

Abstract
Broad-based, targeted metabolite profiling using mass spectrometry (MS) has become a major platform used in the field of metabolomics for a variety of applications. However, quantitative MS analysis is challenging owing to numerous factors including (1) the need for, ideally, isotope-labeled internal standards for each metabolite, (2) the fact that such standards may be unavailable or prohibitively costly, (3) the need to maintain the standards’ concentrations close to those of the target metabolites, and (4) the alternative use of time-consuming calibration curves for each target metabolite. Here, we introduce a new method in which metabolites from a single serum specimen are quantified on the basis of a recently developed NMR method [Nagana Gowda et al. Anal. Chem. 2015, 87, 706] and then used as references for absolute metabolite quantitation using MS. The MS concentrations of 30 metabolites thus derived for test serum samples exhibited excellent correlations with the NMR ones (R2 > 0.99) with a median CV of 3.2%. This NMR-guided-MS quantitation approach is simple and easy to implement and offers new avenues for the routine quantification of blood metabolites using MS. The demonstration that NMR and MS data can be compared and correlated when using identical sample preparations allows improved opportunities to exploit their combined strengths for biomarker discovery and unknown-metabolite identification. Intriguingly, however, metabolites including glutamine, pyroglutamic acid, glucose, and sarcosine correlated poorly with NMR data because of stability issues in their MS analyses or weak or overlapping signals. Such information is potentially important for improving biomarker discovery and biological interpretations. Further, the new quantitation method demonstrated here for human blood serum can in principle be extended to a variety of biological mixtures.
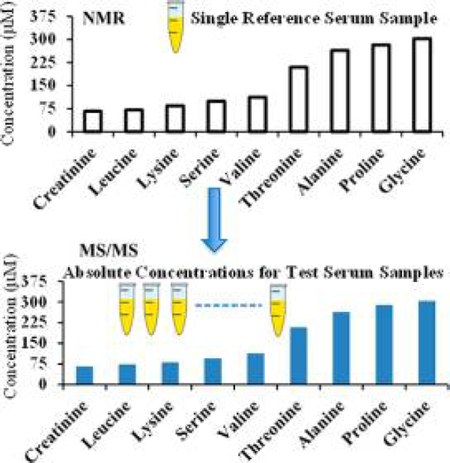
Graphical Abstract
A large number of investigations in metabolomics are focused on profiling human blood serum and plasma, owing to their relevance in diagnosing, managing, and understanding virtually all human diseases.1–3 Mass spectrometry (MS) and nuclear-magnetic-resonance (NMR) spectroscopy are the two major analytical platforms used in the field for the analysis of blood serum and plasma. They are complementary methods; MS is highly sensitive, and NMR is highly quantitative. However, while NMR enables the absolute quantitation of metabolites in a biological mixture using a single internal standard, MS lacks such a capability, which is due to a number of factors, including ionization efficiencies, ion suppression, and matrix effects. Common methods for absolute quantitation using MS include the use of internal isotope-labeled standards or structural analogues, external calibration using standard curves, and standard addition. However, numerous factors, including the often high costs or unavailability of internal standards and the need to maintain the standards’ concentrations close to those of their target metabolites or alternatively the requirement of calibration curves for each target metabolite, pose a major challenge, especially for routine metabolomics applications. There have been numerous developments in the exploitation of metabolites isotopically labeled in vivo using bacteria, algae, or fungi for quantitation in metabolomics and lipidomics.4–10 However, the use of uniformly labeled compound mixtures is in a developmental phase, and their utility for blood-metabolite analysis has not been practical so far. The limited ability to quantitate a large number of metabolites routinely has often restricted MS-based metabolomics studies to the comparison of relative peak areas and rendered the vast body of published MS data difficult to compare across multiple studies. Comparisons and correlations of metabolite data generated using MS and NMR in order to exploit their combined strength in the metabolomics field has also been limited as a result.
