.1 | 12.3 | 16.2 |
| Rfree (%)e | 22.4 | 14.4 | 19.0 |
AU, asymmetric unit.
, where I = observed intensity, <I> = average intensity over symmetry equivalent; values in parentheses are for the highest resolution shell.
RMSD, root mean square deviation.
Rfree was calculated from 5% of reflections, chosen randomly, which were omitted from the refinement process.
Danoprevir resistance of the Y56H/D168A variant is solely due to the D168A substitution
Danoprevir is a P1-P3 macrocyclic NS3/4A inhibitor with an isoindoline P2 group. We previously elucidated the resistance mechanisms to this inhibitor due to RASs by analyzing highresolution crystal structures of danoprevir bound to WT and mutant proteases.25 Comparing the structures of danoprevir bound to WT and Y56H proteases revealed that the binding mode and active site residues Arg155 and Asp168 are unchanged (Figure 4A). These results reinforced the inhibition data indicating danoprevir is not susceptible to Y56H, likely because the molecular contacts needed for efficient inhibitor binding are unaltered. In contrast, D168A substitution (Figure S1) resulted in a 0.8 Å shift of the Arg155 side chain away from residue Asp168, disrupting the cation-n interaction critical for danoprevir binding. Superposition of the D168A- and Y56H/D168A-protease structures showed that the active sites are almost identical (Figure 4B), in agreement with no further significant loss due to addition of Y56H and lack of coupling between the two substitutions for this inhibitor. Thus the D168A substitution alone underlies danoprevir resistance of the Y56H/D168A protease variant.
Figure 4: Crystal structures of danoprevir and grazoprevir bound to WT and mutant proteases.
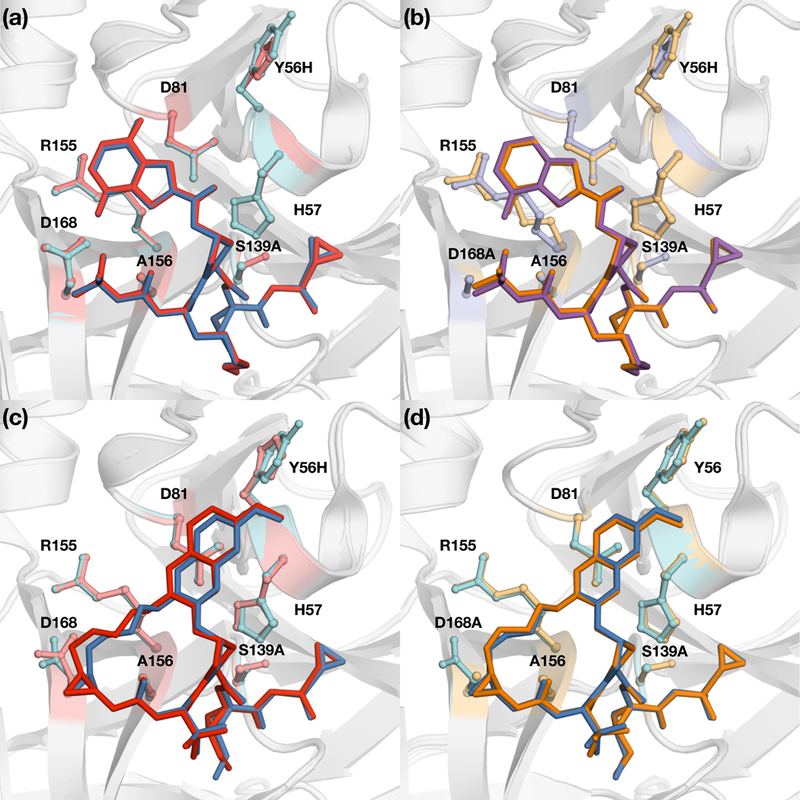
Superposition of danoprevir bound to (a) WT (blue) and Y56H (red) and (b) D168A (orange) and Y56H/D168A (purple) proteases. Superposition of grazoprevir bound to (c) WT (blue) and Y56H (red) and (d) D168A (orange) proteases. Danoprevir’s P2 isoindoline moiety occupies two conformations in the protease active site. Drug resistance residues and the catalytic triad residues His57, Asp81 and S139A are shown in sticks. See also Figure S1.
To assess the molecular details of inhibitor packing, van der Waals (vdW) contact energies were calculated for danoprevir and protease active site for each crystal structure. Total vdW contact energies were conserved between WT and Y56H proteases (−83.6 and −82.6 kcal/mol, respectively) as well as D168A and Y56H/D168A proteases (−81.0 and −81.7 kcal/mol, respectively). Compared to WT protease, Y56H contact energy landscape was highly conserved (Figure S2), but disrupted in the D168A and Y56H/D168A complexes with reduced interactions in the S2-subsite around residue Asp168. Thus, structural changes due to substitution at residue Asp168 underlie danoprevir resistance of the Y56H/D168A variant, which is expected to be valid for other inhibitors that primarily interact with S2 subsite residues.
Resistance mechanism of grazoprevir and analogs against single and double mutants
Grazoprevir is a P2-P4 macrocyclic inhibitor with a P2 quinoxaline moiety. Given that grazoprevir and analogs share a binding mode whereby the P2 quinoxaline moiety predominately interacts with the catalytic residues (His57 and Asp81), the mechanism of grazoprevir resistance for the Y56H/D168A protease was expected to differ from danoprevir.
The P1-P3 macrocyclic analogs of grazoprevir, JZ01–15, 5172-mcP1P3 and AM-07, exhibited resistance profiles similar to grazoprevir against the single and double substitution variants. JZ01–15 exhibited the best profile with similar activity against the D168A substitution as grazoprevir. We recently showed that P1-P3 macrocyclic inhibitors with small substitutions at the 3-position of the P2 quinoxaline, such as JZ01–15 and 5172-mcP1P2, maintain potency against single RASs (Matthew et al., 2017). In contrast, AM-07 has a larger thiophene- substituted quinoxaline that makes cation-π interactions with Arg155 and hydrophobic interactions with Tyr56. As a result of this binding mode, this inhibitor is highly susceptible to D168A, resulting in a higher loss in potency against the double substitution variant compared to grazoprevir and the other two analogs. Thus, structural features of the inhibitor, especially the identity of the P2 heterocyclic moiety, determine susceptibility to the Y56H/D168A protease variant.
To investigate the molecular basis of resistance conferred by Y56H substitution, grazoprevir crystal structures bound to WT and mutant protease were analyzed and compared. The comparative structural analysis revealed significant changes in inhibitor packing at the P2 quinoxaline and P2-P4 macrocycle region due to Y56H substitution. The P2 quinoxaline moiety moved away from the catalytic residue His57 toward the S2 subsite residues, weakening the critical π-π interactions between the quinoxaline and His57 ring (Figure 4C). His57 side chain adopted an alternate conformation relative to WT, further affecting the stacking interactions with the P2 moiety. Moreover, Asp168 shifted closer to the Arg155 residue, exhibiting a conformation similar to other inhibitor-protease co-complexes (Romano et al., 2012). Thus unlike danoprevir, the active site of protease and stacking of grazoprevir were perturbed by the Y56H substitution.
