

Abstract
The σ1 receptor is a poorly understood membrane protein expressed throughout the human body. Ligands targeting the σ1 receptor are in clinical trials for treatment of Alzheimer’s disease, ischemic stroke, and neuropathic pain. Despite this, relatively little is known regarding the σ1 receptor’s molecular function. Here, we present crystal structures of human σ1 receptor bound to the antagonists haloperidol and NE-100, and the agonist (+)-pentazocine, at crystallographic resolutions of 3.1 Å, 2.9 Å, and 3.1 Å respectively. These structures reveal a unique binding pose for the agonist. The structures and accompanying molecular dynamics (MD) simulations identify agonist-induced structural rearrangements in the receptor. Additionally, we show that ligand binding to σ1 is a multistep process, rate-limited by receptor conformational change. We use MD simulations to reconstruct a ligand binding pathway involving two major conformational changes. These data provide a framework for understanding the molecular basis for σ1 agonism.
Pharmacological studies of opioid compounds and their chemical analogs in the 1970s led to the identification of several opioid receptor “subtypes”, one of which was termed the σ receptor1. Subsequent pharmacological characterization showed that the σ receptor is distinguished from the true opioid receptors by a divergent ligand binding profile2,3. Later pharmacological studies further divided the receptor into σ1 and σ2 subtypes4. Molecular cloning of the σ1 receptor5 and later the σ2 receptor6 showed that these proteins are genetically unrelated to each other and have no similarity to the true opioid receptors. In fact, the σ1 receptor subtype shows no sequence similarity to any other human protein5.
The σ1 receptor continues to be of pharmacological interest because it binds a host of structurally dissimilar pharmacologically active compounds with high affinity (Fig. 1a). These include benzomorphans, antipsychotics, psychosis-inducing drugs, the antifungal agent fenpropimorph, sterols such as progesterone, and numerous other compounds7. These molecules contain few shared features, although most include a basic nitrogen atom flanked on two sides by longer hydrophobic moieties (typically phenyl rings), representing a minimal σ1-binding pharmacophore (Fig. 1a)8. The σ1 receptor’s nearest homolog is the yeast Δ8-Δ7 sterol isomerase, ERG2p, although the σ1 receptor itself has no detectable isomerase activity5. Human genetic data have linked point mutants in σ1 receptor to inherited motor neuron diseases9–11, and animal models implicate the receptor in Parkinson’s disease12, addiction13, and pain14. A σ1 receptor antagonist is currently in clinical trials for the treatment of neuropathic pain15, and agonists are in clinical trials for Alzheimer’s disease16 and ischemic stroke17.
Fig. 1. Crystal structures of human σ1 receptor bound to the classical antagonists haloperidol and NE-100.
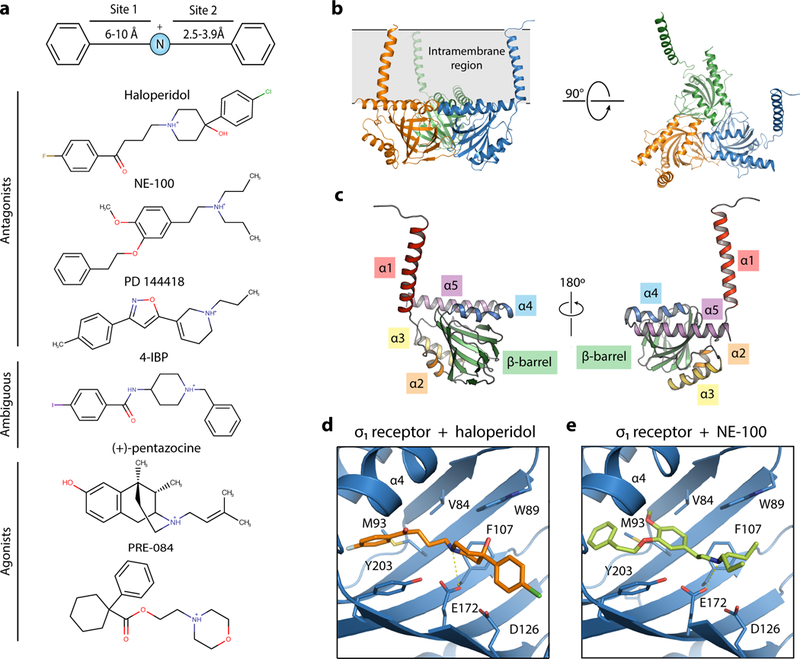
| a, σ1 ligand pharmacophore, based on the work of Glennon et al.8 Representative σ1 ligands are shown below. b, The overall structure of the human σ1 receptor (PDB 5HK1). c, The structure of a single σ1 monomer, with the secondary structural elements labeled. d and e, The binding pocket of the human σ1 receptor (blue) binding in complex with haloperidol (orange, PDB ID: 6DJZ) (d) and NE-100 (light green, PDB ID: 6DK0) (e).