For many years, a major factor that affected the comparison and correlation of NMR and MS data was the fact that NMR-based metabolomics analysis traditionally involved the use of intact blood serum and plasma, whereas MS analysis involved the prior removal of abundant serum and plasma proteins. Major drawbacks of intact-serum analysis using NMR are that (1) the peak intensities of many low- to moderate-concentration metabolites are hard to distinguish from the large and variable protein and lipid signal background, and (2) the concentrations of many metabolites are grossly underestimated because of attenuation caused by binding to serum and plasma proteins,11–15 which makes comparisons with MS data very challenging. More recently, we developed an optimized protein-removal method, resulting in the quantitation of a large number of metabolites, including amino acids, organic acids, carbohydrates, and heterocyclic compounds.14,15 The use of an identical sample-preparation method for both NMR and MS provided a common platform for the analysis of blood metabolites, such that data could be compared directly. This is important because the performance of sample processing for MS analysis is typically evaluated on the basis of the total number of ions detected and not on the comprehensive quantitative analysis of metabolites.16–23
In the present study, we describe a new NMR-guided method for the absolute quantitation of blood metabolites using MS. Here, NMR-derived concentrations for a single serum sample were used as reference values for the quantitation of the metabolites in the rest of the samples using MS. Excellent correlations were observed between NMR-guided-MS concentrations and those derived from NMR, and the results were duplicated on a second targeted LC-MS platform. The NMR-guided-MS quantitation approach is simple and easy to implement and, importantly, by obviating the need for internal standards and tedious procedures, offers new avenues for MS-based quantitative blood metabolomics. A few metabolites, however, correlated poorly with the NMR data, and interestingly, such results challenge the implicit assumption that metabolites are largely stable during MS analysis. This approach therefore opens new avenues for the identification of potentially unstable metabolites during MS analysis, the knowledge of which may be critical for biomarker discovery and biological interpretations.
MATERIALS AND METHODS
Methanol; sodium phosphate, monobasic (NaH2PO4); sodium phosphate, dibasic (Na2HPO4); and 3-(trimethylsilyl)propionic acid-2,2,3,3-d4 sodium salt (TSP) were obtained from Sigma-Aldrich (St. Louis, MO). Deuterium oxide (D2O) and a mixture of uniformly 13C,15N-labeled (97–99% enrichment) standard amino acids that included leucine, methionine, tryptophan, and tyrosine were obtained from Cambridge Isotope laboratories, Inc. (Andover, MA). Human-serum samples from eight healthy subjects (four male and four female, Table S1) were obtained from Innovative Research, Inc. (Novi, MI). A commercial, pooled human serum was also obtained from Innovative Research, Inc. Deionized (DI) water was purified using an in-house Synergy Ultrapure Water System from Millipore (Billerica, MA). All chemicals were used with no further purification.
Preparation of the Phosphate Buffer.
A buffer solution (0.1 M, pH = 7.4) was prepared by dissolving 249.9 mg of anhydrous NaH2PO4 and 1124.0 mg of anhydrous Na2HPO4 in 100 g of D2O and used without further pH correction.
Serum-Protein Precipitation.
Frozen serum samples were thawed at room temperature (25 °C) and homogenized using a vortex mixer, and then 550 μL of each was pipetted into 2 mL Eppendorf vials (Fisher Scientific). For the protein removal, the serum samples were mixed with methanol in a 1:2 (v/v) ratio, which was recently shown by NMR to extract metabolites optimally.14,15 The resulting mixtures were vortexed for 30 s, incubated at −20 °C for 20 min, and centrifuged at 13 400 rcf for 30 min to pellet the proteins. The supernatants were filtered using 0.45 μm PVDF filters (Phenomenex, Torrance, CA), and the filtrate for each sample was divided into two parts. One part (1050 μL) was dried using an Eppendorf Vacufuge-Plus vacuum concentrator; the resulting residue was mixed with 300 μL of phosphate buffer (0.1 M) in D2O containing 50 μM TSP. The solution volume was increased to 600 μL using phosphate buffer in D2O (0.1 M) with no TSP and transferred to a 5 mm NMR tube for analysis. The other part (150 μL) was diluted to 500 μL using a mixture of deionized water and methanol (1:2, v/v) and used for targeted LC-MS/MS analysis.