Compared to WT protease, the structure and binding mode of grazoprevir to D168A variant was minimally altered (Figure 4D). However, as we previously reported, grazoprevir resistance against the D168A variant is due to disruption of the ionic network between Arg155 and Asp168 (Romano et al., 2012). As indicated by the structural analysis, the individual Y56H and D168A substitutions have different mechanisms in conferring resistance, and combination of these substitutions cause a significant reduction in grazoprevir potency. However, whether Y56H and D168A substitutions together further alter inhibitor packing, or exacerbate the loss of critical interactions with the His57 ring observed in the Y56H co-complex is unclear.
Altered dynamics correlate with structural mechanisms of resistance
To interrogate the resistance mechanism against the double substitution variant further and complement our experimental data we turned to computational methods. Molecular dynamics simulations were performed to investigate the dynamic mechanism of resistance for both danoprevir and grazoprevir, as we have previously shown that changes in HCV protease dynamics underlie susceptibility to resistance (Soumana et al., 2016b).
To assess inhibitor packing against the protease active site, inter-molecular vdW interactions were assessed over the MD trajectories for each residue at the active site (Figure 5, S3). Both single substitution and the double substitution variants showed an overall decrease in total vdW contact energies (5 and 10 kcal/mol, respectively) relative to WT protease for grazoprevir and danoprevir, consistent with the experimental loss in binding affinity. As expected, D168A substitution decreased contacts of this residue with grazoprevir in both the single and double substitution proteases (Figure 5A-D, S3a). Interestingly, the Y56H substitution not only decreased grazoprevir interactions at this position but also at other binding site residues, especially at the catalytic His57. This loss was compounded when D168A was combined with Y56H, with considerable loss of contacts not only locally at the site of mutation but distributed throughout the binding site, including at residues Thr42, Gly58, and Gly137. There was a striking ~2.5 kcal/mol loss in vdW interactions of grazoprevir with His57 relative to WT protease, indicating that the addition of Y56H substitution to the D168A background causes severe loss of interactions with the neighboring catalytic His57 residue.
Figure 5: Packing of inhibitors in the NS3/4A protease active site during MD trajectories.
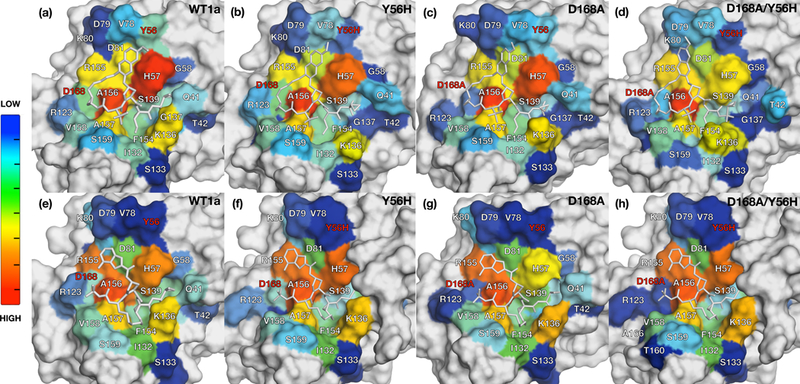
The van der Waals (vdW) contact potentials averaged from MD simulations of protease active site residues for grazoprevir bound to (a) WT1a, (b) Y56H, (c) D168A, (d) Y56H/D168A and danoprevir bound to (e) WT1a, (f) Y56H, (g) D168A, (h) and Y56H/D168A proteases, respectively. The warmer (red) and cooler (blue) colors indicate more and less contacts with the inhibitor, respectively. Drug resistance residues are highlighted in red. See also Figures S2 and S3.
In contrast to grazoprevir, danoprevir binding to the active site was similar to WT when bound to each single and double substitution variant, with more localized loss of interactions due to the substitutions (Figure 5E-5H, S3b). While the D168A substitution caused decreased interactions with both this residue and the catalytic His57, the addition of Y56H did not cause any further considerable changes in the double substitution variant, in direct contrast to the case of grazoprevir. Overall, the binding landscape of danoprevir was relatively unaltered in the variants relative to WT protease in the dynamic analysis (Figure 5), similar to what was seen in the crystal structures (Figure S3).
The loss of correlation of motions between the inhibitor and protease can be an indicator of resistance, as we have previously shown for GT3 HCV NS3/4A protease (Soumana et al., 2016a). To examine the coupling of atomic fluctuations between the inhibitor and protease, the correlation coefficients between the inhibitor and the protease backbone were calculated (Figure 6). In the WT complex, the dynamics of grazoprevir were highly correlated with the motions of the residues in the active site (Figure 6A). This coupling was the most pronounced for active site residues 134–136 and 156–157, displaying correlations with all inhibitor moieties. The dynamics of the P1 and P1 ‘ moieties of grazoprevir were highly coupled to the dynamics of residue His57.
Figure 6: Cross-correlation coefficients of inhibitors and atomic fluctuations of active site residues mapped onto protease surface.
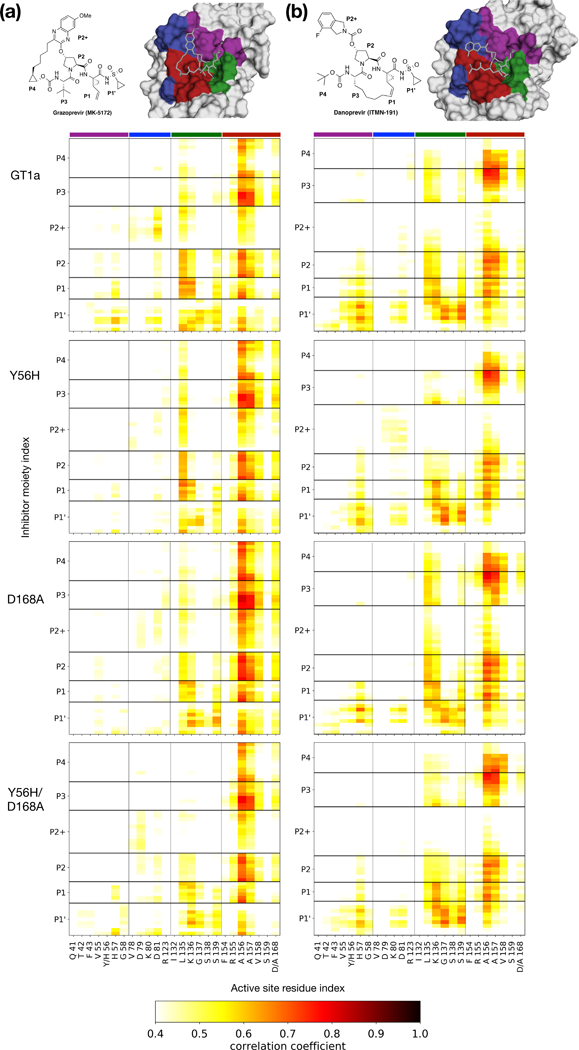
Protease-inhibitor dynamic coupling of (a) grazoprevir and (b) danoprevir bound to GT1a, Y56H, D168A, and Y56H/D168A proteases. Warm colors in the matrices indicate increased correlations. Residues are colored on the surface to indicate their location in the active site.
