

Abstract
This perspective showcases our development of benzylic and allylic amine and alcohol derivatives as electrophiles for stereospecific, nickel-catalyzed cross-coupling reactions, as well as the prior art that inspired our efforts. The success of our effort has relied on the use of benzyl ammonium triflates as electrophiles for cross-couplings via C–N bond activation and benzylic and allylic carboxylates for cross-couplings via C–O bond activation. Our work, along with others’ exciting discoveries, has demonstrated the potential of stereospecific, nickel-catalyzed cross-couplings of alkyl electrophiles in asymmetric synthesis, and enables efficient generation of both tertiary and quaternary stereocenters.
Graphical Abstract
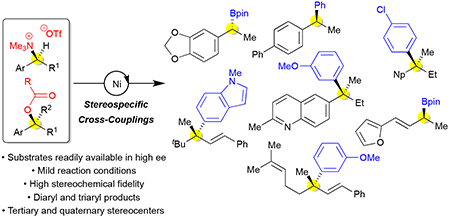
Stereospecific, nickel-catalyzed cross-couplings of alkyl ammonium salts and carboxylates enable preparation of highly enantioenriched products with tertiary and quaternary stereocenters.
1. Introduction
Transition metal-catalyzed cross-couplings have revolutionized organic chemistry, allowing creative and nonobvious disconnections in organic synthesis. Compared to cross-couplings of sp2-hybridized carbons,1 cross-couplings of sp3-hybridized carbons are much less developed due to challenging oxidative addition and reductive elimination steps, along with competitive side reactions.2 However, cross-couplings of sp3-hybridized carbons are gaining increased attention.3
In particular, the use of alkyl reagents in transition metal-catalyzed cross-coupling reactions provides opportunities for the incorporation of stereochemical information. As complementary approaches, enantioselective and stereospecific cross-couplings offer distinct advantages (Scheme 1a).4 Enantioselective cross-couplings rely on catalyst-controlled asymmetric induction and allow for the first installation of stereochemistry in a molecule. Stereospecific reactions are substrate-controlled with respect to product stereochemistry, and conserve stereochemical information while building complexity. Because a stereocenter has been set previously, stereospecific cross-couplings do not require steric or electronic differentiation to afford high enantiospecificity (es).5 This makes stereospecific cross-couplings particularly useful for late-stage synthesis and for generating stereocenters where enantioselective catalysis fails.
Scheme 1.
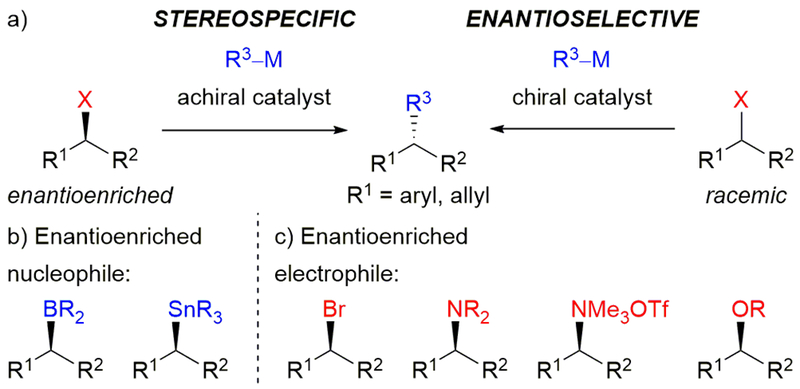
Alkyl reagents in transition metal-catalyzed cross-coupling reactions: (a) comparison of stereospecific and enantioselective reactions illustrated with alkyl electrophiles; (b) use of enantioenriched nucleophiles for stereospecific cross-couplings; and (c) use of enantioenriched electrophiles for stereospecific cross-couplings
When forming C(sp3)–C(sp2) bonds, either the electrophile or nucleophile can be enantioenriched. A number of methods have been developed for the use of secondary and tertiary alkylmetal reagents in stereospecific reactions (Scheme 1b), which have been recently reviewed.6 This perspective will focus instead on our use of enantioenriched electrophiles in stereospecific transition metal-catalyzed cross-coupling reactions, as well as precedent that inspired our work and related concurrent examples (Scheme 1c).
Although the SN2 reaction is of historical and pedagogical importance, the stereospecific reaction of a secondary electrophile to generate a tertiary carbon stereocenter is an atypical disconnection. Elimination (E2) competes with stereospecific substitution. This challenge is even more pronounced for the preparation of all-carbon quaternary stereocenters from tertiary electrophiles.7 By offering an alternative mechanism, transition metal catalysis offers an opportunity to expand the utility of stereospecific substitutions to secondary and tertiary enantioenriched electrophiles, while also enabling the use of mild and functional group-tolerant nucleophilic partners. Palladium-catalyzed cross-couplings of alkyl halides are typically stereospecific and proceed with inversion at the stereogenic center.8 However, as the steric hindrance of the electrophile increases, the energy barrier for oxidative addition increases, making palladium-catalyzed cross-couplings of secondary and tertiary halides more challenging.9 On the other hand, first-row transition metal complexes are highly reactive toward oxidative addition and slow to undergo β-hydride elimination,4c,9 which makes them ideal catalysts for cross-coupling reactions involving alkyl electrophiles. However, oxidative addition of nickel complexes with alkyl halides typically proceeds through radical intermediates, resulting in stereoablative reactions.10
Instead, non-traditional electrophiles—namely, amine and alcohol derivatives—provide a complementary alternative to alkyl halides and an entry into stereospecific reactions to afford tertiary and quaternary stereocenters. Amines and alcohols are generally air- and moisture-stable and have low toxicity. Additionally, although few methods exist for the preparation of enantiomerically enriched alkyl halides, amines and alcohols are readily available in high enantiopurity.3a Furthermore, amine and alcohol derivatives can engage in polar, two-electron oxidative addition with low-valent nickel complexes, thereby enabling stereospecific transformations.11 With this review, we aim to showcase the work that we and others have conducted to expand the synthetic utility of enantioenriched amine- and alcohol-based electrophiles in transition metal-catalyzed cross-coupling reactions to prepare tertiary and quaternary stereocenters.
2. C(sp3)–N Bond Activation of Amine Derivatives
2.1. Secondary Allylic and Benzylic Amines
The carbon–nitrogen (C–N) bond is ubiquitous, present in many organic and biological molecules.12 Although C–N bonds are typically considered inert due to high bond dissociation energies and poor leaving group ability,13 significant attention has been given to the activation and use of C(sp2)–N bonds in transition metal-catalyzed cross-coupling reactions.14 In contrast, the use of alkyl amines as electrophiles in cross-coupling reactions has been much less examined, and much of this focus has been on allylic amine substrates. In a seminal report in 1995, Trost and coworkers reported a nickel-catalyzed cross-coupling of tertiary allylic amines with boronic acids wherein the boronic acid served as both the nucleophile and a Lewis acid activating group for the amine.15 Tian and coworkers utilized this strategy for the activation of allylic amines in palladium-catalyzed cross-couplings with boronic acids16 and substitutions with other nucleophiles.17
Subsequently, Tian and coworkers proposed that the presence of a sulfonyl group would activate allylic amines toward C(sp3)–N bond cleavage.18 Reaction of enantioenriched allylic sulfonamide 1 with catalytic copper(I) iodide and phenylmagnesium bromide afforded 2 as a single regioisomer in 84% yield (Scheme 2a). This transformation proceeded with complete inversion of configuration at the allylic stereocenter. In applying this strategy to benzylic amines, a second tosyl group was necessary to activate the more stubborn benzylic C–N bond. Arylation of enantioenriched benzylic sulfonimide 3 with 4-methoxyphenylmagnesium bromide also proceeded with inversion of configuration to afford diarylethane 4 (Scheme 2b), albeit in modest stereochemical fidelity. This pivotal work demonstrated that a benzylic C–N bond could indeed be converted to a C–C bond stereospecifically, but that considerable activation of the nitrogen was necessary.
Scheme 2.
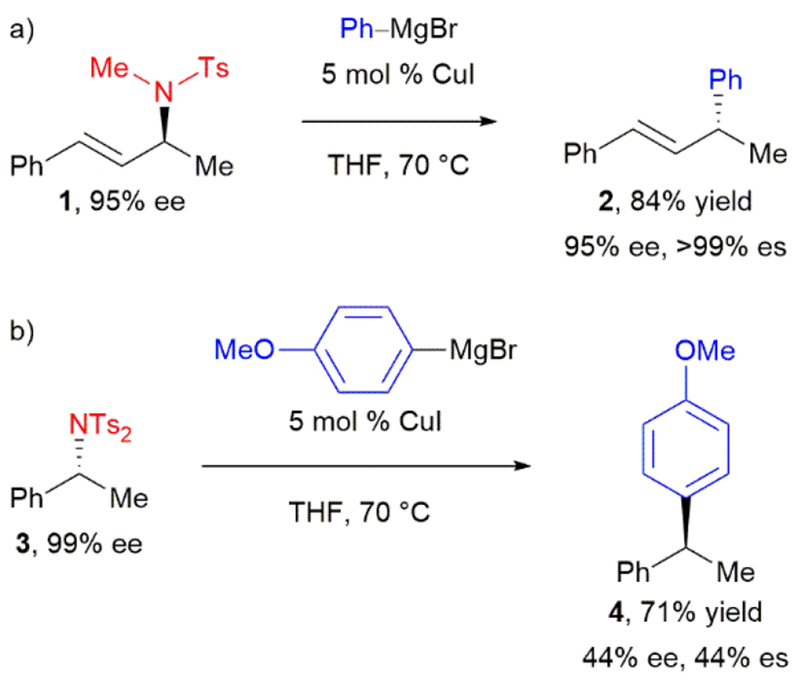
Tian’s stereospecific, copper-catalyzed arylation of enantioenriched amines with aryl Grignard reagents
2.2. Secondary and Tertiary Aziridines
At the time we began our work, aziridines were the only other electrophiles that had been demonstrated in cross-couplings via C–N bond activation. Like allylic and benzylic amines, aziridines are versatile synthetic intermediates, especially for the preparation of valuable β-substituted amines. Like epoxides, aziridines experience significant ring strain (26–27 kcal/mol),19 which renders them susceptible to nucleophilic ring-opening. Transition metal catalysis offers an alternative to classic aziridine alkylation and arylation, which require strong nucleophiles and often afford poor regioselectivity.20 However, the development of transition metal-catalyzed cross-coupling reactions of aziridines faces challenging oxidative addition and competitive β-hydride elimination.21 In seminal reports, Hillhouse22 and Wolfe23 demonstrated that aliphatic N-sulfonyl aziridines undergo oxidative addition with stoichiometric nickel or palladium species to form isolable azametallocyclobutanes, wherein the metal has inserted into the less hindered C–N bond. Alper and coworkers also demonstrated that carbonyl insertion could outcompete β-hydride elimination in rhodium-catalyzed reactions of styrenyl aziridines to form β-lactams with high regioselectivity for insertion into the benzylic C–N bond.24
Building on this precedent, Doyle and Huang reported the first catalytic activation of an aziridine C(sp3)–N bond in a cross-coupling reaction.25 Under nickel catalysis, styrene-derived N-tosyl aziridines underwent a Negishi cross-coupling with alkylzinc reagents. Use of dimethylfumarate 6 (Scheme 3a) as ligand was essential; this electron-deficient olefin is proposed to accelerate the challenging reductive elimination.26 The reaction went with complete regioselectivity for cleavage of the benzylic C–N bond. With respect to stereospecificity, when enantiopure aziridine 5 was subjected to the cross-coupling conditions, amine 7 was generated in only 11% ee. The enantiomeric excess of recovered 5 was unchanged. The authors thus proposed that oxidative addition is irreversible with a subsequent stereoablative step.
Scheme 3.
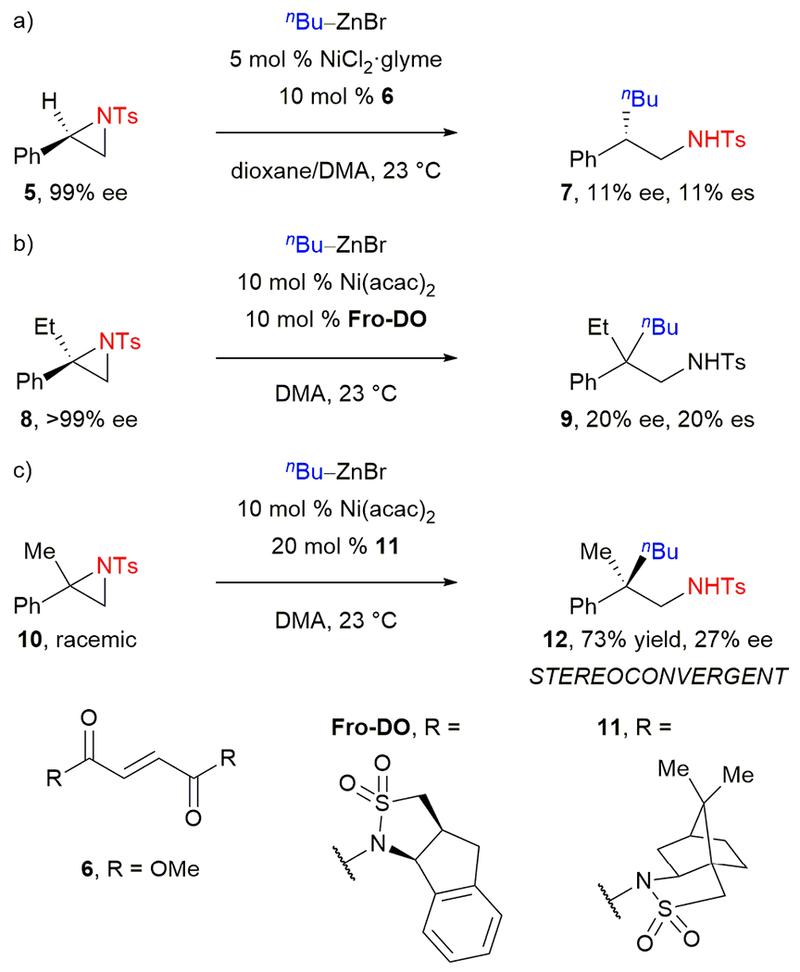
Doyle’s nickel-catalyzed Negishi cross-coupling reactions of aziridines.