NMR Spectroscopy.
NMR experiments were performed at 25 °C on a Bruker Avance III 800 MHz spectrometer equipped with a cryogenically cooled probe and Z-gradients suitable for inverse detection, as described previously.15 Briefly, the CPMG (Carr–Purcell–Meiboom–Gill) pulse sequence with water suppression using presaturation was used for the 1H 1D NMR experiments. To enable the absolute quantitation of metabolites using the internal reference, TSP, the CPMG experiments were performed with 128 transients and a sufficiently long recycle delay (D1 = 15 s). Chemical shifts were referenced to the internal TSP signal. The raw data was Fourier transformed after zero filling once to a total spectrum size of 32K points. Bruker Topspin software packages versions 3.0 or 3.1 were used for NMR data acquisition, processing, and analyses.
NMR-Peak Assignment and Metabolite Quantitation.
The assignment of NMR peaks to specific metabolites was based on the recent study on blood-metabolite quantitation using NMR, in which the characteristic peaks for metabolites are annotated to enable their easy identification and routine analysis.15 The Chenomx NMR Suite Professional Software package (version 5.1; Chenomx Inc., Edmonton, AB) was used to quantitate metabolites. This software allows the fitting of spectral lines using the standard metabolite library for 800 MHz 1H NMR spectra and the determination of concentrations. Peak fitting with reference to the internal TSP signal enabled the determination of absolute concentrations for the metabolites.15
Mass Spectrometry.
Targeted LC-MS Analysis.
Blood-serum metabolites were analyzed by mass spectrometry using an AB Sciex QTrap 5500 or AB Sciex QTrap 6500+ mass spectrometer (AB Sciex, Toronto, ON). The AB Sciex QTrap 5500 spectrometer was coupled to an LC system composed of two Agilent 1260 binary pumps, an Agilent 1260 autosampler, and an Agilent 1290 column compartment containing a column-switching valve (Agilent Technologies, Santa Clara, CA). For chromatography using the Agilent LC system, the mobile phase was composed of solvent A, 5 mM ammonium acetate in 90% H2O/10% acetonitrile with 0.2% acetic acid, and solvent B, 5 mM ammonium acetate in 90% acetonitrile/10% H2O with 0.2% acetic acid. The AB Sciex QTrap 6500+ mass spectrometer was coupled to an LC system composed of four Shimadzu Nexera LC-20 pumps, an AB Sciex autosampler, and an AB Sciex column compartment containing a column-switching valve (AB Sciex, Toronto, ON). For chromatography using the Shimadzu LC system, the mobile phase was composed of solvent A, 10 mM ammonium acetate in 95% H2O/3% acetonitrile/2% methanol/0.2% acetic acid, and solvent B, 10 mM ammonium acetate in 93% acetonitrile/2% methanol/5% H2O/0.2% acetic acid. The eluted metabolites were ionized using a Sciex Turbo electrospray-ionization (ESI) source, and targeted data acquisition was performed in the multiple-reaction-monitoring (MRM) mode using optimized parameters, as described earlier.24 The metabolites were analyzed in positive- or negative-ionization mode by injecting each sample twice, 2 μL (for Qtrap 5500) or 5 μL (for Qtrap 6500+) for analysis using the positive-ionization mode and 10 μL for analysis using the negative-ionization mode. The samples for Qtrap 6500+ were diluted by a factor of 2.3 relative to those used for Qtrap 5500. Two hydrophilic-interaction-chromatography (HILIC) columns (SeQuant ZIC-cHILIC, 150 × 2.1 mm, 3.0 μm particle size, Merck KGaA, Darmstadt, Germany for Agilent LC; Waters XBridge Amide, 150 × 2.1 mm, 2.5 μm particle size for Shimadzu LC) connected in parallel were run under identical conditions for the positive- and negative-ionization modes. MRM peaks for metabolites were integrated using MultiQuant 2.1 or 3.02 software (AB Sciex). To ensure the instrument’s stability during the analysis, a quality-control sample prepared using a pooled serum was analyzed under identical conditions, before and after the analyses of the serum samples. Separately, MS analysis was also performed using the AB Sciex QTrap 5500 mass spectrometer after spiking the samples with an 13C,15N-isotope-labeled amino acid mixture (10 μL; 30.55 mg in 50 mL of 1:1, v/v, water/methanol).