Additionally, the dynamics of P2 quinoxaline, and P1 and P1’ moieties were coupled with catalytic residue Asp81, and Leu82. Neither of these correlations was present when the Tyr56 was mutated to a histidine. Apart from this loss of correlations, Y56H substitution had only little effect on intra-molecular dynamics.
In contrast, the D168A substitution severely reduced the dynamic coupling of grazoprevir with the protease active site. In addition to the loss of P1 and P1’ coupling with catalytic residues His57 and Asp81, correlation between the P2 quinoxaline and residues 132–138 were severely reduced. In the Y56H/D168A protease, the correlation of motions between grazoprevir and residues 132–138 was completely lost, in addition to severe weakening of the coupled dynamics between residue 157 and the inhibitor moieties. Thus, the loss of coupling between inhibitor and protease dynamics correlated with reduced inhibitor potency against both the single mutant and the Y56H/D168A double substitution variant.
The dynamics of danoprevir-protease complexes were also in agreement with the structural mechanisms of resistance revealed from crystal structures (Figure 6B). In the WT complex, danoprevir dynamic coupling with active site residues were most pronounced at residues 56–58, 135–136 and 156–157. The correlations of danoprevir with the Y56H protease were essentially unaltered relative to WT complex. This similarity is in agreement with structural analysis, as the Tyr56 substitution does not impact binding of danoprevir (Figure 4). In the D168A complex, correlations of the P1’ moiety with residues 55–58 were reduced, while P2+ isoindoline moiety had increased correlations with residue K136, in agreement with increased intermolecular vdW contacts (Figure 5). In the Y56H/D168A variant, the correlations of motions were similar to those seen in the D168A variant. Thus, for inhibitors with binding modes similar to danoprevir, substitutions at Asp168 are the primary cause of resistance to the double substitution variant Y56H/D168A.
Discussion
Although drug resistance has been a major problem in the efficacy of anti-HCV therapeutics, especially for NS3/4A PIs, newer generation inhibitors are robust against single-site substitutions that were once detrimental to PI clinical viability. More importantly, there are currently two all-oral regimens that have pan-genotypic HCV activity including against the evasive GT3 (Ng et al., 2017). Though much progress has been made in anti-HCV therapeutics, a new challenge that may threaten the success of PIs is the emergence of viral variants with more than one substitution in the protease (Gane et al., 2016; Ng et al., 2017). In this study we reveal the structural and dynamic mechanisms of drug resistance for the Y56H/D168A protease, a double-site RAS variant that has been identified in patients who failed therapy with PI-containing regimens (Gane et al., 2016).
The Y56H/D168A variant was resistant to all tested PIs, and the inhibitor binding mode determined the molecular mechanism of resistance. Prior to the development of grazoprevir, PIs typically contained large heterocyclic P2 moieties that strongly interacted with S2 subsite residues (Ali et al., 2013; Romano et al., 2012). This binding mode resulted in detrimental loss of potency against single-site RASs in the protease active site, especially at residues Arg155 and Asp168 (Ali et al., 2013; Romano et al., 2012). In the inhibitor-bound state, HCV NS3/4A protease has an extensive active site electrostatic network that spans the catalytic triad residues His57 and Asp81 all the way to S2 subsite residues Arg155, Asp168 and Arg123. Residues Arg155 and Asp168, located beyond the substrate envelope, form a salt bridge that is critical to inhibitor binding and disrupted upon substitution at either residue (Romano et al., 2010). For inhibitors with a binding mode similar to danoprevir, disruption of this electrostatic network due to an Asp168 substitution causes rearrangement of the Arg155 side chain, resulting in the loss of favorable cation-π interactions between the P2 isoindoline and Arg155 guanidinium group (O’Meara et al., 2013; Romano et al., 2012). For such inhibitors that do not stack on the catalytic residues and have no physical interactions with Tyr56, resistance to the double substitution is predominantly due to D168A. Thus the addition of Y56H substitution to D168A protease does not cause any further active site changes or loss in potency.
Grazoprevir and newer generation inhibitors have a P2 quinoxaline moiety that makes extensive interactions with the catalytic triad, reducing susceptibility to single-site RASs at the S2 subsite (Soumana et al., 2016b). However, this binding mode causes vulnerability to substitutions that result in loss of critical π-π stacking interactions with the catalytic residue His57. The crystal structure of grazoprevir bound to the Y56H protease determined here reveals that this interaction is weakened when neighboring Tyr56 mutates to a smaller His. This loss is compounded in the double substitution Y56H/D168A variant, thereby destabilizing the binding of grazoprevir. The alterations due to Y56H and the double substitution Y56H/D168A are unlikely to impact the recognition and processing of the viral substrates (Table S1), as unlike grazoprevir the substrates do not stack on the aromatic surface of His57 (Romano et al., 2012; Romano et al., 2010; Romano et al., 2011). The substrates also make no direct contact with Tyr56, so substitutions at Tyr56 are unlikely to either directly or indirectly impact substrate turnover. As a general rule, the reliance of an inhibitor on interactions with the target that are not essential for biological function creates an opportunity for resistance causing substitutions (Romano et al., 2012; Soumana et al., 2016b; Yilmaz et al., 2016). When these interactions are within the substrate envelope or with catalytic residues, the chance of resistance emerging is minimized. Nevertheless, in this study we observe an indirect mechanism of resistance that disrupts interactions with the catalytic residue through substitution in a neighboring residue not involved in substrate recognition.
Robustness of grazoprevir against single-site RASs has led to drug design efforts by pharmaceutical companies to pursue PIs with similar scaffolds to grazoprevir. Pan-genotypic PIs voxilaprevir and glecaprevir have a P2 quinoxaline and P2-P4 macrocycle as in grazoprevir. Considering the high similarity in the scaffolds of these latest-generation inhibitors, there is a danger that all PIs currently in clinic might be susceptible to the same resistant variants, including Y56H/D168A. While these inhibitors have low susceptibility to single-site substitutions at residues Arg155 and Asp168, they have selected for double substitution variants in in vitro studies (Ng et al., 2017). In fact, in vitro resistance testing of glecaprevir selected for resistance against GT3 double substitution variant Y56H/Q168R losing almost 1400-fold in potency (Ng et al., 2017). This suggests that double substitution variants containing Y56H may emerge in other genotypes and reduce the clinical effectiveness of PIs.
Under increased drug pressure, more protease variants with more than one substitution will likely become clinically relevant. The accumulation of additional substitutions can allow RAS variants to emerge that alone are not viable, but in combination can rescue the viral fitness. We previously demonstrated that inhibitors with a P2-P4 macrocycle are highly susceptible to substitutions at Ala156, as a change to a larger side chain results in steric clashes with the inhibitor’s macrocycle. Ala156 substitutions cause low replicative capacity, but additional changes at other positions in the NS3/4A protease can improve enzymatic activity and thus viral fitness, leading to clinically relevant variants. Voxilaprevir and glecaprevir also select for substitutions at Ala156 in vitro, which causes a large fold shift in inhibitor potency. Moreover, Ala156-Asp168 double substitutions have been selected in vitro, which improve fitness. Although not yet observed clinically, the A156T substitution if coupled with such a fitness- rescuing second substitution could cause resistance to all P2-P4 macrocyclic PIs with a P2 quinoxaline moiety. In fact, the additional substitution does not have to occur at the active site. We have shown in HIV-1 protease that active site and distal substitutions often occur in combination to confer resistance (Ragland et al., 2014). Similarly, glecaprevir selected substitutions at Ala156 in combination with Gln/Pro89 in GT 1a/b, which is located outside of the active site. This additional substitution at position 89 appears to have improved replicative efficiency to 100% (Ng et al., 2017). Understanding the molecular mechanisms of resistance and enzymatic fitness of these multi-substituted variants will be necessary to improve potency of PIs against emerging resistant variants.