Expanding on these studies, Doyle and Huang then reported a nickel-catalyzed Negishi cross-coupling of 1,1-disubstituted aziridines, affording β-substituted phenethylamines.27 The electron-deficient olefin ligand Fro-DO was crucial in achieving C–C bond formation over β-hydride elimination. To test the stereospecificity of the cross-coupling reaction, the authors subjected enantioenriched aziridine 8 to the reaction conditions with n-butylzinc bromide (Scheme 3b). However, product 9 was obtained in only 20% ee. Although this was the first example of a transition metal-catalyzed cross-coupling of a benzylic electrophile forming an all-carbon quaternary stereocenter with any stereospecificity, the low stereochemical fidelity indicated that a stereoablative or epimerization step was competitive with the stereospecific pathway. Recovered 8 was enantiopure, which is consistent with the transformation proceeding through an irreversible oxidative addition, at or after which a stereoablative step occurs. The authors proposed that the most likely mechanism for oxidative addition is single-electron transfer (SET) from a nickel(I) species to generate a stabilized benzylic radical intermediate. They also proposed that by using a chiral ligand, this cross-coupling could be stereoconvergent. Indeed, when racemic aziridine 10 was subjected to the reaction conditions with chiral electron-deficient olefin ligand 11, β-substituted phenethylamine 12 was formed in 73% yield and 27% ee (Scheme 3c). Notably, this result is the first example of a stereoselective cross-coupling with a tertiary, non-allylic electrophile. Based on this result, Doyle and coworkers developed an enantioselective nickel-catalyzed reductive cross-coupling of styrenyl aziridines and aryl iodides, which proceeds with good yields and enantioselectivities.28
In a complementary example, Minakata and coworkers demonstrated a stereospecific, palladium-catalyzed Suzuki–Miyaura cross-coupling of styrenyl aziridines and aryl boronic acids.29 N-heterocyclic carbene (NHC) ligands efficiently promoted the cross-coupling while suppressing β-hydride elimination. Reaction of enantioenriched aziridine 5 with p-tolylboronic acid afforded β-substituted amine 15 in 74% yield and complete stereochemical fidelity (Scheme 4). The cross-coupling proceeded with inversion of configuration, consistent with oxidative addition occurring via an SN2- or SN2’-type mechanism to give intermediates 13 or 14.
Scheme 4.
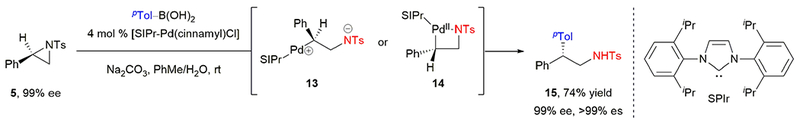
Minakata’s stereospecific, palladium-catalyzed Suzuki–Miyaura cross-coupling of enantioenriched aziridines.
Three transition metal-catalyzed cross-coupling reactions of unactivated aliphatic N-sulfonyl aziridines have also been reported. Doyle30 and Jamison31 reported nickel-catalyzed Negishi cross-couplings employing aryl- and alkylzinc reagents, respectively. Michael also reported a palladium-catalyzed Suzuki–Miyaura cross-coupling of alkyl aziridines and arylboronic acids.32 Unlike the cross-coupling reactions of benzylic aziridines, these cross-couplings of unactivated alkyl aziridines displayed complete regioselectivity for oxidative addition into the least substituted C(sp3)–N bond to afford linear products.
2.3. Secondary Benzylic Ammonium Salts
The work by Tian, Doyle, Minakata, and others impressively demonstrates the potential of using benzylic amine derivatives and azirdines as substrates for stereospecific cross-couplings to yield highly enantioenriched diarylalkanes. This prior art also taught us that both the nitrogen activating group and the catalyst components would be crucial in achieving high stereochemical fidelity in such cross-couplings. We needed to identify a nitrogen leaving group and conditions that would not lead to radical intermediates, epimerization, or β-hydride elimination. We proposed that the C(sp3)–N bond of benzyl amines, when activated as an ammonium salt, would undergo nickel-catalyzed cross-coupling reactions in a stereospecific fashion. Cross-coupling reactions of aryl ammonium salts were known,33 and the trimethylammonium group was unlikely to undergo SET to form alkyl radical intermediates due to the lack of an available π* orbital. However, although allylic and benzylic ammonium salts had been utilized as electrophiles in reactions with organometallic nucleophiles,34 their use in transition metal-catalyzed cross-coupling reactions was limited to a single example. Csákÿ and coworkers demonstrated the rhodium-catalyzed cross-coupling of gramine-derived ammonium iodide 16 with phenylboronic acid, affording 3-benzylindole 17 in 85% yield (Scheme 5).35 Notably, this gramine-derived substrate benefits from weakening of the C–N bond by the indole, and no other benzylic ammonium salts were included.
Scheme 5.
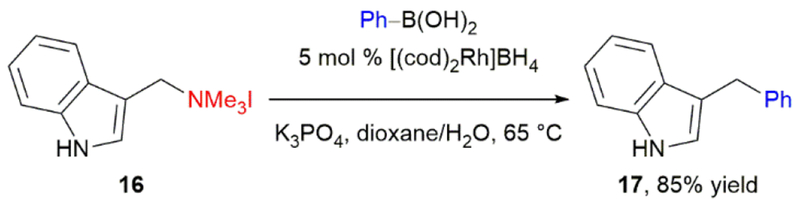
Csákÿ’s rhodium-catalyzed Suzuki–Miyaura cross-coupling of a benzylic ammonium iodide.
Our goal was to develop a general method for the stereospecific cross-coupling of benzylic electrophiles and functional group-tolerant coupling partners.36 Although great progress had been made with Grignard18,37–39 and organozinc coupling partners,8c,40–41 no enantioselective cross-couplings of benzylic electrophiles were known with arylboronic reagents at the time we began our work, and there was only a single stereospecific example of a benzylic α-cyanohydrin mesylate.8b Highly enantioenriched benzylic amines are ideal electrophile precursors, because they are readily prepared, are stable to long-term storage, and offer a functional group handle orthogonal to halides and ethers.12,42 Enantioenriched ammonium triflates 18 were readily prepared in quantitative yield via methylation of the corresponding chiral tertiary amines and did not require chromatographic purification.
The combination of Ni(cod)2 and a monodentate phosphine, P(o-Tol)3, proved optimal for catalyzing the cross-coupling, affording enantioenriched diarylmethanes in good yields, high stereochemical fidelity, and inversion of configuration at the benzylic stereocenter (Scheme 6). The weakly coordinating triflate counterion gave the best reactivity. We hypothesize that this may be due to more facile coordination of the boronate to a more electrophilic Ni(II) intermediate. Either K3PO4 or CsF could serve as the base; however, if base-sensitive functional groups were present, the use of CsF proved advantageous. The mild conditions tolerated a wide range of functional groups, including ether 20, alkene 21, ester 22, and nitrile 23, highlighting the advantage of an arylboronic acid over a Grignard partner. In addition to arylboronic acids, a vinylboronic acid underwent the cross-coupling to give 24 in 96% yield and >99% es. To expand the ammonium triflate scope beyond those with napthyl substitution—a common limitation in stereospecific cross-couplings37–40,43–44—higher catalyst loadings and a change of ligand to tBu-XantPhos were required. Using these modified conditions, diarylethanes 25 and 26 were obtained with excellent stereochemical fidelity albeit lower yields. Additionally, these conditions allowed selective C–N bond activation in the presence of ethers, highlighting the orthogonal functionality of ammonium salts.
Scheme 6.
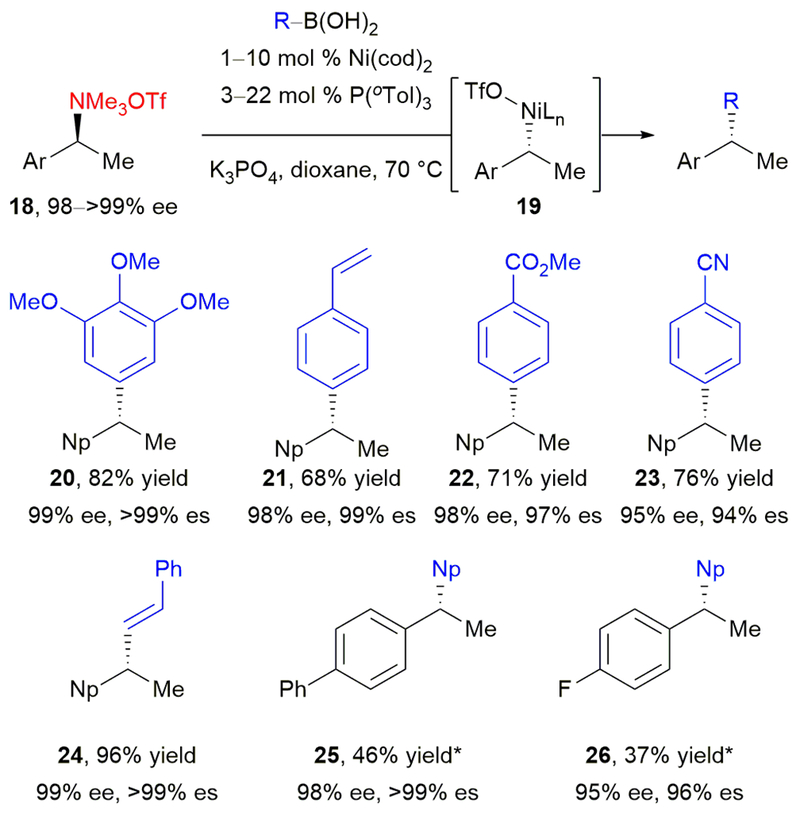
Our stereospecific, nickel-catalyzed arylation of benzylic ammonium triflates with boronic acids to form enantioenriched diarylethanes. *tBu-XantPhos used as ligand in place of P(oTol)3.
In 2014, we reported improved conditions for the stereospecific cross-coupling of secondary benzylic ammonium salts.45 By conducting the cross-coupling of ammonium triflates 27 and arylboronic acids in the presence of Ni(cod)2 without exogenous ligand, diarylalkanes were obtained in higher yields than under our first-generation conditions (Scheme 7). As before, the reaction proceeds with inversion at the benzylic stereocenter with high levels of stereochemical fidelity. Under our original conditions, heteroaromatic boronic acids gave only modest yields and enantiospecificities. Under the “phosphine-less” reaction conditions, heteroaromatic groups were well tolerated, including benzofuran 28. These conditions also tolerated bulky groups at R, including isopropyl (29). In addition to aryl boronic acids, vinyl boronic acids underwent the cross-coupling to afford products 30 and 31 in good yields and excellent stereochemical fidelity. Finally, although the stereochemical fidelity was slightly diminished, non-naphthyl-substituted ammonium triflates underwent the reaction with moderate to good yields. Electron-poor substrates were most efficient (32), but the cross-coupling also afforded p-methoxyphenyl-substituted 33 in 53% yield.
Scheme 7.
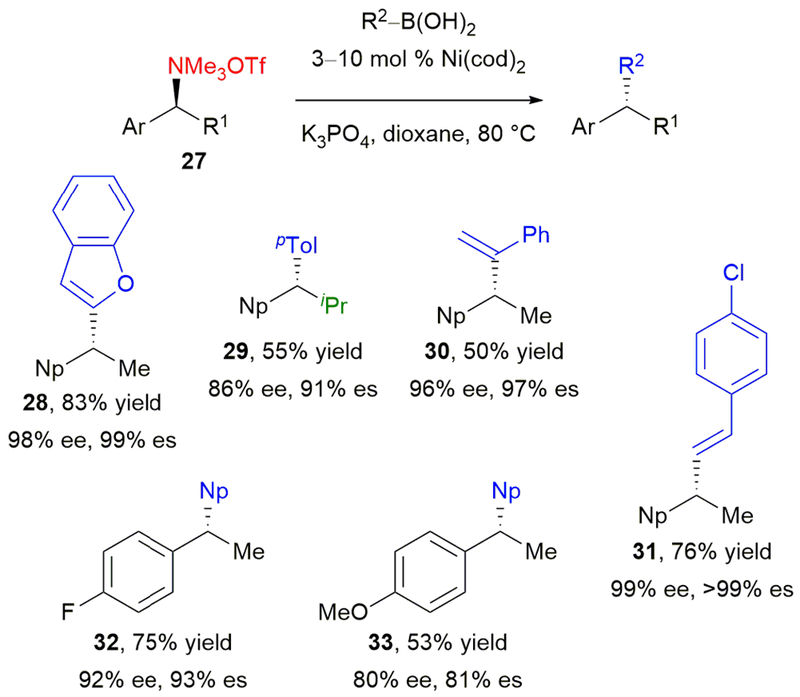
Our phosphine-less stereospecific, nickel-catalyzed cross-coupling of benzylic ammonium triflates.