Absolute Metabolite Quantitation Using Targeted LC-MS/MS Guided by NMR.
One of the serum samples (Table S1) was randomly designated as the reference sample. The absolute concentration of each metabolite in the reference serum obtained by NMR was set as the MS concentration for the corresponding metabolite’s MRM peak. Absolute concentrations for a metabolite in the remaining (test) serum samples were then derived by multiplying the MRM peak area of the metabolite by the ratio of the absolute concentration to the MRM peak area of the same metabolite in the reference serum, as shown in eq 1.
| (1) |
where C(MS)mn is the absolute concentration of metabolite m in sample n derived from NMR-guided mass spectrometry, MRMmn is the MS peak area of metabolite m in sample n, C(NMR)mr is the absolute concentration of metabolite m derived from NMR in reference sample r, and MRMmr is the MS peak area of metabolite m in reference sample r.
Furthermore, to comprehensively evaluate the thus-derived concentrations on the basis of a single reference sample, the concentrations for each metabolite were recalculated by switching the reference serum to a different sample. This procedure was repeated until each of the eight serum samples (Table S1) was used as the reference once. Separately, the metabolite concentrations for the eight serum samples were obtained using a serum pooled from all eight samples or a commercial pooled serum sample as a reference. The absolute concentrations thus derived using MS were correlated with the NMR concentrations for the same samples to evaluate the performance of NMR-guided MS for absolute quantitation. Errors in concentrations across the sample set, computed using the MS/NMR data, were used to evaluate relative matrix effects across samples.
RESULTS AND DISCUSSION
Protein precipitation using methanol provided well-resolved 1HNMR spectra for all the serum samples. Mass spectrometry analyses in MRM mode combining positive- and negative-ionization modes targeted 198 metabolites. Thirty-eight metabolites were detected by both MS and NMR and were selected as the initial set of compounds for quantitation (Table S2). However, the NMR/MS peaks for eight metabolites overlapped in the NMR spectrum, coeluted off the LC column and had the same MRM transition in MS, or displayed poor chromatographic peak shapes, which made them less reliable in the evaluation of this quantitation approach and hence were omitted from further analysis. The concentrations for the remaining 30 metabolites were obtained by NMR using a single internal reference, TSP, and the obtained values are listed in Table S3 for all the serum samples. Figure 1 shows a typical NMR spectrum of a serum sample with expanded regions and peak annotations for the quantitated metabolites, and Figure S1 shows typical MS/MS ion chromatograms for the metabolites.
Figure 1.

Typical 800 MHz (cryo-probe) 1D CPMG 1H NMR spectrum of human serum obtained after protein precipitation using methanol (1:2, v/v, serum/methanol) with expanded regions and peak annotations for the 30 metabolites, which were quantitated using NMR to guide their absolute quantitation using MS.
The metabolite concentrations corresponding to the MRM peak areas for one of the serum samples (initially, sample 1) were set equal to those obtained by NMR (i.e., the NMR reference concentrations). Using these values, the absolute concentrations corresponding to each metabolite in the other seven samples based on the MRM peak areas were obtained (Table S4). Comparison of the determined concentrations for the same metabolite in the same serum sample using different NMR reference concentrations indicated that most of the metabolite concentrations agreed well irrespective of the reference sample used. Metabolite concentrations thus derived by MS showed median CVs of 4.0, 3.4, 3.5, 4.0, 3.2, 5.8, 3.2, and 5.7% for 25 metabolites when serum samples 1–8 were used as the references, respectively. As an example, Figure 2 shows a comparison of metabolite concentrations derived either by NMR or NMR-guided MS for a typical serum sample. The data shown here indicate the excellent agreement between the two methods of quantitation. Almost identical data were obtained even when the serum pooled from all eight samples or commercial pooled serum was used as the reference sample (Figure S2). Correlations of metabolite concentrations for the eight serum samples are shown in Figures 3 and S3, using sample 5 as the reference; the data show an excellent correlation between the NMR and NMR-guided-MS concentrations (R2 > 0.99) for each of the other seven serum samples. In the figures, serum 5 (Table S3), used as the reference, is also shown for comparison (R2 = 1). Excellent correlations were obtained irrespective of the serum sample was used as the reference. In addition, comparison of the concentrations of the amino acids leucine, methionine, tryptophan, and tyrosine obtained by the NMR-guided approach and the isotope-labeled-internal-standard approach showed good agreement, with a median CV of 6.2% between the two approaches (Figure 4).