One strategy used in rational drug design of PIs to avoid drug resistance is exploiting interactions with the catalytic triad residues. The catalytic triad residues are critical for the biological function of the protease and thus almost always invariant. However, we find that even though the catalytic residues themselves cannot mutate to confer resistance, substitutions at other locations can disrupt critical inhibitor interactions of these residues. While exploiting interactions with evolutionarily constrained residues is still one of the best strategies for inhibitor design, diverse PIs need to be considered, preferably with enhanced interactions with the catalytic residues that cannot be disrupted by nearby changes. As new drugs and combinations are developed, in addition to targeting multiple viral proteins or mechanisms, drug resistance needs to be considered at the outset of inhibitor design to minimize the emergence of resistance.
Additionally, the arsenal of PIs needs to be diversified, as the similarity in scaffold is likely to lead to cross-resistance and susceptibility to multi-substituted variants.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Celia A. Schiffer (Celia.Schiffer@umassmed.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Expression and Purification of NS3/4A Constructs
The HCV GTla NS3/4A protease gene described in the Bristol Myers Squibb patent was synthesized by GenScript and cloned into a PET28a expression vector (Wittekind et al., 2002). The D168A, Y56H and Y56H/D168A genes were engineered using the Q5 site-directed mutagenesis protocol from New England Biolabs. Protein expression and purification were carried out as previously described with minor modifications (Romano et al., 2012). Briefly, transformed Escherichia coli BL21 (DE3) cells were grown in TB media containing 30 μg/mL of kanamycin antibiotic at 37 °C. After reaching an OD600 of 0.8, cultures were induced with 1 mM IPTG and harvested after 3 h of expression. Cells were pelleted by centrifugation, resuspended in Resuspension buffer (RB) [50 mM phosphate buffer, 500 mM NaCl, 10% glycerol, 2 mM β-ME, pH 7.5] and frozen at −80 °C for storage.
Cell pellets were thawed and lysed via cell disruptor (Microfluidics Inc.) two times to ensure sufficient DNA shearing. Lysate was centrifuged at 19,000 rpm, for 25 min at 4 °C. The soluble fraction was applied to a nickel column (Thermo Scientific) pre-equilibrated with RB. The beads and soluble fraction were incubated at 4 °C for 1.5 h and the lysate was allowed to flow through. Beads were washed with RB supplemented with 20 mM imidazole and eluted with RB supplemented with 200 mM imidazole. The eluent was dialyzed overnight (MWCO 10 kD) to remove the imidazole, and the His-tag was simultaneously removed with thrombin treatment.
The eluate was judged >90% pure by polyacrylamide gel electrophoresis, concentrated, flash frozen, and stored at −80 °C.
Protein expression and purification were carried out in transformed Escherichia coli BL21(DE3) cells were grown in TB media containing 30 μg/mL of kanamycin antibiotic at 37 °C. After reaching an OD600 of 0.8, cultures were induced with 1 mM IPTG and harvested after 3 h of expression. Cells were pelleted by centrifugation, resuspended in Resuspension buffer (RB) [50 mM phosphate buffer, 500 mM NaCl, 10% glycerol, 2 mM β-ME, pH 7.5] and frozen at −80 °C for storage.
METHOD DETAILS
Enzyme Inhibition Assays
For each assay, 2 nM of NS3/4A protease (GTla, Y56H, D168A and Y56H/D168A) was preincubated at room temperature for 1 h with increasing concentration of inhibitors in assay buffer (50 mM Tris, 5% glycerol, 10 mM DTT, 0.6 mM LDAO, and 4% dimethyl sulfoxide, pH 7.5). Inhibition assays were performed in non-binding surface 96-well black half-area plates (Corning) in a reaction volume of 60 μL. The proteolytic reaction was initiated by the injection of 5 μL of HCV NS3/4A protease substrate Ac-DE-D(Edans)-EE-Abu-ψ-[COO]-AS-K(Dabcyl)-NH2 (AnaSpec), to a final concentration of 200 nM and kinetically monitored using a Perkin Elmer EnVision plate reader (excitation at 485 nm, emission at 530 nm). The initial velocities were fit to the Morrison equation to obtain the Ki value. Gibbs free energy of binding was calculated using the following equation: ΔG = RTlniKi
Determination of the Inner Filter Effect
The inner filter effect (IFE) for the NS3/4A protease substrate was determined using a previously described method (Liu et al., 1999). Briefly, fluorescence end-point readings were taken for substrate concentrations between 0 μM and 20 μM. Afterward, free 5-FAM fluorophore was added to a final concentration of 25 μM to each substrate concentration and a second round of fluorescence end-point readings was taken. The fluorescence of free 5-FAM was determined by subtracting the first fluorescence end point reading from the second round of readings. IFE corrections were then calculated by dividing the free 5-FAM florescence at each substrate concentration by the free 5-FAM florescence at zero substrate.
Determination of Michaelis-Menten (Km) Constant
Km constants for GT1 and D168A protease were previously determined (Ali et al., 2013). The Km of Y56H and Y56H/D168A proteases were determined using the following method. A 20 μM concentration of substrate [Ac-DE-Dap(QXL520)- EE-Abu-γ-[COO]AS-C(5-FAMsp)-NH2] (AnaSpec) was serially diluted into assay buffer [50 mM Tris, 5% glycerol, 10 mM DTT, 0.6 mM LDAO, and 4% dimethyl sulfoxide] and proteolysis was initiated by rapid injection of 10 μL protease (final concentration 20 nM) in a reaction volume of 60 μL. The fluorescence output from the substrate cleavage product was measured kinetically using an EnVision plate reader (Perkin-Elmer) with excitation wavelength at 485 nm and emission at 530 nm. Inner filter effect corrections were applied to the initial velocities (Vo) at each substrate concentration. Vo versus substrate concentration graphs were globally fit to the Michaelis-Menten equation to obtain the Km value.
Cell-Based Drug Susceptibility Assays
Cell-based assays using an HCV Conl (genotype lb) reporter replicon containing the H77 (genotype la) NS3 protease region were used to assess HCV protease inhibitor susceptibility. Expression of HCV non-structural genes leads to RNA replication and maintenance of luciferase expression. Mutations (Y56H, D168A, and Y56H/D168A) were introduced into the wildtype H77 NS3 protease region of the Con1 (genotype 1b) replicon using the “megaprimer” method of site-directed mutagenesis (Sarkar and Sommer, 1990). Huh7 cells, previously cured for optimal HCV replicon replication, were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 0.1 mM non-essential amino acids (DMEM - 10% FBS). Huh7 cells were transfected with replicon RNA transcripts using electroporation and maintained in the absence or presence of serially diluted protease inhibitors. Replicon replication was then assessed by measuring luciferase activity (relative light units) 96 h post electroporation. The drug concentrations required to inhibit replicon replication by 50% (EC50) were calculated directly from the drug inhibition curves.