Expanding the utility of benzylic ammonium salts in cross-coupling reactions, we also developed a stereospecific, nickel-catalyzed Miyaura borylation.46 As the first example of a cross-coupling utilizing a benzylic electrophile to afford highly enantioenriched organoboranes, this provides a complementary method to previous methods for the asymmetric synthesis of benzylic boronate esters.47 Reaction of ammonium triflates 34 with B2pin2, Ni(cod)2, and PPh3 afforded enantioenriched benzylic pinacol boronates in good yields and stereochemical fidelity (Scheme 8a). Heteroaryl substitution was well tolerated, affording benzofuran 35 in 64% yield. Increased substitution adjacent to the benzylic stereogenic center was also tolerated, with 36 forming in 50% yield. Notably, products with such branched substituents are not accessible under asymmetric hydroboration conditions.48 Other diboranes also successfully underwent the cross-coupling reaction to afford boronate esters (37). With the more electron-donating PPh2Cy and increased reaction temperature, benzylic ammonium triflates without naphthyl substitution engaged in the cross-coupling (Scheme 8b). Enantioenriched boronate products 39, 40, and 41 were obtained in good yields and stereospecificities.
Scheme 8.
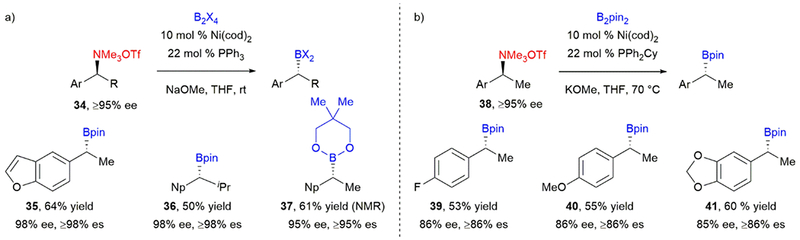
Our stereospecific, nickel-catalyzed Miyaura borylation of benzylic ammonium triflates to afford enantioenriched benzylic boronates using (a) naphthyl-substituted ammonium triflates, and (b) non-naphthyl-substituted ammonium triflates.
In analogy to stereospecific cross-couplings of benzylic ethers, we hypothesize that the stereospecific cross-couplings of benzylic ammonium salts 42 proceed via oxidative addition of an electron-rich Ni(0) complex into the C–N bond, generating either η1- or η3-bound nickel(II) intermediate 43 or 44 (Scheme 9a).49 Transmetalation with the activated boronate to form intermediate 45 (or its η3 analogue) and subsequent reductive elimination then delivers cross-coupled product 46. Consistent with oxidative addition into the C–N bond, benzylnickel(II) triflate 48 was produced in 51% isolated yield upon reaction of ammonium triflate with stoichiometric Ni(cod)2 and PPh2Cy. The structure of 48 was confirmed by X-ray crystallography (Scheme 9b), and 48 proved reactive as a substrate and catalytically competent in the Suzuki–Miyaura arylation. Because retention of configuration during transmetalation and reductive elimination is precedented for alkyl metal species,50 we propose that inversion of configuration occurs during oxidative addition of the nickel catalyst into the benzylic C–N bond. Additionally, because the cross-couplings afford higher yields for substrates with naphthyl substituents instead of phenyl substituents, we propose that oxidative addition likely occurs via an SN2’-type mechanism (TS-1). Partially breaking the aromaticity of the naphthyl group is far less endothermic than fully breaking the aromaticity of a phenyl substituent in this step. An analogous mechanism had been previously proposed by Jarvo for her stereospecific cross-couplings of benzylic ethers.43 To test this hypothesis, we compared the borylation of ammonium triflates 49 and 52 (Scheme 9c). For ammonium triflate 49, if the methoxy group sterically blocks addition at C1, SN2’-type attack of nickel at C3 would result in complete loss of aromaticity. Indeed, no desired product 50 was observed. However, SN2’-type attack on ammonium triflate 52 maintains some aromaticity in intermediate 53, resulting in product 54 in 49% yield.
Scheme 9.
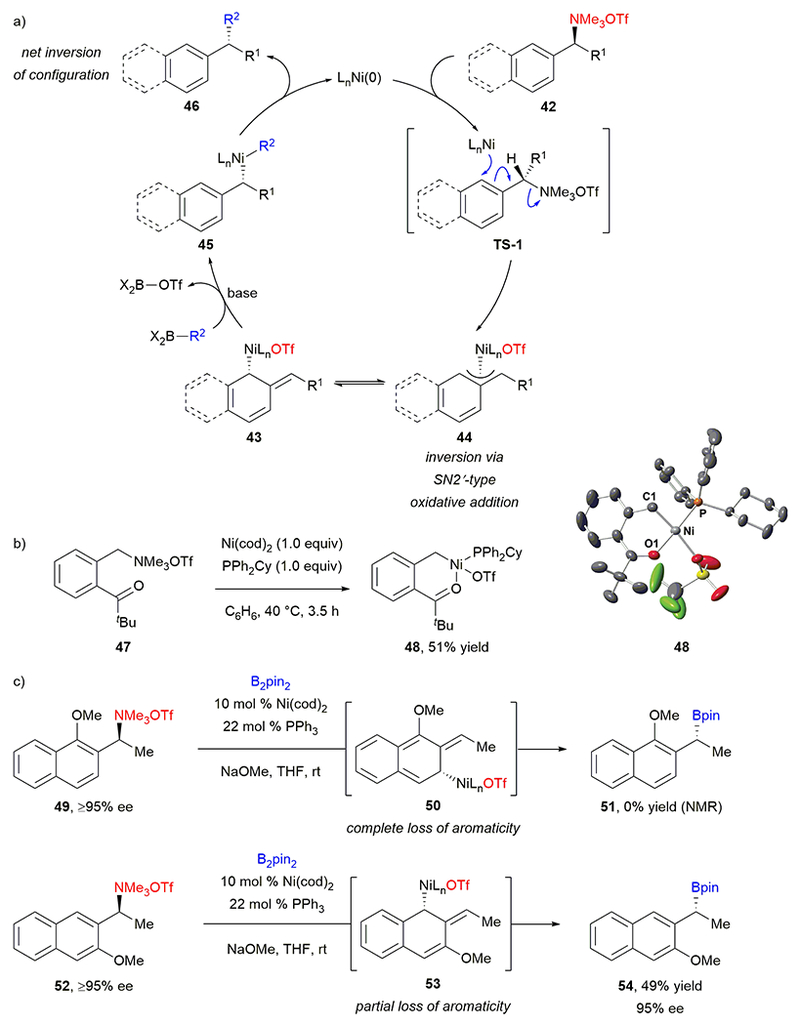
Stereospecific, nickel-catalyzed cross-coupling mechanism: (a) proposed catalytic cycle; (b) synthesis and crystal structure of oxidative addition complex 48; (c) experiments demonstrating oxidative addition via an SN2’ mechanism.
Excitingly, our efforts seem to have reinvigorated interest in utilizing ammonium salts as electrophiles in transition metal-catalyzed cross-coupling reactions, including the development of nickel-catalyzed carboxylation51 and reduction52 of benzylic ammonium triflates. Additionally, non-metal-catalyzed stereospecific reactions of benzylic ammonium triflates have been reported subsequent to our work.53 Further, Tortosa and coworkers have demonstrated that copper catalysts can also be used, specifically in their stereospecific arylation of propargylic ammonium salts.54 Reaction of enantioenriched propargylic ammonium triflate 55 with aryl Grignard reagent affords 56 in 98% yield (Scheme 10). The reaction proceeds with α-regioselectivity and inversion of configuration in excellent stereochemical fidelity. A subsequent report from this group has also utilized enantioenriched propargylic ammonium triflates to generate allenes in high ee’s.55 Additionally, the use of benzylic ammonium salts in cross-coupling reactions has led to the development of alternative activating groups for amines, including pyridinium salts, reported by us56 and others.57 Overall, we are excited to have identified a novel mode of C(sp3)–N bond activation, which allows for the stereospecific formation of new C–C bonds.
Scheme 10.
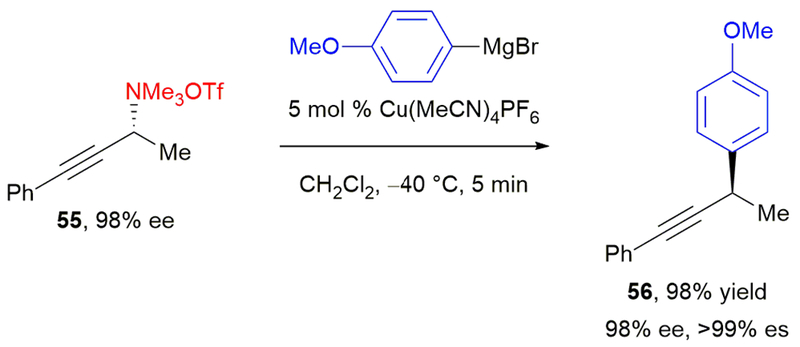
Tortosa’s stereospecific, copper-catalyzed Negishi cross-coupling of enantioenriched propargylic ammonium triflates.
3. C(sp3)–O Bond Activation of Benzylic Alcohol Derivatives
3.1. Secondary Benzylic Ethers
Alcohols are readily available substrates, making them ideal candidates to participate as electrophiles in cross-coupling reactions. Indeed, nickel-catalyzed C(sp2)–O bond activation of phenols, enols, and their derivatives is well developed.58 However, productive activation of C(sp3)–O bonds is more challenging; they undergo slow oxidative addition due to their high bond dissociation energies.59 Overcoming this barrier, Shi and coworkers reported the first nickel-catalyzed C(sp3)–O bond activation of benzylic ethers.60 Reaction of ether 57 with methylmagnesium bromide under nickel catalysis afforded product 58 quantitatively (Scheme 11). Complete selectivity for the benzylic ether over an aryl ether was observed.
Scheme 11.
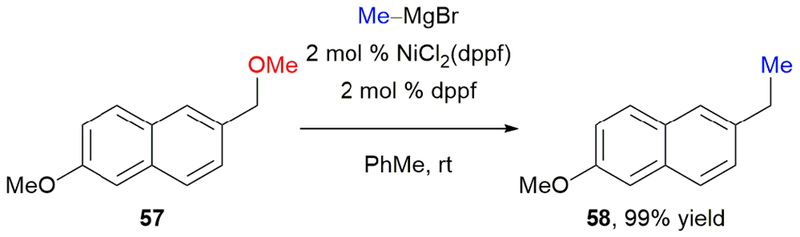
Shi’s nickel-catalyzed Negishi cross-coupling of benzylic methyl ethers.
In 2011, Jarvo and coworkers reported the first stereospecific, nickel-catalyzed cross-coupling of secondary benzylic ethers.39 Reaction of enantioenriched ether 59 with methylmagnesium iodide afforded 60 in 72% yield (Scheme 12a). Optimization of the ligand was key to promoting the desired reactivity by accelerating oxidative addition and minimizing β-hydride elimination. This cross-coupling proceeded with excellent stereochemical fidelity for inversion at the benzylic stereocenter. The authors demonstrated the utility of this method by synthesizing a diarylethane with potent tubulin polymerization inhibition activity. They subsequently utilized enantioenriched secondary methyl ethers as electrophiles in additional stereospecific, nickel-catalyzed cross-couplings, including a Kumada cross-coupling with alkyl Grignard reagents61 and an intramolecular Heck reaction.62
Scheme 12.
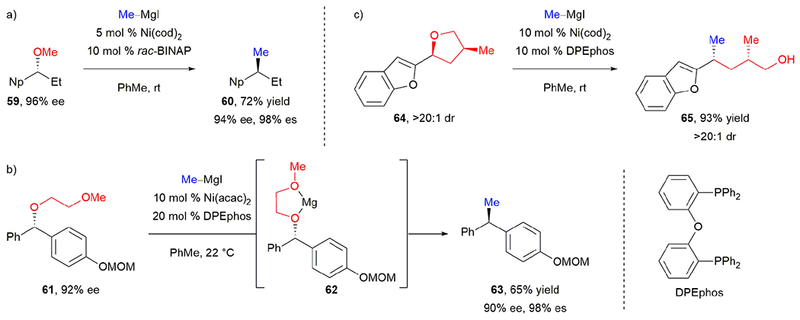
Jarvo’s stereospecific, nickel-catalyzed Negishi cross-couplings of (a) secondary benzylic methyl ethers, (b) secondary benzylic ethers using a traceless directing group, and (c) tetrahydrofurans.