Figure 2.

Comparison of the absolute concentrations of 28 metabolites derived from NMR and NMR-guided MS for the same blood-serum sample (average CV = 4.1%). MS concentrations were derived on the basis of one of the serum samples that was used as the reference sample. Similar results were obtained irrespective of the serum sample that was used as the reference (see text). Glutamine and pyroglutamic acid were omitted because of errors arising from glutamine cyclization.15,25,26 Unless otherwise noted, the MS data were obtained using an AB Sciex QTrap 5500 mass spectrometer.
Figure 3.

Correlations of concentrations for metabolites derived from NMR and NMR-guided MS for eight healthy-human-serum samples. Here, MS concentrations for one serum sample were set equal to those derived from NMR for the same serum sample (serum 5, Table S3, R2 = 1; a); these concentrations, along with their MRM peak areas were used as references for the quantitation of the same metabolites in the remaining serum samples (b–h). Virtually identical results were obtained irrespective of the serum sample that was used as the reference. Glutamine and pyroglutamic acid were omitted because of errors arising from glutamine cyclization;15,25,26 lactate and glucose were also omitted for clarity (see Figure S3 for data for all 30 metabolites).
Figure 4.

Comparison of absolute concentrations for four metabolites derived from NMR-guided MS and targeted MS using 13C,15N-isotope-labeled (uniformly) internal standards. Here, the NMR-guided-MS concentration for each metabolite was determined separately using each of the eight serum samples as references, and slight variation in concentration values due to different references is shown with an error bar. The median CV between the values from the NMR-guided-MS method and isotope-labeled-internal-standard MS was 6.2%.
To further evaluate this quantitation approach, the concentrations for the same metabolites in all the serum samples derived by NMR and NMR-guided MS were compared, individually. As shown in Figures 5 and S4, most of the metabolites exhibited very good correlations between NMR and MS (R2 > 0.9). However, metabolites including glutamine, pyroglutamic acid, glucose, and sarcosine showed very poor correlations (Figure 6). For glutamine and pyroglutamic acid, the observed correlations were in accordance with the known phenomena of glutamine cyclization to pyroglutamic acid.15,25,26 Glutamine cyclization is shown to occur during sample preparation.25 Cyclization also occurs in the electrospray-ionization (ESI) source during LC-MS analysis; the magnitude of cyclization in the ESI source can range from 33 to 100% and depends on the fragmentor voltage.26 In view of such effects by the ESI source, as well as those caused by sample preparation,15,25 it is important to be careful with the general assumption that the vast majority of metabolites are stable when evaluated in metabolomics. These findings also open new avenues for the identification of potentially unstable metabolites during MS analysis, the knowledge of which is potentially important for improved biomarker discovery and biological interpretations. The poor correlation of glucose may be because it exhibits multiple MRM peaks (Figure S1), and sugars such as mannose with identical molecular weights and similar retention times overlap. On the other hand, the poor correlation of sarcosine may arise from measurement errors due to its weak MRM peak (Figure S1).
Figure 5.

Correlations of absolute concentrations for metabolites derived from NMR and NMR-guided MS for eight healthy human (four male and four female) serum samples. Here, MS concentrations for one serum sample (serum 1, Table S3) were set equal to those derived from NMR and used as the references for the quantitation of the same metabolites in the remaining serum samples. Virtually identical results were obtained irrespective of the serum sample used as the reference.