Inhibitor Synthesis
The P1-P3 macrocyclic NS3/4A protease inhibitors with flexible P2 quinoxaline moieties were synthesized using the reaction sequence outlined in Scheme S1. The key tert-butoxycarbonyl (Boc)-protected P2 intermediates 1a-d were prepared from the corresponding 3-substituted quinoxalin-2-ones by a cesium carbonate-mediated nucleophilic substation reaction with the activated cis-hydroxyproline derivative as described previously (Matthew et al., 2017). The target macrocyclic inhibitors were assembled from the P2 intermediates 1a-d using a sequence of deprotection and peptide coupling steps followed by the ring-closing metathesis (RCM) reaction. Briefly, removal of the Boc group in 1a-d using 4 N hydrochloric acid in 1,4-dioxane provided the amine salts 2a-d, which were coupled with the (S)-2-((tert-butoxycarbonyl)amino)non-8- enoic acid 3 in the presence of 1-[bis(dimethylamino)-methylene]-1H-1,2,3-triazolo[4,5- b]pyridinium 3-oxid hexafluorophosphate (HATU) and diisopropylethylamine (DIEA) in dimethylformamide to yield the P2-P3 ester intermediates 4a-d. The esters 4a-d were treated with lithium hydroxide in a mixture of tetrahydrofuran and water to afford the carboxylic acids 5a-d. The required P1-P1’ acylsulfonamide intermediates 6 and 7 were prepared following reported methods (Wang et al., 2006) (Rudd et al., 2015) and were reacted with the P2-P3 acid intermediates 5a-d under HATU/DIEA coupling conditions to provide the bis-olefin intermediates 8b-d and 9a. Finally, cyclization of the bis-olefin intermediates was accomplished using a highly efficient RCM catalyst 1,3-bis(2,4,6-trimethylphenyl)-4,5-dihydroimidazol-2- ylidene[2-(i-propoxy)-5-(N,N-dimethylaminosulfonyl)-phenyl]methyleneruthenium(II) dichloride (Zhan Catalyst-1B) and provided the target inhibitors 11b-d (5172-mcP1P3, AM-07, and MG-28, respectively) and 12a (JZ01–15). Details of experimental procedures and characterization data for intermediates and final compounds have been reported previously (Matthew et al., 2017).
Scheme S1.
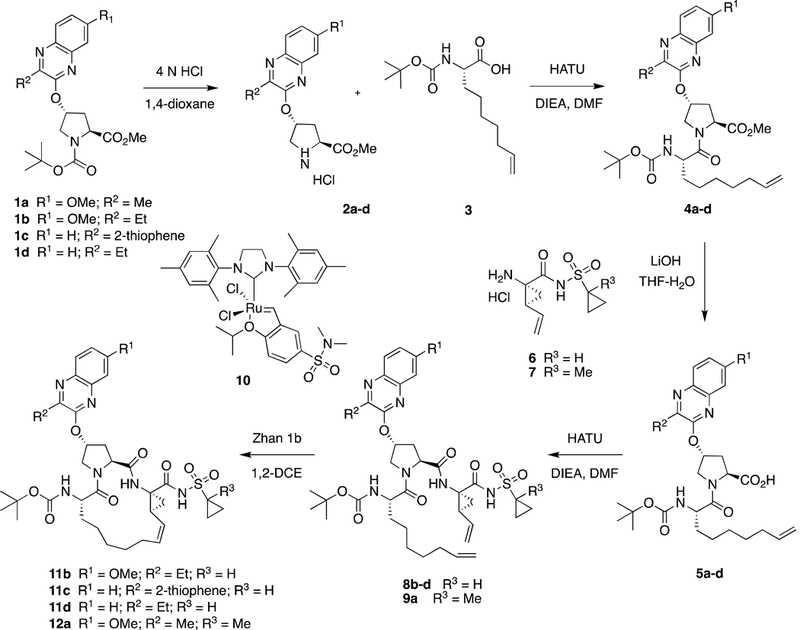
Synthesis of HCV NS3/4A protease inhibitors
Paritaprevir (ABT-450) and danoprevir (ITMN-191) were synthesized from the corresponding P2 proline derivatives using a similar convergent reaction sequence as described previously (Romano et al., 2010) (Ali et al., 2013). Briefly, the P2 and P1-P1’ fragments were preassembled and the macrocyclic drug compound was generated by a four-step reaction sequence, including P2-P3 amide coupling, ester hydrolysis, coupling with the P1-P1’ fragment, and RCM reaction. The P2-P4 macrocyclic inhibitor grazoprevir (MK-5172) was prepared following the synthetic methods reported by Harper et al. (Harper et al., 2012).
Crystallization and structure determination
Protein expression and purification were carried out as previously described (Romano et al., 2012). The Ni-NTA purified WT1a protein was thawed, concentrated to 3 mg/mL, and loaded on a HiLoad Superdex75 16/60 column equilibrated with gel filtration buffer (25 mM MES, 500 mM NaCl, 10% glycerol, and 2 mM DTT, pH 6.5). The protease fractions were pooled and concentrated to 25 mg/mL with an Amicon Ultra-15 10 kDa filter unit (Millipore). The concentrated samples were incubated for 1 h with 3:1 molar excess of inhibitor. Diffraction- quality crystals were obtained overnight by mixing equal volumes of concentrated protein solution with precipitant solution (20–26% PEG-3350, 0.1 M sodium MES buffer, 4% ammonium sulfate, pH 6.5) at RT or 15 °C in 24-well VDX hanging drop trays. Crystals were harvested and data was collected at 100 K. Cryogenic conditions contained the precipitant solution supplemented with 15% glycerol or ethylene glycol.
X-ray diffraction data were collected at Advance Photon Source beamline 23-ID-B or our in-house Rigaku X-ray system with a Saturn 944 detector. All datasets were processed using HKL- 3000 (Otwinowski and Minor, 1997). Structures were solved by molecular replacement using PHASER (McCoy et al., 2007). Model building was performed manually using Coot (Emsley and Cowtan, 2004) and refinement was carried out using phenix.refine (Adams et al., 2010) with four cycles automatically generating all hydrogens in the model and updating waters. Under refinement settings XYZ coordinates, Real-space and individual B-factors were chosen with “optimize X-ray/stereochemistry weight” and “optimize X-ray/ADP weight” options. The final structures were evaluated with MolProbity (Davis et al., 2007) prior to deposition in the PDB. Structure analysis, superposition and figure generation were done using PyMOL. X-ray data collection and crystallographic refinement statistics are presented in Table 1.
System Preparation for Molecular Dynamics Simulations
Crystal structures of HCV protease bound to grazoprevir were taken from the Protein Databank (PDB IDs 3SUD and 3SUF) (Romano et al., 2012). When there were multiple copies of the protease-inhibitor complex in the crystallographic unit, the complex with the lowest overall B- Factors was chosen. In the case of the double substitution variant, the complex was modeled in silico using the Prime structure prediction wizard. Protein structures were then prepared for simulation using the Protein Preparation Wizard from the Schrodinger Suite (Sastry et al., 2013), keeping all co-crystallized water molecules. Missing atom were added using Prime (Jacobson et al., 2002). The protonation state of the protein side chains at pH 7.0 was determined using PROPKA (Olsson et al., 2011; Sondergaard et al., 2011). The hydrogen bond network was optimized by exhaustive sampling of the orientation of crystallographic waters and side chains of polar amino acids. Finally, the structure was subjected to gradient minimization with convergence criterion 0.5 Å using Impref (Banks et al., 2005).