By changing the alcohol activating group from a methyl ether to a 2-methoxyethyl ether, Jarvo and coworkers expanded the stereospecific, nickel-catalyzed methylation scope to secondary dibenzylic ethers without naphthyl substituents.37 Under nickel catalysis, cross-coupling of enantioenriched dibenzylic ether 61 and methylmagnesium iodide afforded diarylethane 63 in 65% yield and 98% es (Scheme 12b). The authors proposed that the traceless directing group accelerates oxidative addition by forming five-membered chelate 62 with magnesium salts,10h,44b,63 thereby allowing the use of less reactive electrophiles. In subsequent publications, Jarvo and coworkers used this traceless directing group for stereospecific, nickel-catalyzed cross-coupling reactions of secondary benzylic ethers with aryl Grignard reagents to afford enantioenriched triarylmethanes.38,64 Additionally, they utilized a similar directing group for the stereospecific, nickel-catalyzed Negishi cross-coupling of secondary benzylic esters with dimethylzinc.40
Benzylic tetrahydropyrans, tetrahydrofurans, and lactones also undergo ring-opening under similar nickel-catalyzed methylation conditions.65 When multiple stereogenic centers are present, the ring opening occurs with inversion at the benzylic stereogenic center without affecting or being affected by the other stereogenic centers. Reaction of cis-64 afforded syn-65 in 93% yield and excellent diastereoselectivity (Scheme 12c). When the trans diastereomer was used, anti product was obtained in the same yield and diastereoselectivity. Jarvo has also demonstrated this C(sp3)–O bond activation and ring-opening strategy in a stereospecific, nickel-catalyzed cross-electrophile reductive coupling to efficiently synthesize cyclopropanes from tetrahydropyrans.66 Overall, the work by Jarvo and coworkers demonstrated that nickel-catalyzed cross-couplings of enantioenriched benzylic ethers with Grignard and organozinc reagents occur with excellent levels of stereochemical fidelity. Their seminal reports certainly showed the power of stereospecific cross-couplings of readily available enantioenriched electrophiles, and inspired much of our effort with benzylic ammonium triflates, as well as benzylic pivalates as discussed below.
3.2. Secondary Benzylic Carboxylates and Carbamates
In the stereospecific cross-couplings discussed above, Grignard reagents were used as the nucleophilic coupling partners. Although Grignard reagents are less expensive than their boronic acid counterparts, we imagined that the convenience and functional group tolerance offered by using an organoboron reagent would often be an attractive alternative. Based on our previous success with the activation of the C(sp2)–O bond of aryl pivalates,67 we focused our attention on developing a stereospecific, nickel-catalyzed Suzuki–Miyaura cross-coupling of benzylic pivalates.68 Predicting that boronate coordination to an electrophilic Ni(II) intermediate may be a key step in transmetallation,69 we hypothesized that the nickel(II) pivalate may be able to undergo transmetalation with an aryl boronate due to the weaker coordination of the pivalate versus the alkoxides generated with benzylic ether substrates. The weaker C–O bond may also lead to a higher concentration of the oxidative addition intermediate, further facilitating a more difficult transformation. Enantioenriched secondary pivalates were prepared via Corey–Bakshi–Shibata (CBS) reduction of the corresponding ketones, followed by acylation.70 In the presence of Ni(cod)2 and electron-rich PCy2Ph, cross-coupling of enantioenriched pivalate 66 and phenylboronic acid afforded diarylethane (R)-67 in 93% yield and 54% es with retention of configuration (Scheme 13a). In contrast, conditions without exogenous ligand afforded (S)-67 in improved yield and stereochemical fidelity with inversion of configuration. Stereoretention had not been previously reported in any stereospecific cross-coupling of a benzylic electrophile, and we were intrigued by this result.
Scheme 13.
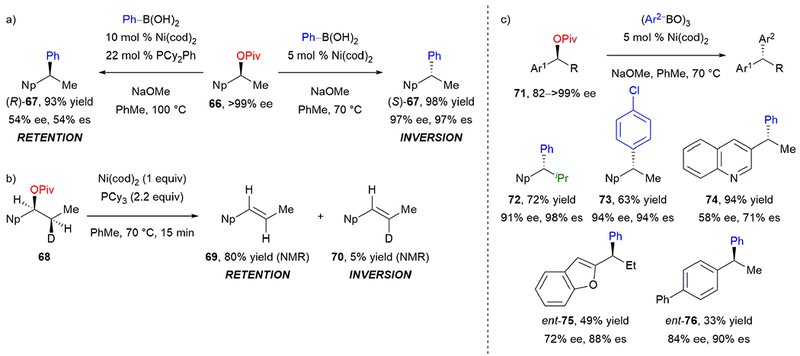
Our stereospecific, nickel-catalyzed Suzuki–Miyaura cross-coupling of secondary benzylic pivalates: (a) retention or inversion of stereochemistry depends on ligand; (b) mechanistic experiment; and (c) abbreviated scope.
In analogy to our benzylic ammonium triflates (see Scheme 9 above), we hypothesized that the stereochemical outcome is dictated by the oxidative addition step. To confirm that the overall stereoretention was indeed due to the oxidative addition step, we wanted to isolate the oxidative addition from the subsequent transmetallation and reductive elimination. Inspired by a similar experiment by Fu,50b we subjected deuterated pivalate 68 to stoichiometric Ni(cod)2 and PCy3, and allowed β-hydride elimination to take place. Alkene 69 was the major product with alkene 70 as a minor byproduct (Scheme 13b). Formation of alkene 69 is consistent with β-hydride elimination (via synperiplanar C–Ni and C–D bonds) of the intermediate resulting from stereoretentive oxidative addition. This suggested that oxidative addition in the presence of phosphine ligand proceeds via a distinct mechanism from the precedented SN2’-type oxidative addition of nickel complexes into ammonium salts and ether electrophiles. Without phosphine ligand, oxidative addition likely follows the SN2’-type mechanism, consistent with the observed inversion of configuration at the benzylic stereocenter.
Under the “phosphine-less” conditions, reaction of enantioenriched 71 with arylboroxine had good functional group tolerance (Scheme 13c). The use of boroxine resulted in better yields and higher levels of chirality transfer, indicating that water had a detrimental effect on the reaction. Notably, aryl chloride is tolerated (73), providing a functional group handle for further cross-coupling reactions. Steric hindrance was also well tolerated, as evidenced by the i-Pr group in 72. Electron-poor (74) and -rich (75) heteroaryl-substituted pivalates underwent the cross-coupling, although the yield was diminished for 75. Additionally, biphenyl-substituted pivalate underwent the reaction to afford 76 in good stereochemical fidelity, albeit diminished yield.
Concurrent with our report of this work, Jarvo and coworkers reported a stereospecific, nickel-catalyzed Suzuki–Miyaura cross-coupling of dibenzylic carbamates 77a (Scheme 14).43 Excitingly, they also observed that the ligand determined whether the cross-coupling proceeds with retention or inversion of configuration. Furthermore, they were able to optimize conditions to achieve excellent stereochemical fidelity in both pathways. The ability to access both product enantiomers in high ee from a single enantiomer of starting material overcomes the common limitation of stereospecific reactions. This work, along with stereochemical flips observed in allylic substrates, set the stage for advancing the field’s understanding of what reaction parameters can be used for stereodivergency in stereospecific cross-coupling reactions.
Scheme 14.
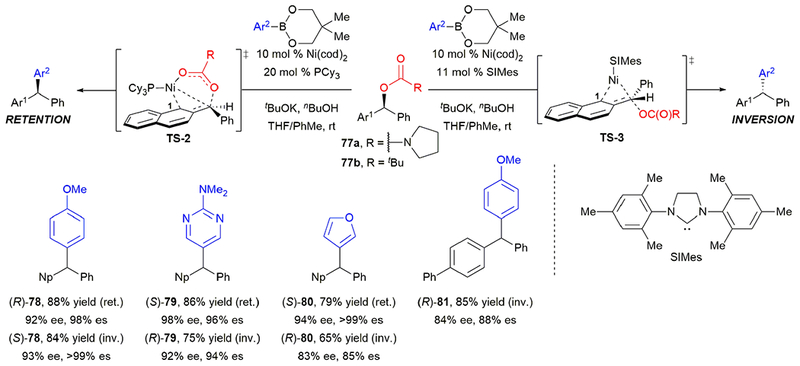
Jarvo’s stereospecific, nickel-catalyzed Suzuki–Miyaura cross-coupling of secondary benzylic carbamates.
Towards this goal of mechanistic understanding, subsequent computations by Jarvo, Houk, and Hong studied the transformation of pivalate 77b to form 78. Their calculations suggest that with phosphine ligand PCy3, there is a preference for oxidative addition via cyclic transition state TS-2, resulting in retention of stereochemistry.71 When using N-heterocyclic carbene (NHC) ligand SIMes, there is a preference for oxidative addition by SN2’ back-side attack through an open transition state (TS-3), leading to inversion of configuration. They propose that the major factor contributing to this change in mechanism is the difference in energy caused by bending the C1–Ni–ligand angle to accommodate the formation of the Ni–O bond in TS-2. For PCy3, the nickel–ligand interaction involves mainly σ-donation, so there is less of an energy penalty for C1–Ni–ligand angle distortion than when using SIMes, which has a more rigid nickel–ligand bond due to additional d–p back-donation.
Functional group tolerance was good for both reaction conditions, including reaction of heteroaryl boronic esters 79 and 80. Although naphthyl substitution was not required, this report is limited to dibenzylic carbamates. This requirement results from coordination of the nickel to the aryl group prior to oxidative addition, which is consistent with both computed mechanistic pathways and is similar to the previously proposed SN2’-type mechanism. Jarvo and coworkers later used this method to synthesize enantioenriched diarylalkanes and trialkylmethanes.72
The use of enantioenriched alcohol derivatives has continued to gain increasing attention in transition metal catalysis. Benzylic pivalates have been used in subsequent reports of nickel-catalyzed reactions, including a Suzuki–Miyaura cross-coupling to form triarylmethanes73 a stereospecific intramolecular cross-electrophile coupling,74 and a stereospecific Miyaura borylation.75 C(sp3)–O activating groups now include 2-pyridyl etherates76 and vinyl dioxanones.77 In a related reaction, Tunge and Mendis reported a stereospecific, palladium-catalyzed decarboxylative alkynylation of diarylcarbonates.78 Additionally, Tang and coworkers demonstrated a transition metal-free stereospecific addition of vinyl boronic acids to enantioenriched benzylic mesylates.79 Palladium-catalyzed dynamic kinetic resolution of dibenzylic carboxylates has also been recently reported.80 By activating widely available enantioenriched alcohols as carboxylates and carbamates, cross-couplings with organoboranes have been enabled, greatly increasing the functional group tolerance and convenience of these stereospecific reactions.
3.3. Tertiary Benzylic Carboxylates
Based on the proposed SN2’ oxidative addition and the high degree of steric hindrance tolerated in the stereospecific cross-coupling reactions of secondary benzylic carboxylates, we envisioned that a stereospecific, nickel-catalyzed cross-coupling of tertiary benzylic electrophiles may be possible. If the nickel catalyst was adding in an SN2’ fashion, increased steric hindrance at the benzylic position should be tolerated. This reaction would allow access to benzylic, all-carbon quaternary stereocenters in high enantiopurity. By using a stereospecific cross-coupling to accomplish this challenging transformation, there would be no need to differentiate similar alkyl groups with a chiral catalyst. Thus, when coupled with enantioselective ketone alkylation, we believed that this method would offer a powerful asymmetric synthesis of benzylic quaternary stereocenters from readily available, achiral ketone precursors. Notably, the only examples of metal-catalyzed cross-couplings of alkyl electrophiles to give quaternary centers in high enantioenrichment utilized allylic substrates;81 no tertiary benzylic electrophiles had yet been demonstrated.
Capitalizing on this idea, we developed a stereospecific, nickel-catalyzed arylation of tertiary benzylic acetates with boronate esters.82 Enantioenriched tertiary alcohols were readily prepared via Walsh’s enantioselective addition of organozinc reagents to acetophenones.83 Although pivalates could not be easily prepared likely due to steric congestion of the tertiary alcohol, acetates 82 were readily accessible and proved to be excellent substrates (Scheme 15).
Scheme 15.
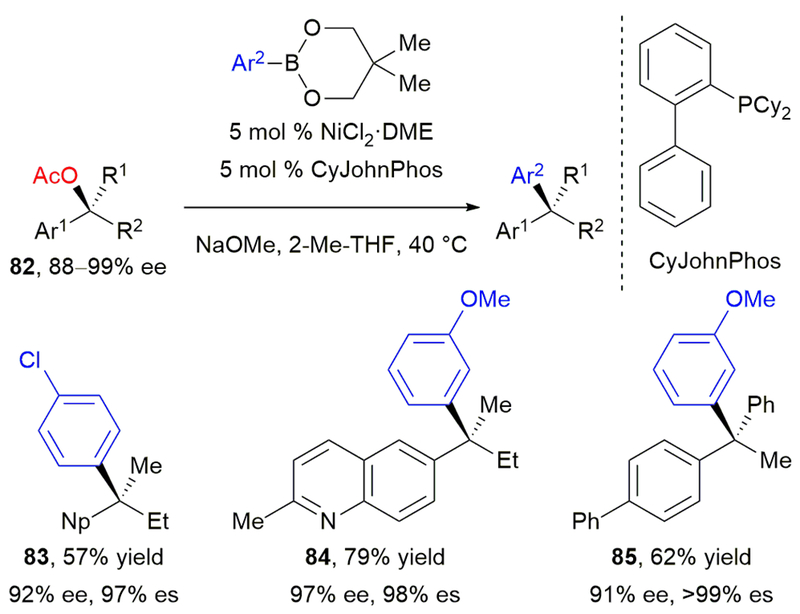
Our stereospecific, nickel-catalyzed Suzuki–Miyaura cross-coupling of tertiary benzylic acetates.
The success of this reaction relied upon significant optimization of the catalyst system. The reaction conditions optimized for the cross-coupling of secondary benzylic pivalates afforded high yield of cross-coupled product but with low stereochemical fidelity. Addition of a monodentate phosphine ligand improved stereochemical fidelity, but also led to elimination byproducts. Hypothesizing that these were due to competitive β-hydride elimination, we investigated bidentate ligands, including those with hemilabile arms. By switching to a Buchwald ligand, CyJohnPhos, elimination products were reduced and the enantiospecificity was much improved, affording products with retention of stereochemistry.