Figure 6.

Correlations of concentrations for individual metabolites derived from MS and NMR for eight healthy-human-serum samples. Here, MS concentrations for one serum sample were set equal to those derived from NMR (serum 1, Table S3) and used as the references for the quantitation of the same metabolites in the remaining serum samples. The poor correlations of glutamine and pyroglutamic acid are as anticipated on the basis of massive glutamine cyclization discovered recently by NMR15,25 as well as MS.26
In order to assess the relative magnitude of the matrix effect on MS ionization across samples, concentrations of metabolites determined separately from the same sample using different NMR-reference samples were evaluated. As shown in Figure S5, the average CV for more than 50% of the metabolites was 5.6%, and the average CV for all metabolites except three was 7.2%. Three metabolites, glutamine, pyroglutamic acid, and sarcosine, however, each exhibited a CV of more than 15%, which is in accordance with the poor correlations shown between the NMR and NMR-guided-MS concentrations (Figure 6). As an additional evaluation for the matrix effect, the concentrations of the metabolites in the eight serum samples obtained using the serum pooled from all eight samples or the commercial pooled serum as the reference showed almost identical data to those that used one of the eight serum samples as the reference (Figure S2). These results indicate that the sample-matrix effect on quantitation is tolerable.
Absolute quantitation of blood metabolites using MS on a routine basis is impeded by numerous factors and challenges, as described in the introduction. Metabolites isotopically labeled in vivo, especially using yeast (Pichia pastoris), is promising as a reliable tool for quantitation in metabolomics and lipidomics.7–9 More than 99% labeling efficiency has been achieved for hundreds of already identified metabolites using this approach, the extracts show high dynamic ranges for use in quantitative experiments, and the addition of this extract to samples has demonstrated that it does not alter untargeted metabolomics analyses. This approach, however, is still in a developmental phase, and its utility for blood-metabolite analysis has not been demonstrated.
NMR spectroscopy draws its strength from being highly reproducible and quantitative; a single internal reference can in principle be used to determine absolute concentrations for all NMR-measurable metabolites in a biological mixture. In this study, we have exploited this strength of NMR to guide the absolute quantitation of metabolites in biological samples with similar matrices using MS. In particular, the study exploits recent advances in blood-metabolite quantitation using NMR, which provides an optimized and common protocol for the analysis of the same sample using both NMR and MS.15 Using this approach and identical sample processing for both NMR and MS methods, linear correlations between NMR and MS data could be achieved for metabolites in all the serum samples (Figures 2, 3, and 5). Because of this linear relationship, NMR-derived metabolite concentrations for a single serum is sufficient to obtain absolute concentrations for the same metabolites in other serum samples using MS.
In this proof-of-concept study, we used human serum to demonstrate NMR-guided metabolite quantitation in MS because absolute quantitation of human blood metabolites is of great interest in biomedicine. It may be interesting to note that blood serum or plasma is particularly suited for NMR-guided quantitation because variation among samples is very small (typically less than a factor of 2). Application of this approach to biological systems that exhibit greater variation among samples than serum or plasma will likely be more challenging. In particular, factors that contribute to ion suppression as well as MS linearity can become key issues to address when using the NMR-guided approach for the measurement of such biological systems.
In conclusion, we present a new NMR-guided-MS method for absolute metabolite quantitation in human blood serum using mass spectrometry, which takes advantage of recent advances in NMR-based blood-metabolite quantitation. The method employs quantitative data for a single serum specimen derived from NMR for MS-based absolute quantitation of the same metabolites in other serum samples. It uses an optimized protein-removal method common to both MS and NMR to ensure that the detected metabolites retain a linear relationship between NMR and MS. Absolute quantitation of blood metabolites provides the capability to compare data across metabolomics studies and to incorporate clinical data obtained using other platforms. The only requirement is to use the same NMR-calibrated reference sample, periodically, to correct for changes in the MS peak intensities. From the data presented, relative sample-matrix effects across samples appear to be limited, at least in this small study. Further validation of this observation needs to be performed, and the use of internal standards when available and cost-effective will normally provide the most accurate results. However, as a potential replacement for time-consuming external calibration using multiple standards, this approach may be appealing. Additionally, it is both interesting and a cause for concern that some metabolites correlate very poorly between NMR and MS. Because such correlations were not anticipated and challenge the widely accepted implicit assumption that the vast majority of metabolites are generally stable during MS analysis, this approach provides a mechanism to identify potentially unstable metabolites. This knowledge is highly useful for biomarker discovery and biological interpretations.