Molecular Dynamics Simulations
Molecular dynamic simulations of protein-inhibitor complexes were carried out as previously described (Ragland et al., 2014). The protein-inhibitor complex was placed in a cubic solvent box maintaining at least 1.5 nm spacing between any solute atom and the box boundaries. The system’s net charge was neutralized by adding chloride ions; additionally, sodium and chloride ions were added to a total salt concentration of 0.15 M.
MD simulations were carried out using the Desmond software suite.(Bowers et al.) Protein and ligand were parameterized using the OPLS3 force field (Harder et al., 2015). For the water molecules, TIP3P force field parameters were used (Jorgensen et al., 1983). During simulations, long-range electrostatic forces were calculated using the Particle Mesh Ewald method (Essmann et al., 1995). Short-range non-bonded forces were truncated smoothly at 1.2 nm. The RESPA integrator was used with a 2 fs time step for bonded and short-range non-bonded forces and a 6 fs time step for long-range electrostatic forces (Tuckerman et al., 1992).
Before running MD simulations, the solvated system was energy minimized using a stepwise protocol. In the first iteration, all solvent molecules were minimized using 10 steepest decent steps followed by up to 5000 L-BSFG minimization steps. The convergence criterion was an energy gradient of 0.5 kcal mol−1 Å−1 while applying solute heavy atoms a force constant of 1000 kcal mol−1 Å-2. In the second iteration only the backbone restraints were kept and the system was subjected to the same minimization procedure as in the first iteration. In the third step, the restraints on the backbone heavy atoms were lowered to 5 kcal mol−1 Å-2. For the final minimization step, all restraints were removed and the system was minimized using the L-BFSG method until an energy gradient of 0.05 kcal mol−1 Å−1 was reached.
Following minimization, a number of short MD simulations were performed to equilibrate the system. Initially a 12 ps simulation in the NVT ensemble using the Berendsen thermostat at 10 K was performed. The backbone position was restrained using a force constant of 50 kcal mol−1 Å-2. This was followed by a 24 ps simulation in the NPT ensemble maintaining the restraints on the protein backbone. Subsequently a 50 ps unrestrained NPT simulation was run during which the temperature was increased from 10 K to the target temperature of 300 K. This was followed by a 500 ps NPT simulation at 300 K allowing the system to thermalize. The final production stage consisted of a 100 ns simulation in the NPT ensemble at 300 K and lbar. Atomic coordinates were recorded every 5 ps.
Correlated Motions
To measure both linear and non-linear correlated protein dynamics, the Linear Mutual Information between atom pairs was calculated. This is based on the approach proposed by Lange and Grubmueller (Lange and Grubmuller, 2006). Briefly, correlated dynamics were measured by calculating their mutual information where x and y are two sets of atomic coordinates, p(x) and p(y) are there marginal probability distributions and p(x;y) is the joint probability distribution. If the two atoms x and y are fully independent the joint probability distribution p(x;y) equals the sum of its marginal probability distributions, thus I[x;y] becomes 0. If the two atoms are not completely independent I[x;y] assumes a positive value. To improve interpretability the mutual information I(x;y) is subsequently transformed into the generalized correlation coefficient with d=3 since x and y are Euclidean coordinates. has a value of 0 for fully independent motions and a value of 1 for fully coupled dynamics. To calculate the generalized correlation coefficient, snapshots were taken from a trajectory at 5ps intervals. Global, translational and rotational motions were minimized by aligning all snapshots to the first frame of the trajectory.
QUANTIFICATION AND STATISTICAL ANALYSIS
In enzyme kinetics assays, three independent data sets were collected for each experiment, which were globally fit to the appropriate equation. The reported are the errors from the fit.
Crystal structure model building and refinement were performed using Coot (Emsley and Cowtan, 2004) and PHENIX (Adams et al., 2010), respectively. The final structures were evaluated with MolProbity (Davis et al., 2007) prior to deposition in the PDB. To limit the possibility of model bias throughout the refinement process, 5% of the data were reserved for the free R-value calculation (Brunger, 1992). X-ray data collection and crystallographic refinement statistics are presented in Table 1.
All MD simulations were run in triplicate using the aforementioned protocol. In calculation of the correlated motions from MD simulations, pairwise and marginal probability distributions were estimated by a Gaussian distribution. This approach has the advantage to capture both linear and non-linear correlated motions while avoiding computationally more demanding density estimates.
Supplementary Material
Highlights.
Y56H/D168A variant causes high levels of resistance
The two mutations have interdependency in conferring resistance
Y56H causes disruption of stacking interactions with the catalytic His57
Double mutations may confer cross-resistance to current PIs in clinical development
Acknowledgements
This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02–06CH11357. We thank beam line specialist at 23-ID-B for their help in data collection. This work was supported by a grant from the National Institute of Allergy and Infectious Diseases of the NIH (R01 AI085051). ANM was also supported by the National Institute of General Medical Sciences of the NIH (F31 GM119345). We thank Profs. Brian Kelch, Paul Thompson, William Royer and Dan Bolon for helpful discussions.
Footnotes
DATA AND SOFTWARE AVAILABILITY
The crystal structures have been deposited in the Protein Data Bank under ID codes 6C2M, 6C2O, 6C2N.