A wide range of functional groups were well tolerated, including chloride 83. An example of heteroaryl substitution was also demonstrated, affording 84 in 79% yield. As with the cross-coupling of secondary alcohol and amine derivatives, either an aryl substituent with an extended π-system or diaryl substitution (85) was required on the electrophile. Despite this limitation, this reaction was the first example of a transition metal-catalyzed cross-coupling of a benzylic tertiary electrophile to give products in high enantiomeric excess, and overcame traditional limitations in utilizing tertiary electrophiles in substitution reactions. It highlights the advantage of combining a stereospecific cross-coupling with known catalytic asymmetric reactions, and offers a highly efficient strategy for asymmetric synthesis of diaryl and triaryl alkanes with quaternary stereocenters.
4. C(sp3)–O Bond Activation of Allylic Alcohol Derivatives
4.1. Secondary Allylic Alcohol Derivatives
Stereospecific arylation of readily accessible 1,3-disubstituted secondary allylic electrophiles enables facile construction of enantioenriched products equipped with vinyl-substituted benzylic carbon stereocenters. Kobayashi was the first to report a stereospecific, nickel-catalyzed Suzuki–Miyaura cross-coupling of an allylic alcohol derivative.84 Cyclic allylic acetate 86 underwent cross-coupling with phenylzinc borate reagent to afford product 87 with inversion of configuration (Scheme 16a). Sawamura and coworkers then developed stereospecific, palladium- and copper-catalyzed Suzuki–Miyaura cross-coupling reactions of allylic esters and phosphates (Scheme 16b).85 These γ-selective reactions afforded products with retention of stereochemistry (89). Zhang,86 Tian,87 and Bäckvall88 developed stereospecific cross-coupling reactions of allylic electrophiles that proceed with α-selectivity (90). However, no stereospecific, nickel-catalyzed cross-couplings of acyclic allylic electrophiles and arylboron reagents to deliver highly enantioenriched products had been reported.
Scheme 16.
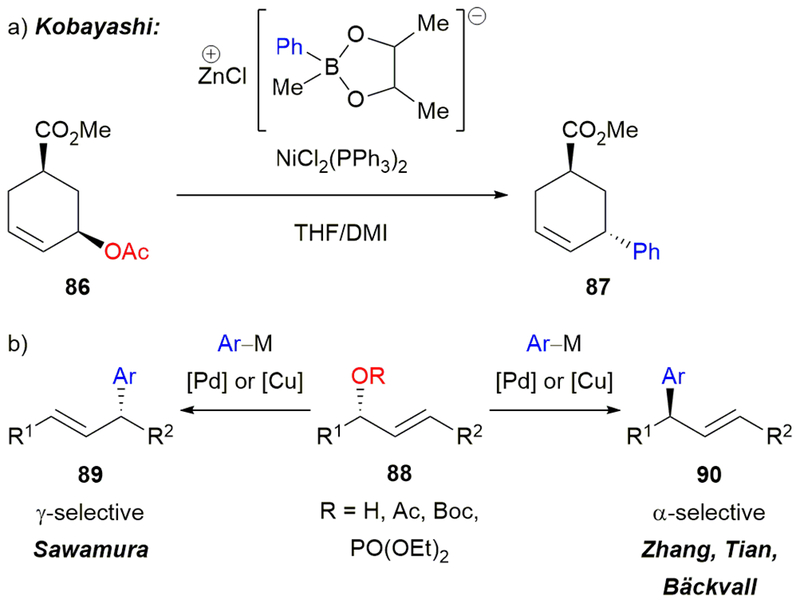
(a) Kobayashi’s nickel-catalyzed arylation of cyclic allylic acetates. (b) Examples of stereospecific, transition metal-catalyzed arylations of 1,3-disubstituted allylic electrophiles.
Based on our studies of stereospecific, nickel-catalyzed cross-couplings of benzylic carboxylates and arylboronates, we envisioned that nickel-based catalysts may also serve as efficient, nonprecious metal catalysts for highly stereospecific and regioselective cross-couplings of 1,3-disubstituted allylic pivalates and arylboronates. In 2014, we reported the first stereospecific, nickel-catalyzed cross-coupling of an acyclic allylic electrophile to deliver a highly enantioenriched product.89 Pivalates were readily prepared in high enantiomeric excess via CBS reduction and pivalation.70 Reaction of pivalates 91 and arylboroxines with Ni(cod)2 and BnPPh2 afforded products with excellent yields, regioselectivities, and enantiospecificities (Scheme 17a). This method is mild and tolerates a wide scope of functional groups including heteroaryl substitution. Halides were well tolerated on the allylic pivalate (93) and arylboroxine (94), highlighting the orthogonality of the reaction to this group. Additionally, the cross-coupling had a high tolerance for steric hindrance at the benzylic position (95). We proposed that the reaction proceeds through a π-allylnickel intermediate with selectivity for forming the conjugated alkene in the product. Consistent with the intermediacy of a π-allylnickel complex, the reaction of alkene regioisomer 96 resulted in product 97 (Scheme 17b).
Scheme 17.
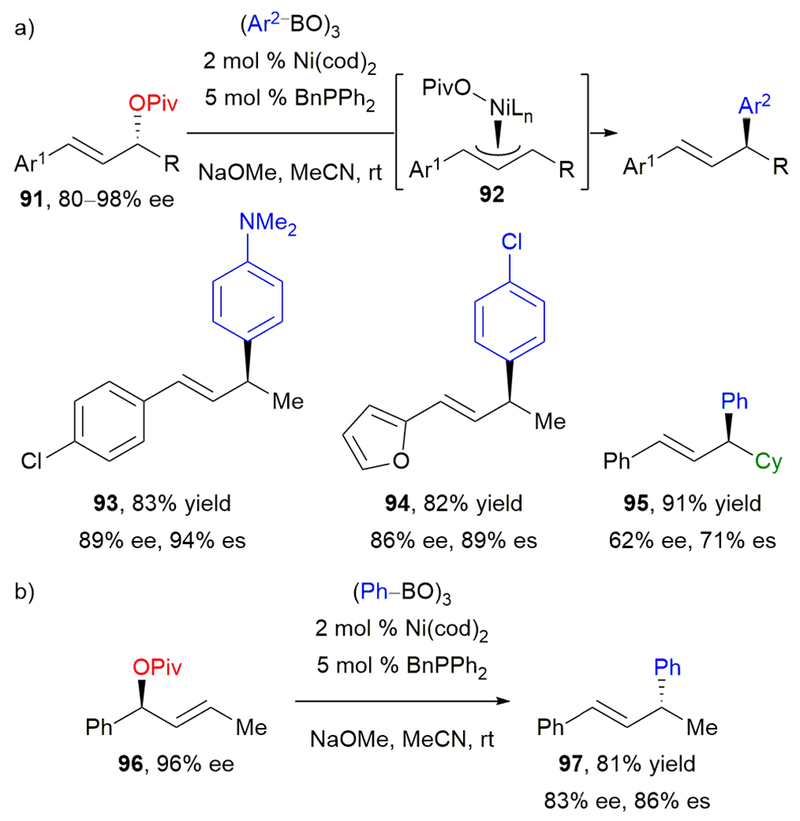
Our stereospecific, nickel-catalyzed Suzuki–MIyaura cross-coupling of allylic pivalates: (a) abbreviated scope, and (b) evidence for π-allylnickel intermediate.
We also recognized that allylic boronates are useful intermediates in organic synthesis.90 In 2005, Ito, Kawakami, and Sawamura reported a stereospecific, copper-catalyzed borylation of allylic carbonates to deliver highly enantioenriched γ-alkyl allylic boronates.91 However, no stereospecific, transition metal-catalyzed borylation for the preparation of γ-aryl α-chiral allylic boronates had been reported. We envisioned that a stereospecific, nickel-catalyzed Miyaura borylation of γ-aryl allylic pivalates would deliver highly enantioenriched γ-aryl α-stereogenic boronates.92 The optimal conditions for our stereospecific allylic arylation gave high yields and stereochemical fidelity for the Miyaura borylation, affording product (S)-101 with inversion of stereochemistry (Scheme 18). However, when toluene was used as the solvent, product (R)-101 formed with retention of configuration. Further optimization of ligand and other conditions led to high stereochemical fidelity and regioselectivity under these stereoretentive conditions. Notably, this was the first example of solvent as the predominant factor in a stereochemical flip.93
Scheme 18.
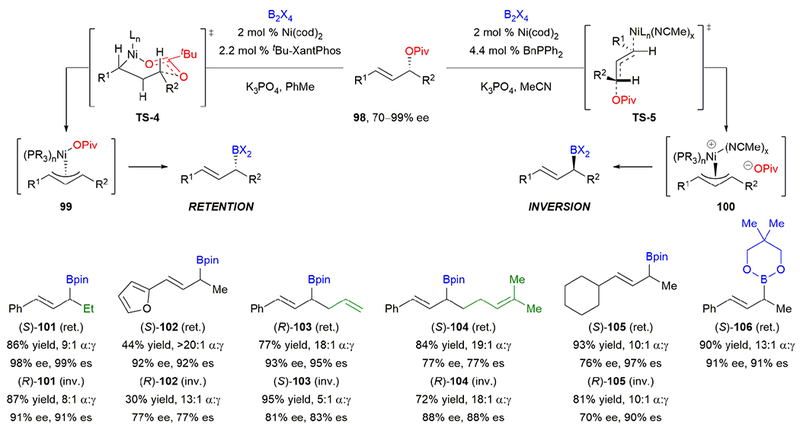
Our stereospecific, nickel-catalyzed Miyaura borylation of allylic pivalates.
Heteroaryl groups could be incorporated (102). Tether length of pendant olefins affected the stereochemical fidelity of the cross-coupling (103, 104), potentially indicating a change in mechanism or epimerization pathway. In addition to aryl allylic boronates, γ-alkyl allylic boronate 105 was formed in high yield, regioselectivity, and enantiospecificity. Diborane coupling partners other than B2pin2 could be used; for example, boronate 106 was produced in 90% yield and 91% es.
Based on a series of mechanistic studies, we proposed that when the cross-coupling is run in nonpolar solvents, the pivalate leaving group directs the nickel. Oxidative addition occurs via closed transition state TS-4. Nickel adds to the re face of the alkene, thereby allowing R1 and R2 to be pseudoequatorial, which leads to retention of stereochemistry. More strongly coordinating carboxylates lead to higher stereochemical fidelity, consistent with carboxylate coordination to the nickel catalyst.
When conducted in more polar solvents, oxidative addition via an open transition state to give a charge-separated intermediate is competitive. Acetonitrile also appears to act as a ligand, coordinating nickel and prohibiting coordination of the pivalate. Favored transition state TS-5 minimizes A1,3-strain, affording inversion of configuration. Under the conditions optimized for stereoretention, p-substituted benzonitriles were added. There was excellent correlation between the electron-donating ability of the added nitrile and the stereospecificity, with more electron-poor benzonitriles leading to higher stereochemical fidelity under the retention conditions. This mechanistic insight offers exciting new options for stereodivergent synthesis and adds solvent as an additional parameter to accomplish stereochemical flips.
4.2. Tertiary Allylic Alcohol Derivatives
With our success in setting benzylic quaternary centers and allylic substitution, we targeted quaternary center formation via allylic arylation. In particular, we were excited to form products with the additional vinyl handle for further elaboration. The use of allylic halides and phosphate esters in enantioselective cross-coupling reactions to afford molecules containing all-carbon quaternary stereocenters with terminal alkenes is well developed (Scheme 19a).81f,94 However, the preparation of enantioenriched products with all-carbon quaternary stereocenters and internal alkenes using enantioenriched allylic electrophiles was limited to reactions using stoichiometric copper (Scheme 19b).81e,95 In an umpolung approach, Morken and coworkers developed a stereospecific, palladium-catalyzed cross-coupling of allylic boronate esters with aryl halides.96
Scheme 19.
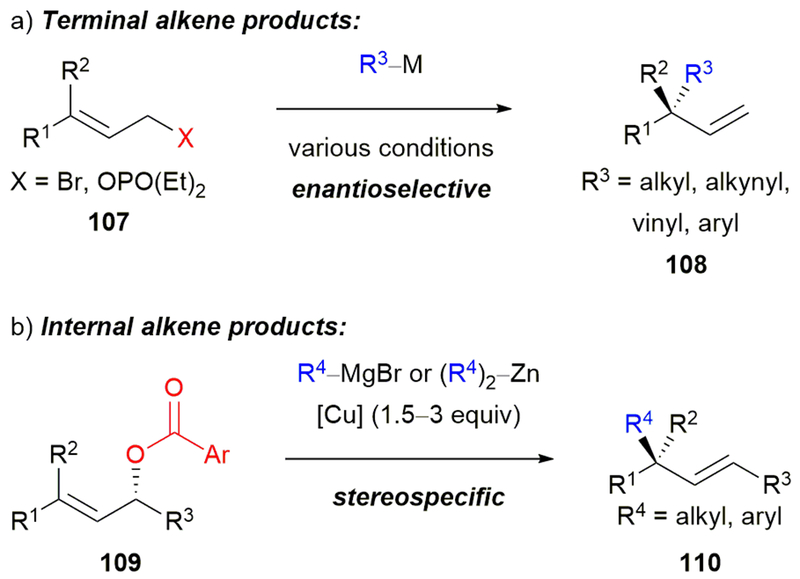
(a) Enantioselective, transition metal-catalyzed allylic substitution reactions to form products with terminal alkenes. (b) Stereospecific allylic substitution reactions to prepare products with internal alkenes.