In sum, the described NMR-guided quantitative method is simple and easy to implement, and importantly, it can reduce or obviate the need for internal standards and tedious procedures, thereby offering new avenues for doing MS-based quantitative blood metabolomics on a routine basis. These findings provide an opportunity for the quantitative analysis of an enhanced pool of metabolites in blood and ultimately a variety of other biological mixtures. Although this work constitutes a proof-of-concept study with a relatively small number of quantified metabolites, the use of sensitivity-enhancement methods, including smaller sample-volume probes, enhanced sample concentrations, and signal averaging, can improve the limit of quantitation for NMR and expand significantly the metabolite pool quantifiable by NMR-guided MS. It should be noted that for both NMR and MS, a weak or overlapping peak or a peak with a poor line shape can deleteriously affect peak integration and thereby affect reliable quantitation. Hence, high-quality spectra are critical for accurate quantitation. In addition, the CPMG NMR experiment used in this study, which provides a much cleaner baseline compared with one-pulse or 1D NOESY experiments, can underestimate serum-metabolite concentrations, albeit marginally, because of relaxation effects. A comparison of metabolite concentrations in the two experiments showed an underestimation in CPMG experiment by an average of 4.6%.14 Such relaxation effects, therefore, need to be accounted for to improve quantitation accuracy in this kind of experiment.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge financial support from the NIH (National Institute of General Medical Sciences 2R01GM085291).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.7b04089.
Demographic parameters for the serum samples, common metabolites detected, NMR-derived metabolites concentrations, MS-derived concentrations, typical MS/MS ion chromatograms, comparisons of absolute concentrations obtained using NMR-guided MS, correlations of concentrations of all metabolites derived from NMR and NMR-guided MS, correlations of absolute concentrations of individual metabolites derived from NMR and NMR-guided MS, and coefficients of variation of concentrations of metabolites (PDF)
The authors declare the following competing financial interest(s): Daniel Raftery reports holding equity and an executive position at Matrix Bio, Inc.
REFERENCES
- (1).Beckonert O; Keun HC; Ebbels TM; Bundy J; Holmes E; Lindon JC; Nicholson JK Nat. Protoc 2007, 2 (11), 2692–2703. [DOI] [PubMed] [Google Scholar]
- (2).Psychogios N; Hau DD; Peng J; Guo AC; Mandal R; Bouatra S; Sinelnikov I; Krishnamurthy R; Eisner R; Gautam B; Young N; Xia J; Knox C; Dong E; Huang P; Hollander Z; Pedersen TL; Smith SR; Bamforth F; Greiner R; McManus B; Newman JW; Goodfriend T; Wishart DS PLoS One 2011, 6 (2), e16957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Nagana Gowda GA; Raftery D Curr. Metabolomics 2013, 1(3), 227–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Bennett BD; Yuan J; Kimball EH; Rabinowitz JD Nat. Protoc 2008, 3, 1299–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Vielhauer O; Zakhartsev M; Horn T; Takors R; Reuss M J. Chromatogr. B: Anal. Technol. Biomed. Life Sci 2011, 879 (32), 3859–3870. [DOI] [PubMed] [Google Scholar]