Declaration of Interest
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- PyMOL: The PyMOL Molecular Graphics System, Version 1.8, Schrödinger, LLC. [Google Scholar]
- Schrödinger Release 2017–4: Prime, Schrödinger, LLC, New York, NY, 2017. [Google Scholar]
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali A, Aydin C, Gildemeister R, Romano KP, Cao H, Özen A, Soumana D, Newton A, Petropoulos CJ, Huang W, et al. (2013). Evaluating the role of macrocycles in the susceptibility of hepatitis C virus NS3/4A protease inhibitors to drug resistance. ACS Chem. Biol. 8, 1469–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asselah T, Boyer N, Saadoun D, Martinot-Peignoux M, and Marcellin P (2016). Direct- acting antivirals for the treatment of hepatitis C virus infection: optimizing current IFN-free treatment and future perspectives. Liver Int. 36, 47–57. [DOI] [PubMed] [Google Scholar]
- Banks JL, Beard HS, Cao Y, Cho AE, Damm W, Farid R, Felts AK, Halgren TA, Mainz DT, and Maple JR (2005). Integrated modeling program, applied chemical theory (IMPACT). J. Comput. Chem. 26, 1752–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blach S, Zeuzem S, Manns M, Altraif I, Duberg A-S, Muljono DH, Waked I, Alavian SM, Lee M-H, and Negro F (2017). Global prevalence and genotype distribution of hepatitis C virus infection in 2015: a modelling study. Lancet Gastroenterol. Hepatol. 2, 161–176. [DOI] [PubMed] [Google Scholar]
- Bourliere M, Gordon SC, Flamm SL, Cooper CL, Ramji A, Tong M, Ravendhran N, Vierling JM, Tran TT, Pianko S, et al. (2017). Sofosbuvir, velpatasvir, and voxilaprevir for previously treated HCV infection. N. Engl. J. Med. 376, 2134–2146. [DOI] [PubMed] [Google Scholar]
- Bowers KJ, Chow E, Xu H, Dror RO, Eastwood MP, Gregerson BA, Klepeis JL, Kolossvary I, Moraes MA, Sacerdoti FD, et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. Paper presented at: Proceedings of the ACM/IEEE Conference on Supercomputing (SC06) (Tampa, Florida, United States). [Google Scholar]
- Brunger AT (1992). Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature 355, 472–475. [DOI] [PubMed] [Google Scholar]
- Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, Snoeyink J, Richardson JS, et al. (2007). MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucl. Acids Res. 35, W375–W383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, and Pedersen LG (1995). A smooth particle mesh ewald method. J. Chem. Phys. 103, 8577–8593. [Google Scholar]
- Falade-Nwulia O, Suarez-Cuervo C, Nelson DR, Fried MW, Segal JB, and Sulkowski MS (2017). Oral direct-acting agent therapy for hepatitis c virus infection: a systematic review. Ann. Intern. Med. 166, 637–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fried MW, Shiffman ML, Reddy KR, Smith C, Marinos G, Goncales FL Jr., Haussinger D, Diago M, Carosi G, Dhumeaux D, et al. (2002). Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N. Engl. J. Med. 347, 975–982. [DOI] [PubMed] [Google Scholar]
- Gane E, Ben Ari Z, Mollison L, Zuckerman E, Bruck R, Baruch Y, Howe AY, Wahl J, Bhanja S, Hwang P, et al. (2016). Efficacy and safety of grazoprevir + ribavirin for 12 or 24 weeks in treatment-naive patients with hepatitis C virus genotype 1 infection. J. Viral Hepat. 23, 789–797. [DOI] [PubMed] [Google Scholar]
- Ghany MG, Nelson DR, Strader DB, Thomas DL, and Seeff LB (2011). An update on treatment of genotype 1 chronic hepatitis C virus infection: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 54, 1433–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gower E, Estes C, Blach S, Razavi-Shearer K, and Razavi H (2014). Global epidemiology and genotype distribution of the hepatitis C virus infection. J. Hepatol. 61, S45–S57. [DOI] [PubMed] [Google Scholar]
- Guo Z, Black S, Hu Y, McMonagle P, Ingravallo P, Chase R, Curry S, and Asante- Appiah E (2017). Unraveling the structural basis of grazoprevir potency against clinically relevant substitutions in hepatitis C virus NS3/4A protease from genotype 1a. J. Biol. Chem. 292, 6202–6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder E, Damm W, Maple J, Wu C, Reboul M, Xiang JY, Wang L, Lupyan D, Dahlgren MK, and Knight JL (2015). OPLS3: a force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 12, 281–296. [DOI] [PubMed] [Google Scholar]
- Harper S, McCauley JA, Rudd MT, Ferrara M, DiFilippo M, Crescenzi B, Koch U, Petrocchi A, Holloway MK, Butcher JW, et al. (2012). Discovery of MK-5172, a macrocyclic hepatitis C virus NS3/4a protease inhibitor. ACS Med. Chem. Lett. 3, 332–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horovitz A (1996). Double-mutant cycles: a powerful tool for analyzing protein structure and function. Fold Des. 1, R121–126. [DOI] [PubMed] [Google Scholar]
- Jacobson MP, Friesner RA, Xiang Z, and Honig B (2002). On the role of the crystal environment in determining protein side-chain conformations. J. Mol. Biol. 320, 597–608. [DOI] [PubMed] [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, and Klein ML (1983). Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935. [Google Scholar]
- Kwo PY, Poordad F, Asatryan A, Wang S, Wyles DL, Hassanein T, Felizarta F, Sulkowski MS, Gane E, Maliakkal B, et al. (2017). Glecaprevir and pibrentasvir yield high response rates in patients with HCV genotype 1–6 without cirrhosis. J. Hepatol. 67, 263–271. [DOI] [PubMed] [Google Scholar]
- Lange OF, and Grubmuller H (2006). Generalized correlation for biomolecular dynamics. Proteins 62, 1053–1061. [DOI] [PubMed] [Google Scholar]
- Lawitz E, Gane E, Pearlman B, Tam E, Ghesquiere W, Guyader D, Alric L, Bronowicki J-P, Lester L, Sievert W, et al. (2014). Efficacy and safety of 12 weeks versus 18 weeks of treatment with grazoprevir (MK-5172) and elbasvir (MK-8742) with or without ribavirin for hepatitis C virus genotype 1 infection in previously untreated patients with cirrhosis and patients with previous null response with or without cirrhosis (C-WORTHY): a randomised, open-label phase 2 trial. Lancet 385, 1075–1086 [DOI] [PubMed] [Google Scholar]
- Lawitz E, Yang JC, Stamm LM, Taylor JG, Cheng G, Brainard DM, Miller MD, Mo H, and Dvory-Sobol H (2017). Characterization of HCV resistance from a 3-day monotherapy study of voxilaprevir, a novel pangenotypic NS3/4A protease inhibitor. Antivir. Ther. [DOI] [PubMed] [Google Scholar]
- Lawitz EJ, O’Riordan WD, Asatryan A, Freilich BL, Box TD, Overcash JS, Lovell S, Ng TI, Liu W, Campbell A, et al. (2015). Potent antiviral activities of the direct-acting antivirals ABT-493 and ABT-530 with three-day monotherapy for hepatitis C virus genotype 1 infection. Antimicrob. Agents Chemother. 60, 1546–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Kati W, Chen CM, Tripathi R, Molla A, and Kohlbrenner W (1999). Use of a fluorescence plate reader for measuring kinetic parameters with inner filter effect correction. Anal. Biochem. 267, 331–335. [DOI] [PubMed] [Google Scholar]
- Lontok E, Harrington P, Howe A, Kieffer T, Lennerstrand J, Lenz O, McPhee F, Mo H, Parkin N, Pilot-Matias T, et al. (2015). Hepatitis C virus drug resistance-associated substitutions: state of the art summary. Hepatology 62, 1623–1632. [DOI] [PubMed] [Google Scholar]