As we considered an efficient and convenient approach to the synthesis of quaternary stereocenters substituted with internal alkenes, we were inspired to develop a stereospecific, nickel-catalyzed Suzuki–Miyaura cross-coupling of allylic pivalates and arylboronates (Scheme 20).97 The secondary pivalates 111 were readily prepared in highly enantioenriched form using a CBS reduction.98 Although electron-rich phosphine dppf gave high stereochemical fidelity, it afforded low yield, which we hypothesized was due to decomposition of starting material from redox activity with the ferrocene. Changing the ligand to BISBI, which has a wide bite angle and a semi-rigid backbone,99 afforded products in high yield and stereochemical fidelity. The use of aryl boronic acid in place of arylboroxine resulted in lower yields with significant hydrolysis of pivalate. Heteroaryl-substituted boroxines were well tolerated, affording products 112 and 113 in good yields. Steric hindrance at the stereogenic center was also well tolerated, affording 114 in 84% yield. As expected in MeCN solvent, the reaction proceeds with inversion of configuration. Additionally, the geometry of the starting alkene affects the stereochemistry. The allylic pivalate synthesized from nerol gives the opposite absolute configuration of product 115 as the allylic pivalate synthesized from geraniol (116).
Scheme 20.
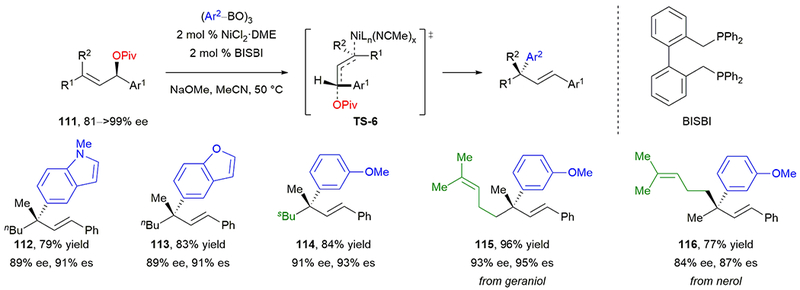
Our stereospecific, nickel-catalyzed Suzuki–Miyaura cross-coupling of allylic pivalates to form all-carbon quaternary stereocenters.
We propose that oxidative addition proceeds through open transition state TS-6, where the nickel adds to the opposite face of the allylic system as the pivalate leaving group. This conformation minimizes any developing A1,3 interactions. Acetonitrile may favor this open transition state by coordinating with the nickel catalyst and blocking coordination with the pivalate. Transmetalation and reductive elimination then deliver the product where the alkene is conjugated with the adjacent aryl group. This reaction offers an entry into the preparation of all-carbon quaternary stereocenters adjacent to internal alkenes in high regioselectivity and enantiospecificity.
5. Conclusions and Outlook
Inspired by the prior art from a range of groups, we have developed a series of stereospecific cross-couplings of benzylic and allylic amine and alcohol derivatives. The success of this effort has relied on the design of benzylic ammonium triflates as substrates for cross-couplings via C–N bond activation, and the development of benzylic and allylic carboxylates for cross-couplings via C–O bond activation. Our efforts, along with others’ exciting discoveries, have demonstrated that stereospecific, transition metal-catalyzed cross-coupling reactions using enantioenriched electrophiles are useful in asymmetric synthesis, particularly when combined with highly efficient asymmetric reactions to generate the enantioenriched intermediates. We have also begun to uncover detailed understanding of how to manipulate catalyst systems and other reaction conditions to enable stereodivergency in these stereospecific reactions. Despite the clear potential of these reactions in asymmetric synthesis, challenges remain. The scope of benzylic ammonium salts and carboxylates remains limited by the need for polycyclic aryl substituents (e.g., naphthyl) or dibenzylic substrates, and few heteroaryl groups have been demonstrated. Continued efforts to understand mechanism are also needed to allow prediction of stereodivergent reaction conditions broadly across the full range of stereospecific cross-couplings. We are excited to continue to contribute to the development of this field, and ultimately hope that these stereospecific cross-couplings will advance into indispensible reactions for asymmetric synthesis.
Acknowledgements
We thank NIH (R01 GM111820) for generous support of this work.
Footnotes
Conflicts of interest
There are no conflicts to declare.
Reprinted with permission from P. Maity, D. M. Shacklady-McAtee, G. P. A. Yap, E. R. Sirianni and M. P. Watson, J. Am. Chem. Soc., 2013, 135, 280–285. Copyright 2012 American Chemical Society.
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.(a) Yamaguchi J, Muto K and Itami K, Eur. J. Org. Chem, 2013, 2013, 19–30. [Google Scholar]; (b) Johansson Seechurn CCC, Kitching MO, Colacot TJ and Snieckus V, Angew. Chem., Int. Ed, 2012, 51, 5062–5085. [DOI] [PubMed] [Google Scholar]; (c) Negishi E.-i, Angew. Chem., Int. Ed, 2011, 50, 6738–6764. [DOI] [PubMed] [Google Scholar]; (d) Suzuki A, J. Organomet. Chem, 1999, 576, 147–68. [Google Scholar]; (e) Heck RF, Iin Organic Reactions, ed. Dauben WG, John Wiley & Sons, Inc., Hoboken, NJ, 1982, Vol. 27, pp 345–390. [Google Scholar]
- 2.(a) Luh T-Y, Leung M and Wong K-T, Chem. Rev, 2000, 100, 3187–3204. [DOI] [PubMed] [Google Scholar]; (b) Netherton MR and Fu GC, Adv. Synth. Catal, 2004, 346, 1525–1532. [Google Scholar]
- 3.(a) Jana R, Pathak TP and Sigman MS, Chem. Rev, 2011, 111, 1417–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Swift EC and Jarvo ER, Tetrahedron, 2013, 69, 5799–5817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Cherney AH, Kadunce NT and Reisman SE, Chem. Rev, 2015, 115, 9587–9652. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Glasspoole BW, Keske EC and Crudden CM, in New Trends in Cross-Coupling: Theory and Applications, ed. Colacot TJ, Royal Society of Chemistry, Cambridge, 2014, pp. 521–550. [Google Scholar]; (c) Tasker SZ, Standley EA and Jamison TF, Nature, 2014, 509, 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.es = (eeproduct/eestarting material) x 100%, see:; (a) Denmark SE and Vogler T, Chem. – Eur. J, 2009, 15, 11737–11745. [DOI] [PubMed] [Google Scholar]; (b) Denmark SE, Burk MT and Hoover AJ, J. Am. Chem. Soc, 2010, 132, 1232–1233 [DOI] [PubMed] [Google Scholar]
- 6.(a) Rygus JPG and Crudden CM, J. Am. Chem. Soc, 2017, 139, 18124–18137. [DOI] [PubMed] [Google Scholar]; (b) Leonori D and Aggarwal VK, Angew. Chem., Int. Ed, 2015, 54, 1082–1096. [DOI] [PubMed] [Google Scholar]; (c) Wang C-Y, Derosa J and Biscoe MR, Chem. Sci, 2015, 6, 5105–5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pronin SV, Reiher CA and Shenvi RA, Nature, 2013, 501, 195–199. [DOI] [PubMed] [Google Scholar]
- 8.(a) Stille JK, in The Chemistry of the Metal–Carbon Bond, ed. Hartley FR and Patai S, John Wiley & Sons, New York, 1985, vol. 2, pp. 625–787. [Google Scholar]; (b) He A and Falck JR, J. Am. Chem. Soc, 2010, 132, 2524–2525. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) López-Pérez A, Adrio J and Carretero JC, Org. Lett, 2009, 11, 5514–5517. [DOI] [PubMed] [Google Scholar]; (d) Rudolph A, Rackelmann N and Lautens M, Angew. Chem., Int. Ed, 2007, 46, 1485–1488. [DOI] [PubMed] [Google Scholar]; (e) Rodríguez N, de Arellano CR, Asensio G and Medio-Simón M, Chem. – Eur. J, 2007, 13, 4223–4229. [DOI] [PubMed] [Google Scholar]; (f) Legros J-Y, Toffano M and Fiaud J-C, Tetrahedron, 1995, 51, 3235–3246. [Google Scholar]; (g) Lau KSY, Fries RW and Stille JK, J. Am. Chem. Soc, 1974, 96, 4983–4986. [Google Scholar]
- 9.Rudolph A and Lautens M, Angew. Chem., Int. Ed, 2009, 48, 2656–670. [DOI] [PubMed] [Google Scholar]
- 10.(a) Smith SW and Fu GC, Angew. Chem., Int. Ed, 2008, 47, 9334–9336. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gong H, Sinisi R and Gagne MR, J. Am. Chem. Soc, 2007, 129, 1908–1909. [DOI] [PubMed] [Google Scholar]; (c) Phapale VB, Buñuel E, García-Iglesias M and Cárdenas DJ, Angew. Chem., Int. Ed, 2007, 46, 8790–8795. [DOI] [PubMed] [Google Scholar]; (d) Jones GD, Martin JL, McFarland C, Allen OR, Hall RE, Haley AD, Brandon RJ, Konovalova T, Desrochers PJ, Pulay P and Vicic DA, J. Am. Chem. Soc, 2006, 128, 13175–13183. [DOI] [PubMed] [Google Scholar]; (e) Terao J, Todo H, Watanabe H, Ikumi A and Kambe N, Angew. Chem., Int. Ed, 2004, 43, 6180–6182. [DOI] [PubMed] [Google Scholar]; (f) Zhou JS and Fu GC, J. Am. Chem. Soc, 2003, 125, 14726–14727. [DOI] [PubMed] [Google Scholar]; (g) Giovannini R, Stüdemann T, Dussin G and Knochel P, Angew. Chem., Int. Ed, 1998, 37, 2387–2390. [DOI] [PubMed] [Google Scholar]; (h) Devasagayaraj A, Stüdemann T and Knochel P, Angew. Chem., Int. Ed, 1996, 34, 2723–2725. [Google Scholar]; (i) Stille JK and Cowell AB, J. Organomet. Chem, 1977, 124, 253–261. [Google Scholar]
- 11.Han F-S, Chem. Soc. Rev, 2013, 42, 5270–5298. [DOI] [PubMed] [Google Scholar]
- 12.Nugent TC, Chiral Amine Synthesis: Methods, Developments and Applications, Wiley-VCH Verlag GmbH & Co. KGaA, Bremen, 2010. [Google Scholar]
- 13.Blanksby SJ and Ellison GB, Acc. Chem. Res, 2003, 36, 255–263. [DOI] [PubMed] [Google Scholar]
- 14.(a) Ouyang K , Hao W, Zhang W-X and Xi Z, Chem. Rev, 2015, 115, 12045–12090. [DOI] [PubMed] [Google Scholar]; (b) Wang Q, Su Y, Li L and Huang H, Chem. Soc. Rev, 2016, 45, 1257–1272. [DOI] [PubMed] [Google Scholar]
- 15.Trost BM and Spagnol MD, J. Chem. Soc., Perkin Trans. 1, 1995, 2083–2096. [Google Scholar]
- 16.Li M-B, Wang Y and Tian S-K, Angew. Chem., Int. Ed, 2012, 51, 2968–2971. [DOI] [PubMed] [Google Scholar]
- 17.(a) Wu X-S, Chen Y, Li M-B, Zhou M-G and Tian S-K, J. Am. Chem. Soc, 2012, 134, 14694–14697. [DOI] [PubMed] [Google Scholar]; (b) Li M-B, Li H, Wang J, Liu C-R and Tian S-K, Chem. Commun, 2013, 49, 8190–8192. [DOI] [PubMed] [Google Scholar]; (c) Wang Y, Xu J-K, Gu Y and Tian S-K, Org. Chem. Front, 2014, 1, 812–816. [Google Scholar]; (d) Wang Y, Li M, Ma X, Liu C, Gong Y and Tian SK, Chin. J. Chem, 2014, 32, 741–751. [Google Scholar]; (e) Wang T-T, Wang F-X, Yang F-L and Tian S-K, Chem. Commun, 2014, 50, 3802–3805. [DOI] [PubMed] [Google Scholar]
- 18.Li M-B, Tang X-L and Tian S-K, Adv. Synth. Catal, 2011, 353, 1980–1984. [Google Scholar]
- 19.(a) Sweeney JB, Chem. Soc. Rev, 2002, 31, 247–258. [DOI] [PubMed] [Google Scholar]; (b) Bach RD and Dmitrenko O, J. Org. Chem, 2002, 67, 3884–3896. [DOI] [PubMed] [Google Scholar]