- (6).Mairinger T; Hann S Anal. Bioanal. Chem 2017, 409 (15), 3713–3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Neubauer S; Haberhauer-Troyer C; Klavins K; Russmayer H; Steiger MG; Gasser B; Sauer M; Mattanovich D; Hann S; Koellensperger G J. Sep. Sci 2012, 35 (22), 3091–3105. [DOI] [PubMed] [Google Scholar]
- (8).Rampler E; Coman C; Hermann G; Sickmann A; Ahrends R; Koellensperger G Analyst 2017, 142 (11), 1891–1899. [DOI] [PubMed] [Google Scholar]
- (9).Schwaiger M; Rampler E; Hermann G; Miklos W; Berger W; Koellensperger G Anal. Chem 2017, 89 (14), 7667–7674. [DOI] [PubMed] [Google Scholar]
- (10).Weiner M; Tröndle J; Schmideder A; Albermann C; Binder K; Sprenger GA; Weuster-Botz D Anal. Biochem 2015, 478, 134–140. [DOI] [PubMed] [Google Scholar]
- (11).Nicholson JK; Gartland KP NMR Biomed 1989, 2 (2), 77–82. [DOI] [PubMed] [Google Scholar]
- (12).Chatham JC; Forder JR Biochim. Biophys. Acta, Gen. Subj 1999, 1426 (1), 177–184. [DOI] [PubMed] [Google Scholar]
- (13).Bell JD; Brown JC; Kubal G; Sadler PJ FEBS Lett 1988, 235, 81–86. [DOI] [PubMed] [Google Scholar]
- (14).Nagana Gowda GA; Raftery D Anal. Chem 2014, 86 (11), 5433–5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Nagana Gowda GA; Gowda Y; Raftery D Anal. Chem 2015, 87 (1), 706–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Vuckovic D Anal. Bioanal. Chem 2012, 403 (6), 1523–1548. [DOI] [PubMed] [Google Scholar]
- (17).Dunn WB; Broadhurst D; Begley P; Zelena E; Francis-McIntyre S; Anderson N; Brown M; Knowles JD; Halsall A; Haselden JN; Nicholls AW; Wilson ID; Kell DB; Goodacre R Nat. Protoc 2011, 6 (7), 1060–1083. [DOI] [PubMed] [Google Scholar]
- (18).Ivanisevic J; Zhu ZJ; Plate L; Tautenhahn R; Chen S; O’Brien PJ; Johnson CH; Marletta MA; Patti GJ; Siuzdak G Anal. Chem 2013, 85 (14), 6876–6884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Bruce SJ; Tavazzi I; Parisod V; Rezzi S; Kochhar S; Guy PA Anal. Chem 2009, 81 (9), 3285–3296. [DOI] [PubMed] [Google Scholar]
- (20).A J; Trygg J; Gullberg J; Johansson AI; Jonsson P; Antti H; Marklund SL; Moritz T Anal. Chem 2005, 77 (24), 8086–8094. [DOI] [PubMed] [Google Scholar]
- (21).Zelena E; Dunn WB; Broadhurst D; Francis-McIntyre S; Carroll KM; Begley P; O’Hagan S; Knowles JD; Halsall A; Wilson ID; Kell DB Anal. Chem 2009, 81 (4), 1357–1364. [DOI] [PubMed] [Google Scholar]
- (22).Tulipani S; Llorach R; Urpi-Sarda M; Andres-Lacueva C Anal. Chem 2013, 85 (1), 341–348. [DOI] [PubMed] [Google Scholar]
- (23).Want EJ; O’Maille G; Smith CA; Brandon TR; Uritboonthai W; Qin C; Trauger SA; Siuzdak G Anal. Chem 2006, 78 (3), 743–752. [DOI] [PubMed] [Google Scholar]
- (24).Zhu J; Djukovic D; Deng L; Gu H; Himmati F; Chiorean EG; Raftery D J. Proteome Res 2014, 13 (9), 4120–4130. [DOI] [PubMed] [Google Scholar]
- (25).Nagana Gowda GA; Gowda Y; Raftery D Anal. Chem 2015, 87 (7), 3800–3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Purwaha P; Silva LP; Hawke DH; Weinstein JN; Lorenzi PL Anal. Chem 2014, 86 (12), 5633–5637. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.