- Matthew AN, Zephyr J, Hill CJ, Jahangir M, Newton A, Petropoulos CJ, Huang W, Kurt-Yilmaz N, Schiffer CA, and Ali A (2017). Hepatitis c virus NS3/4A protease inhibitors incorporating flexible P2 quinoxalines target drug resistant viral variants. J. Med. Chem. 60, 5699–5716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messina JP, Humphreys I, Flaxman A, Brown A, Cooke GS, Pybus OG, and Barnes E (2015). Global distribution and prevalence of hepatitis C virus genotypes. Hepatology 61, 77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng TI, Tripathi R, Reisch T, Lu L, Middleton T, Hopkins TA, Pithawalla R, Irvin M, Dekhtyar T, Krishnan P, et al. (2017). In vitro antiviral activity and resistance profile of the next-generation hepatitis c virus NS3/4A protease inhibitor glecaprevir. Antimicrob. Agents Chemother. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Meara JA, Lemke CT, Godbout C, Kukolj G, Lagace L, Moreau B, Thibeault D, White PW, and Llinas-Brunet M (2013). Molecular mechanism by which a potent hepatitis C virus NS3-NS4A protease inhibitor overcomes emergence of resistance. J. Biol. Chem. 288, 5673–5681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson MH, Sondergaard CR, Rostkowski M, and Jensen JH (2011). PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 7, 525–537. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, and Minor W (1997). Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Pawlotsky JM (2016). Hepatitis C virus resistance to direct-acting antiviral drugs in interferon-free regimens. Gastroenterology 151, 70–86. [DOI] [PubMed] [Google Scholar]
- Pilot-Matias T, Tripathi R, Cohen D, Gaultier I, Dekhtyar T, Lu L, Reisch T, Irvin M, Hopkins T, Pithawalla R, et al. (2015). In vitro and in vivo antiviral activity and resistance profile of the hepatitis C virus NS3/4A protease inhibitor ABT-450. Antimicrob. Agents Chemother. 59, 988–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poordad F, Felizarta F, Asatryan A, Sulkowski MS, Reindollar RW, Landis CS, Gordon SC, Flamm SL, Fried MW, Bernstein DE, et al. (2017). Glecaprevir and pibrentasvir for 12 weeks for hepatitis C virus genotype 1 infection and prior direct-acting antiviral treatment. Hepatology 66, 389–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragland DA, Nalivaika EA, Nalam MNL, Prachanronarong KL, Cao H, Bandaranayake RM, Cai Y, Kurt-Yilmaz N, and Schiffer CA (2014). Drug resistance conferred by mutations outside the active site through alterations in the dynamic and structural ensemble of HIV-1 protease. J. Am. Chem. Soc. 136, 11956–11963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Torres M, Glass S, Hill J, Freilich B, Hassman D, Di Bisceglie AM, Taylor JG, Kirby BJ, Dvory-Sobol H, Yang JC, et al. (2016). GS-9857 in patients with chronic hepatitis C virus genotype 1–4 infection: a randomized, double-blind, dose-ranging phase 1 study. J. Viral. Hepat. 23, 614–622. [DOI] [PubMed] [Google Scholar]
- Romano KP, Ali A, Aydin C, Soumana D, Özen A, Deveau LM, Silver C, Cao H, Newton A, Petropoulos CJ, et al. (2012). The molecular basis of drug resistance against hepatitis C virus NS3/4A protease inhibitors. PLoS Pathog. 8, e1002832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano KP, Ali A, Royer WE, and Schiffer CA (2010). Drug resistance against HCV NS3/4A inhibitors is defined by the balance of substrate recognition versus inhibitor binding. Proc. Natl. Acad. Sci. U. S. A. 107, 20986–20991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano KP, Laine JM, Deveau LM, Cao H, Massi F, and Schiffer CA (2011). Molecular mechanisms of viral and host cell substrate recognition by hepatitis C virus NS3/4A protease. J. Virol. 85, 6106–6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong L, Dahari H, Ribeiro RM, and Perelson AS (2010). Rapid emergence of protease inhibitor resistance in hepatitis C virus. Sci. Transl. Med. 2, 30ra–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenquist Å, Samuelsson B, Johansson P-O, Cummings MD, Lenz O, Raboisson P, Simmen K, Vendeville S, de Kock H, Nilsson M, et al. (2014). Discovery and development of simeprevir (TMC435), a HCV NS3/4A protease inhibitor. J. Med. Chem. 57, 1673–1693. [DOI] [PubMed] [Google Scholar]
- Sarkar G, and Sommer SS (1990). The “megaprimer” method of site-directed mutagenesis. Biotechniques 8, 404–407. [PubMed] [Google Scholar]
- Sarrazin C, and Zeuzem S (2010). Resistance to direct antiviral agents in patients with hepatitis C virus infection. Gastroenterology 138, 447–462. [DOI] [PubMed] [Google Scholar]
- Sastry GM, Adzhigirey M, Day T, Annabhimoju R, and Sherman W (2013). Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 27, 221–234. [DOI] [PubMed] [Google Scholar]
- Smith DB, Bukh J, Kuiken C, Muerhoff AS, Rice CM, Stapleton JT, and Simmonds P (2014). Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment web resource. Hepatology 59, 318–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Søndergaard CR, Olsson MH, Rostkowski M, and Jensen JH (2011). Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values. J. Chem. Theory Comput. 7, 2284–2295. [DOI] [PubMed] [Google Scholar]
- Soumana DI, Ali A, and Schiffer CA (2014). Structural analysis of asunaprevir resistance in HCV NS3/4A protease. ACS Chem. Biol. 9, 2485–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soumana DI, Kurt Yilmaz N, Ali A, Prachanronarong KL, and Schiffer CA (2016a). Molecular and dynamic mechanism underlying drug resistance in genotype 3 hepatitis C NS3/4A protease. J. Am. Chem. Soc. 138, 11850–11859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soumana DI, Kurt Yilmaz N, Prachanronarong KL, Aydin C, Ali A, and Schiffer CA (2016b). Structural and thermodynamic effects of macrocyclization in HCV NS3/4A inhibitor MK-5172. ACS Chem. Biol. 11, 900–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulkowski M, Hezode C, Gerstoft J, Vierling JM, Mallolas J, Pol S, Kugelmas M, Murillo A, Weis N, Nahass R, et al. (2015). Efficacy and safety of 8 weeks versus 12 weeks of treatment with grazoprevir (MK-5172) and elbasvir (MK-8742) with or without ribavirin in patients with hepatitis C virus genotype 1 mono-infection and HIV/hepatitis C virus co-infection (C-WORTHY): a randomised, open-label phase 2 trial. Lancet 385, 1087–1097. [DOI] [PubMed] [Google Scholar]
- Summa V, Ludmerer SW, McCauley JA, Fandozzi C, Burlein C, Claudio G, Coleman PJ, DiMuzio JM, Ferrara M, Di Filippo M, et al. (2012). MK-5172, a selective inhibitor of hepatitis C virus NS3/4a protease with broad activity across genotypes and resistant variants. Antimicrob. Agents Chemother. 56, 4161–4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuckerman M, Berne BJ, and Martyna GJ (1992). Reversible multiple time scale molecular dynamics. J. Chem. Phys. 97, 1990–2001. [Google Scholar]
- Wittekind M, Weinheirner S, Zhang Y, and Goldfarb V (2002). Modified forms of hepatitis C NS3 protease for facilitating inhibitor screening and structural studies of protease-inhibitor complexes (United States of America). [Google Scholar]
- Yilmaz NK, Swanstrom R, and Schiffer CA (2016). Improving viral protease inhibitors to counter drug resistance. Trends Microbiol. 24, 547–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeuzem S, Jacobson IM, Baykal T, Marinho RT, Poordad F, Bourliere M, Sulkowski MS, Wedemeyer H, Tam E, Desmond P, et al. (2014). Retreatment of HCV with ABT- 450/r-ombitasvir and dasabuvir with ribavirin. N. Engl. J. Med. 370, 1604–1614. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.