- 20.(a) Hu XE, Tetrahedron, 2004, 60, 2701–2743. [Google Scholar]; (b) Lu P, Tetrahedron, 2010, 66, 2549–2560. [Google Scholar]
- 21.Wolfe JP and Ney JE, Org. Lett, 2003, 5, 4607–4610. [DOI] [PubMed] [Google Scholar]
- 22.Lin BL, Clough CR and Hillhouse GL, J. Am. Chem. Soc, 2002, 124, 2890–2891. [DOI] [PubMed] [Google Scholar]
- 23.Ney JE and Wolfe JP, J. Am. Chem. Soc, 2006, 128, 15415–15422. [DOI] [PubMed] [Google Scholar]
- 24.(a) Calet S, Urso F and Alper H, J. Am. Chem. Soc, 1989, 111, 931–934. [Google Scholar]; (b) Roberto D and Alper H, J. Am. Chem. Soc, 1989, 111, 7539–7543. [Google Scholar]; (c) Alper H, Urso F and Smith DJH, J. Am. Chem. Soc, 1983, 105, 6737–6738. [Google Scholar]
- 25.Huang C-Y and Doyle AG, J. Am. Chem. Soc, 2012, 134, 9541–9544. [DOI] [PubMed] [Google Scholar]
- 26.Johnson JB and Rovis T, Angew. Chem., Int. Ed, 2008, 47, 840–871. [DOI] [PubMed] [Google Scholar]
- 27.Huang C-Y and Doyle AG, J. Am. Chem. Soc, 2015, 137, 5638–5641. [DOI] [PubMed] [Google Scholar]
- 28.Woods BP, Orlandi M, Huang C-Y, Sigman MS and Doyle AG, J. Am. Chem. Soc, 2017, 139, 5688–5691. [DOI] [PubMed] [Google Scholar]
- 29.Takeda Y, Ikeda Y, Kuroda A, Tanaka S and Minakata S, J. Am. Chem. Soc, 2014, 136, 8544–8547. [DOI] [PubMed] [Google Scholar]
- 30.Nielsen DK, Huang C-Y and Doyle AG, J. Am. Chem. Soc, 2013, 135, 13605–13609. [DOI] [PubMed] [Google Scholar]
- 31.Jensen KL, Standley EA and Jamison TJ, J. Am. Chem. Soc, 2014, 136, 11145–11152. [DOI] [PubMed] [Google Scholar]
- 32.Duda ML and Michael FE, J. Am. Chem. Soc, 2013, 135, 18347–18349. [DOI] [PubMed] [Google Scholar]
- 33.(a) Wenkert E, Han A-L and Jenny C-J, J. Chem. Soc., Chem. Commun, 1988, 975–976. [Google Scholar]; (b) Reeves JT, Fandrick DR, Tan Z, Song JJ, Lee H, Yee NK and Senanayake CH, Org. Lett, 2010, 12, 4388–4391. [DOI] [PubMed] [Google Scholar]; (c) Guo W-J and Wang Z-X, Tetrahedron, 2013, 69, 9580–9585. [Google Scholar]; (d) Blakey SB and MacMillan DWC, J. Am. Chem. Soc, 2003, 125, 6046–6047. [DOI] [PubMed] [Google Scholar]; (e) Xie L-G and Wang Z-X, Angew. Chem., Int. Ed, 2011, 50, 4901–4904. [DOI] [PubMed] [Google Scholar]; (f) Zhang X-Q and Wang Z-X, J. Org. Chem, 2012, 77, 3658–3663. [DOI] [PubMed] [Google Scholar]; (g) Zhang X-Q and Wang Z-X, Org. Biomol. Chem, 2014, 12, 1448–1453. [DOI] [PubMed] [Google Scholar]
- 34.(a) Langlois Y, Van Bac N and Fall Y, Tetrahedron Lett, 1985, 26, 1009–1012. [Google Scholar]; (b) Dressaire G and Langlois Y, Tetrahedron Lett, 1980, 21, 67–70. [Google Scholar]; (c) Hosomi A, Hoashi K, Tominaga Y, Otaka K and Sakurai H, J. Org. Chem, 1987, 52, 2947–2948. [Google Scholar]; (d) Gupton JT and Layman WJ, J. Org. Chem, 1987, 52, 3683–3686. [Google Scholar]; (e) Hirao T, Yamada N, Ohshiro Y and Agawa T, J. Organomet. Chem, 1982, 236, 409–414. [Google Scholar]; (f) Hosomi A, Hoashi K, Kohra S, Tominaga Y, Otaka K and Sakurai H, J. Chem. Soc., Chem. Commun, 1987, 570–571. [Google Scholar]
- 35.de la Herrán G, Segura A and Csákÿ AG, Org. Lett, 2007, 9, 961–964. [DOI] [PubMed] [Google Scholar]
- 36.Maity P, Shacklady-McAtee DM, Yap GPA, Sirianni ER and Watson MP, J. Am. Chem. Soc, 2013, 135, 280–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Greene MA, Yonova IM, Williams FJ and Jarvo ER, Org. Lett, 2012, 14, 4293–4296. [DOI] [PubMed] [Google Scholar]
- 38.Taylor BLH, Harris MR and Jarvo ER, Angew. Chem., Int. Ed, 2012, 51, 7790–7793. [DOI] [PubMed] [Google Scholar]
- 39.Taylor BLH, Swift EC, Waetzig JD and Jarvo ER, J. Am. Chem. Soc, 2011, 133, 389–391. [DOI] [PubMed] [Google Scholar]
- 40.Wisniewska HM, Swift EC and Jarvo ER, J. Am. Chem. Soc, 2013, 135, 9083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.(a) Arp FO and Fu GC, J. Am. Chem. Soc, 2005, 127, 10482–10483. [DOI] [PubMed] [Google Scholar]; (b) Binder JT, Cordier CJ and Fu GC, J. Am. Chem. Soc, 2012, 134, 17003–17006. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Do H-Q, Chandrashekar ERR and Fu GC, J. Am. Chem. Soc, 2013, 135, 16288–16291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.(a) Mereyala HB and Pola P, Tetrahedron: Asymmetry, 2003, 14, 2683–2685. [Google Scholar]; (b) Savile CK, Janey JM, Mundorff EC, Moore JC, Tam S, Jarvis WR, Colbeck JC, Krebber A, Fleitz FJ, Brands J, Devine PN, Huisman GW and Hughes GJ, Science, 2010, 329, 305–309. [DOI] [PubMed] [Google Scholar]; (c) Vries T, Wynberg H, van Echten E, Koek J, ten Hoeve W, Kellogg RM, Broxterman QB, Minnaard A, Kaptein B, van der Sluis S, Hulshof L and Kooistra J, Angew. Chem., Int. Ed, 1998, 37, 2349–2354. [DOI] [PubMed] [Google Scholar]
- 43.Harris MR, Hanna LE, Greene MA, Moore CE and Jarvo ER, J. Am. Chem. Soc, 2013, 135, 3303–3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.(a) Correa A, León T and Martin R, J. Am. Chem. Soc, 2014, 136, 1062–1069. [DOI] [PubMed] [Google Scholar]; (b) Srogl J, Liu W, Marshall D and Liebeskind LS, J. Am. Chem. Soc, 1999, 121, 9449–9450. [Google Scholar]
- 45.Shacklady-McAtee DM, Roberts KM, Basch CH, Song Y-G and Watson MP, Tetrahedron, 2014, 70, 4257–4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Basch CH, Cobb KM and Watson MP, Org. Lett, 2016, 18, 136–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.(a) Sun C, Potter B and Morken JP, J. Am. Chem. Soc, 2014, 136, 6534–6537. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lee JCH, McDonald R and Hall DG, Nat. Chem, 2011, 3, 894–899. [DOI] [PubMed] [Google Scholar]; (c) Lee JCH and Hall DG, J. Am. Chem. Soc, 2010, 132, 5544–5545. [DOI] [PubMed] [Google Scholar]; (d) Ding J, Lee JCH and Hall DG, Org. Lett, 2012, 14, 4462–4465. [DOI] [PubMed] [Google Scholar]; (e) Morgan JB and Morken JP, J. Am. Chem. Soc, 2004, 126, 15338–15339. [DOI] [PubMed] [Google Scholar]; (f) Feng X, Jeon H and Yun J, Angew. Chem., Int. Ed, 2013, 52, 3989–3992. [DOI] [PubMed] [Google Scholar]; (g) Stymiest JL, Dutheuil G, Mahmood A and Aggarwal VK, Angew. Chem., Int. Ed, 2007, 46, 7491–7494. [DOI] [PubMed] [Google Scholar]; (h) Roesner S, Casatejada JM, Elford TG, Sonawane RP and Aggarwal VK, Org. Lett, 2011, 13, 5740–5743. [DOI] [PubMed] [Google Scholar]; (i) Matteson DS, Sadhu KM, Ray R, Peterson ML, Majumdar D, Hurst GD, Jesthi PK, Tsai DJS and Erdik E, Pure Appl. Chem, 1985, 57, 1741–1748. [Google Scholar]; (j) Beckmann E, Desai V and Hoppe D, Synlett, 2004, 13, 2275–2280. [Google Scholar]; (k) Scott HK and Aggarwal VK, Chem. – Eur. J, 2011, 17, 13124–13132. [DOI] [PubMed] [Google Scholar]; (l) Kim H and Yun J, Adv. Synth. Catal, 2010, 352, 1881–1885. [Google Scholar]; (m) Chea H, Sim H-S and Yun J, Adv. Synth. Catal, 2009, 351, 855–858. [Google Scholar]
- 48.Noh D, Yoon SK, Won J, Lee JY and Yun J, Chem. – Asian J, 2011, 6, 1967–1969. [DOI] [PubMed] [Google Scholar]
- 49.Matsubara R, Gutierrez AC and Jamison TF, J. Am. Chem. Soc, 2011, 133, 19020–19023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.(a) Stille JK, In The Chemistry of the Metal–Carbon Bond, ed. Hartley FR and Patai S, John Wiley & Sons, New York, 1985, vol. 2, pp. 625–787. [Google Scholar]; (b) Netherton MR and Fu GC, Angew. Chem., Int. Ed, 2002, 41, 3910–3912. [DOI] [PubMed] [Google Scholar]
- 51.Moragas T, Gaydou M and Martin R, Angew. Chem., Int. Ed, 2016, 55, 5053–5057. [DOI] [PubMed] [Google Scholar]
- 52.Yi Y-Q, Zhang W-C, Zhai D-D, Zhang X-Y, Li S-Q and Guan B-T, Chem. Commun, 2016, 52, 10894–10897. [DOI] [PubMed] [Google Scholar]
- 53.Gui Y and Tian S-K, Org. Lett, 2017, 19, 1554–1557. [DOI] [PubMed] [Google Scholar]
- 54.(a) Guisán-Ceinos M, Martín-Heras V and Tortosa M, J. Am. Chem. Soc, 2017, 139, 8448–8451. [DOI] [PubMed] [Google Scholar]; (b) For a related enantioselective propargylation, see: Oelke AJ, Sun J and Fu GC, J. Am. Chem. Soc . 2012, 134, 2966–2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guisán-Ceinos M, Martín-Heras V, Soler-Yanes R, Cárdenas DJ and Tortosa M, Chem. Commun, 2018, 54, 83434–8346. [DOI] [PubMed] [Google Scholar]
- 56.(a) Basch CH, Liao J, Xu J, Piane JJ and Watson MP, J. Am. Chem. Soc, 2017, 139, 5313–5316. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Liao J, Guan W, Boscoe BP, Tucker JW, Tomlin JW, Garnsey MR and Watson MP, Org. Lett, 2018, 20, 3030–3033. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Guan W, Liao J and Watson MP, Synthesis, 2018, 50, 3231–3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.(a) Klauck FJR, James MJ and Glorius F, Angew. Chem., Int. Ed, 2017, 56, 12336–12339. [DOI] [PubMed] [Google Scholar]; (b) Ociepa M, Turkowska J and Gryko D, ChemRxiv. Preprint, 2018, Preprint. [Google Scholar]
- 58.(a) Gooßen LJ, Gooßen K and Stanciu C, Angew. Chem., Int. Ed, 2009, 48, 3569–3571. [DOI] [PubMed] [Google Scholar]; (b) Yu D-G, Li B-J and Shi Z-J, Acc. Chem. Res, 2010, 43, 1486–1495. [DOI] [PubMed] [Google Scholar]; (c) Rosen BM, Quasdorf KW, Wilson DA, Zhang N, Resmerita A-M, Garg NK and Percec V, Chem. Rev, 2011, 111, 1346–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Li B-J, Yu D-G, Sun C-L and Shi Z0J, Chem. – Eur. J, 2011, 17, 1728–1759. [DOI] [PubMed] [Google Scholar]; (e) McGlacken GP and Clarke SL, ChemCatChem, 2011, 3, 1260–1261. [Google Scholar]; (f) So CM and Kwong FY, Chem. Soc. Rev, 2011, 40, 4963–4972. [DOI] [PubMed] [Google Scholar]; (g) Sellars JD and Steel PG, Chem. Soc. Rev, 2011, 40, 5170–5180. [DOI] [PubMed] [Google Scholar]; (h) Zeng H, Qiu Z, Domínguez-Huerta A, Hearne Z, Chen Z and Li C-J, ACS Catal, 2017, 7, 510–519. [Google Scholar]
- 59.Handbook of Bond Dissociation Energies in Organic Compounds, ed. Luo Y-R, CRC Press, Boca Raton, 2002. [Google Scholar]
- 60.Guan B-T, Xiang S-K, Wang B-Q, Sun Z-P, Wang Y, Zhao K-Q and Shi Z-J, J. Am. Chem. Soc, 2008, 130, 3268–3269. [DOI] [PubMed] [Google Scholar]
- 61.Yonova IM, Johnson AG, Osborne CA, Moore CE, Morrissette NS and Jarvo ER, Angew. Chem., Int. Ed, 2014, 53, 2422–2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Harris MR, Konev MO and Jarvo ER, J. Am. Chem. Soc, 2014, 136, 7825–7828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.(a) Ni ZJ, Mei NW, Shi X, Tzeng YL, Wang MC and Luh TY, J. Org. Chem, 1991, 56, 4035–4042. [Google Scholar]; (b) Didiuk MT, Morken JP and Hoveyda AH, Tetrahedron, 1996, 54, 1117–1130. [Google Scholar]
- 64.Dawson DD and Jarvo ER, Org. Process Res. Dev, 2015, 19, 1356–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tollefson EJ, Dawson DD, Osborne CA and Jarvo ER, J. Am. Chem. Soc, 2014, 136, 14951–14958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tollefson EJ, Erickson LW and Jarvo ER, J. Am. Chem. Soc, 2015, 137, 9760–9763. [DOI] [PubMed] [Google Scholar]
- 67.Ehle AR, Zhou Q and Watson MP, Org. Lett, 2012, 14, 1202–1205. [DOI] [PubMed] [Google Scholar]
- 68.Zhou Q, Srinivas HD, Dasgupta S and Watson MP, J. Am. Chem. Soc, 2013, 135, 3307–3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.(a) Thomas AA, Zahrt AF, Delaney CP and Denmark SE, J. Am. Chem. Soc, 2018, 140, 4401–4416. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Thomas AA, Wang H, Zahrt AF and Denmark SE, J. Am. Chem. Soc, 2017, 139, 3805–3821. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Thomas AA and Denmark SE, Science, 2016, 352, 329–332. [DOI] [PubMed] [Google Scholar]
- 70.Corey EJ, Bakshi RK, Shibata S, Chen CP and Singh V, J. Am. Chem. Soc, 1987, 109, 7925–7926. [Google Scholar]
- 71.Zhang S-Q, Taylor BLH, Ji C-L, Gao Y, Harris MR, Hanna LE, Jarvo ER, Houk KN and Hong X, J. Am. Chem. Soc, 2017, 139, 12994–13005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Johnson AG, Tranquilli MM, Harris MR and Jarvo ER, Tetrahedron Lett, 2015, 56, 3486–3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen Q, Fan X-H, Zhang L-P and Yang LM, RSC Adv, 2015, 5, 15338–15340. [Google Scholar]
- 74.Konev MO, Hanna LE and Jarvo ER, Angew. Chem., Int. Ed, 2016, 55, 6730–6733. [DOI] [PubMed] [Google Scholar]
- 75.Martin-Montero R, Krolikowski T, Zarate C, Manzano R and Martin R, Synlett, 2017, 28, 2604–2608. [Google Scholar]
- 76.(a) Tobisu M, Zhao J, Kinuta H, Furukawa T, Igarashi T and Chatani N, Adv. Synth. Catal, 2016, 358, 2417–2421. [Google Scholar]; (b) Li J and Wang Z-X, Chem. Commun, 2018, 54, 2138–2141. [DOI] [PubMed] [Google Scholar]
- 77.Guo Y-A, Liang T, Kim SW, Xiao H and Krische MJ, J. Am. Chem. Soc, 2017, 139, 6847–6850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mendis SN and Tunge JA, Org. Lett, 2015, 17, 5164–5167. [DOI] [PubMed] [Google Scholar]
- 79.Li C, Zhang Y, Sun Q, Gu T, Peng H and Tang W, J. Am. Chem. Soc, 2016, 138, 10774–10777. [DOI] [PubMed] [Google Scholar]
- 80.(a) Matsude A, Hirano K and Miura M, Org. Lett, 2018, 20, 3553–3556. [DOI] [PubMed] [Google Scholar]; (b) Najib A, Hirano K and Miura M, Chem. – Eur. J, 2018, 24, 6525–6529. [DOI] [PubMed] [Google Scholar]; (c) Najib A, Hirano K and Miura M, Org. Lett, 2017, 19, 2438–2441. [DOI] [PubMed] [Google Scholar]; (d) Tabuchi S, Hirano K and Miura M, Angew. Chem., Int. Ed, 2016, 55, 6973–6977. [DOI] [PubMed] [Google Scholar]
- 81.(a) Falciola CA and Alexakis A, Eur. J. Org. Chem, 2008, 2008, 3765–. [Google Scholar]; (b) Zhang P, Le H, Kyne RE and Morken JP, J. Am. Chem. Soc, 2011, 133, 9716–. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Jung B and Hoveyda AH, J. Am. Chem. Soc, 2012, 134, 1490–. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Nagao K, Yokobori U, Makida Y, Ohmiya H and Sawamura M, J. Am. Chem. Soc, 2012, 134, 8982–8987. [DOI] [PubMed] [Google Scholar]; (e) Feng C and Kobayashi Y, J. Org. Chem, 2013, 78, 3755–3766. [DOI] [PubMed] [Google Scholar]; (f) Hojoh K, Shido O, Ohmiya H and Sawamura M, Angew. Chem., Int. Ed, 2014, 53, 4954–4958. [DOI] [PubMed] [Google Scholar]
- 82.Zhou Q, Cobb KM, Tan T and Watson MP, J. Am. Chem. Soc, 2016, 138, 12057–12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.(a) Jeon S-J, Li H and Walsh PJ, J. Am. Chem. Soc, 2005, 127, 16416–16425. [DOI] [PubMed] [Google Scholar]; (b) García C, LaRochelle LK and Walsh PJ, J. Am. Chem. Soc, 2002, 124, 10970–10971. [DOI] [PubMed] [Google Scholar]; (c) Li H and Walsh PJ, J. Am. Chem. Soc, 2004, 126, 6538–6539. [DOI] [PubMed] [Google Scholar]; (d) García C and Walsh PJ, Org. Lett, 2003, 5, 3641–3633. [DOI] [PubMed] [Google Scholar]; (e) Waltz KM, Gavenonis J and Walsh PJ, Angew. Chem., Int. Ed, 2002, 41, 3697–3699. [DOI] [PubMed] [Google Scholar]
- 84.(a) Kobayashi Y, Tokoro Y and Watatani K, Tetrahedron Lett, 1998, 39, 7537–7540. [Google Scholar]; (b) Kobayashi Y, Mizojiri R and Ikeda E, J. Org. Chem, 1996, 61, 5391–5399. [Google Scholar]; (c) Kobayashi Y, Watatani K, Kikori Y and Mizojiri R, Tetrahedron Lett, 1996, 37, 6125–6128. [Google Scholar]; (d) Kobayashi Y, Takahisa E and Usmani SB, Tetrahedron Lett, 1998, 39, 597–600. [Google Scholar]; (e) Usmani SB, Takahisa E and Kobayashi Y, Tetrahedron Lett, 1998, 39, 601–604. [Google Scholar]; (f) Kobayashi Y, Tokoro Y and Watatani K, Eur. J. Org. Chem, 2000, 2000, 3825–3834. [Google Scholar]; (g) For a related asymmetric catalytic example, see: Nomura N and RajanBabu TV, Tetrahedron Lett . 1997, 38, 1713–1716. [Google Scholar]
- 85.(a) Ohmiya H, Makida Y, Tanaka T and Sawamura M, J. Am. Chem. Soc, 2008, 130, 17276–17277. [DOI] [PubMed] [Google Scholar]; (b) Ohmiya H, Makida Y, Li D, Tanabe M and Sawamura M, J. Am. Chem. Soc, 2010, 132, 879–889. [DOI] [PubMed] [Google Scholar]; (c) Li D, Tanaka T, Ohmiya H and Sawamura M, Org. Lett, 2010, 12, 3344–3347. [DOI] [PubMed] [Google Scholar]; (d) Makida Y, Ohmiya H and Sawamura M, Chem. – Asian J, 2011, 6, 410–414. [DOI] [PubMed] [Google Scholar]; (e) Ohmiya H, Tokokawa N and Sawamura M, Org. Lett, 2010, 12, 2438–2440. [DOI] [PubMed] [Google Scholar]
- 86.(a) Li C, Xing J, Zhao J, Huynh P, Zhang W, Jiang P and Zhang YJ, Org. Lett, 2012, 14, 390–393. [DOI] [PubMed] [Google Scholar]; (b) Zhao J, Ye J and Zhang YJ, Adv. Synth. Catal, 2013, 355, 491–498. [Google Scholar]
- 87.Wu H-B, Ma X-T and Tian S-K, Chem. Commun, 2014, 50, 219–221. [DOI] [PubMed] [Google Scholar]
- 88.(a) Norinder J and Bäckvall JE, Chem. – Eur. J, 2007, 13, 4094–4102. [DOI] [PubMed] [Google Scholar]; (b) Thalén LK, Sumic A, Bogár K, Norinder J, Persson AKÅ and Bäckvall J-E, J. Org. Chem, 2010, 75, 6842–6847. [DOI] [PubMed] [Google Scholar]
- 89.Srinivas HD, Zhou Q and Watson MP, Org. Lett, 2014, 16, 3596–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lachance H and Hall DG, in Organic Reactions, John Wiley & Sons, Inc., 2009, pp. 1–574. [Google Scholar]
- 91.Ito H, Kawakami C and Sawamura M, J. Am. Chem. Soc, 2005, 127, 16034–16035. [DOI] [PubMed] [Google Scholar]
- 92.Zhou Q, Srinivas HD, Zhang S and Watson MP, J. Am. Chem. Soc, 2016, 138, 11989–11995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.(a) For a ligand-controlled stereochemical switch, see ref. 41.; (b) For a solvent- and base-dependent stereochemical switch, see: ref. 80d; (c) For a solvent-controlled stereochemical switch, see:; Kurosawa H, Ogoshi S, Kawasaki Y, Murai S, Miyoshi M and Ikeda I, J. Am. Chem. Soc, 1990, 112, 2813–2814. [Google Scholar]; (d) For an acidic additive-controlled stereochemical switch, see: Awano T, Ohmura T and Suginome M, J. Am. Chem. Soc, 2011, 133, 20738–20741. [DOI] [PubMed] [Google Scholar]; (e) See also: ref. 66.
- 94.(a) Jackowski O and Alexakis A, Angew. Chem., Int. Ed, 2010, 49, 3346–3350. [DOI] [PubMed] [Google Scholar]; (b) Fañanás-Mastral M, Pérez M, Bos PH, Rudolph A, Harutyunyan SR and Feringa BL, Angew. Chem., Int. Ed, 2012, 51, 1922–1925. [DOI] [PubMed] [Google Scholar]; (c) Kacprzynski MA and Hoveyda AH, J. Am. Chem. Soc, 2004, 126, 10676–10681. [DOI] [PubMed] [Google Scholar]; (d) Larsen AO, Leu W, Oberhuber CN, Campbell JE and Hoveyda AH, J. Am. Chem. Soc, 2004, 126, 11130–11131. [DOI] [PubMed] [Google Scholar]; (e) Van Veldhuizen JJ, Campbell JE, Giudici RE and Hoveyda AH, J. Am. Chem. Soc, 2005, 127, 6877–6882. [DOI] [PubMed] [Google Scholar]; (f) Lee Y, Li B and Hoveyda AH, J. Am. Chem. Soc, 2009, 131, 11625–11633. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Gao F, McGrath KP, Lee Y and Hoveyda AH, J. Am. Chem. Soc, 2010, 132, 14315–14320. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Gao F, Lee Y, Mandai K and Hoveyda AH, Angew. Chem., Int. Ed, 2010, 49, 8370–8374. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Dabrowski JA, Gao F and Hoveyda AH, J. Am. Chem. Soc, 2011, 133, 4778–4781. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Magrez M, le Guen Y, Baslé O, Crévisy C and Mauduit M, Chem. – Eur. J, 2013, 19, 1199–1203. [DOI] [PubMed] [Google Scholar]; (k) Takeda M, Takatsu K, Shintani R and Hayashi T, J. Org. Chem, 2014, 79, 2354–2367. [DOI] [PubMed] [Google Scholar]
- 95.(a) Ibuka T, Tanaka M, Nishii S and Yamamoto Y, J. Chem. Soc., Chem. Commun, 1987, 1596–1598. [Google Scholar]; (b) Ibuka T, Akimoto N, Tanaka M, Nishii S and Yamamoto Y, J. Org. Chem, 1989, 54, 4055–4061. [Google Scholar]; (c) Harrington-Frost N, Leuser H, Calaza MI, Kneisel FF and Knochel P, Org. Lett, 2003, 5, 2111–2114. [DOI] [PubMed] [Google Scholar]; (d) Breit B, Demel P and Studte C, Angew. Chem., Int. Ed, 2004, 43, 3786–3789. [DOI] [PubMed] [Google Scholar]; (e) Feng C, Kaneko Y and Kobayashi Y, Tetrahedron Lett, 2013, 54, 4629–4632. [Google Scholar]
- 96.(a) Potter B, Edelstein EK and Morken JP, Org. Lett, 2016, 18, 3286–3289. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Potter B, Szymaniak AA, Edelstein EK and Morken JP, J. Am. Chem. Soc, 2014, 136, 17918–17921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cobb KM, Rabb-Lynch JM, Hoerrner ME, Manders A, Zhou Q and Watson MP, Org. Lett, 2017, 19, 4355–4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.(a) Morrill C, Beutner GL and Grubbs RH, J. Org. Chem, 2006, 71, 7813–7825. [DOI] [PubMed] [Google Scholar]; (b) Corey EJ and Bakshi RK, Tetrahedron Lett, 1990, 31, 611–615. [Google Scholar]; (c) Corey EJ and Helal CJ, Tetrahedron Lett, 1995, 36, 9153–9156. [Google Scholar]
- 99.Casey CP, Whiteker GT, Melville MG, Petrovich LM, Gavney JA and Powell DR, J. Am. Chem. Soc, 1992, 114, 5535–5543. [Google Scholar]