Despite its potential therapeutic relevance and a wealth of high-affinity ligands, surprisingly little is known about the molecular underpinnings of σ1 receptor function. There is substantial evidence to suggest that the σ1 receptor serves as a modulator for other signaling pathway effectors18–20. Specifically, knockdown or antagonism of σ1 receptor can potentiate G-protein coupled receptor (GPCR) signaling18,19, while agonists of the σ1 receptor result in enhanced IP3 receptor-dependent intracellular calcium flux21 and inhibition of sodium20 and potassium22,23 channel currents. The σ1 receptor exists in multiple oligomeric states, and reports suggest agonists causes a shift to monomeric or low molecular weight species, while antagonists bias the receptor towards high molecular weight species24–27. However, the dominant physiologically relevant oligomeric forms and the precise way in which oligomerization is tied to agonist binding are unknown.
We recently reported the first structures of the human σ1 receptor bound to two different ligands, PD 144418, an antagonist, and 4-IBP, a poorly characterized ligand of ambiguous efficacy class28. The receptor crystallized as a trimer, with each protomer showing a fold including a single transmembrane domain and a β-barrel flanked by α helices28 (Fig. 1b, Fig. 1c, and Supplementary Fig. 1a) While these initial results provided the first structural information on σ1 receptor, neither ligand is commonly used to study σ1 receptor function, and little functional data are available for either.
In order to understand the molecular basis for agonist activity at σ1, we pursued structural studies of three well-characterized classical ligands of the receptor: the antagonists haloperidol and NE-100, and the agonist (+)-pentazocine. Using the lipidic cubic phase method we determined X-ray crystal structures of the receptor in complex with these three compounds at resolutions of 3.1 Å, 2.9 Å, and 3.1 Å, respectively (Table 1, Supplementary Fig. 2).
Table 1.
| Data collection and refinement statistics.
| σ1 bound to haloperidol (PDB 6DJZ)a |
σ1 bound to NE-100 (PDB 6DK0)a |
σ1 bound to (+)-pentazocine (PDB 6DK1)a |
|
|---|---|---|---|
| Data collection | |||
| Space group | P 21 21 2 | P 21 21 2 | P 21 21 2 |
| Cell dimensions | |||
| a, b, c (Å) | 85.1, 126.1, 110.6 | 85.0, 127.0, 110.0 | 85.8, 128.6, 109.2 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 50 – 3.1 (3.30 – 3.10)b | 50 – 2.9 (3.00 – 2.90)b | 46 – 3.1 (3.33 – 3.12)b |
| Rmeas | 28.1 (291.6) | 33.6 (387.7) | 27.4 (208.9) |
| I/σ(I) | 5.9 (0.4) | 5.6 (0.5) | 3.5 (0.6) |
| CC1/2 | 99.4 (16.4) | 98.9 (24.0) | 99.3 (33.4) |
| Completeness (%) | 99.8 (99.6) | 95.3 (96.8) | 93.3 (82.9) |
| Redundancy | 5.7 (5.0) | 7.2 (7.2) | 2.5 (2.2) |
| Refinement | |||
| Resolution (Å) | 40.5 – 3.1 | 46.2 – 2.9 | 45.9 – 3.1 |
| No. reflections | 21454 (1901 in test set) | 25243 (2164 in test set) | 20178 (1673 in test set) |
| Rwork / Rfree | 23.9/27.6 | 25.1/27.3 | 24.7/27.8 |
| No. atoms | |||
| Protein | 5021 | 5052 | 5054 |
| Ligand | 78 | 78 | 63 |
| Solvent ions/lipid | 164 | 155 | 204 |
| Water | 30 | 96 | 86 |
| B factors | |||
| Protein | 102.4 | 83.0 | 78.5 |
| Ligand | 112.4 | 98.0 | 82.0 |
| Solvent ions/lipid | 127.8 | 105.3 | 102.2 |
| Water | 82.9 | 69.3 | 62.6 |
| R.m.s. deviations | |||
| Bond lengths (Å) | 0.003 | 0.002 | 0.002 |
| Bond angles (°) | 0.489 | 0.463 | 0.471 |
the haloperidol and (+)-pentazocine-bound structures were solved from single-crystal datasets, while the NE-100 structure was solved using a merged dataset from seven crystals.
Values in parentheses are for highest-resolution shell.
Results
Structure of human σ1 receptor bound to antagonists
The structures of the σ1 receptor bound to the classical antagonists haloperidol and NE-100 are highly similar to each other and to our previously reported structures of σ1 bound to PD 144418 and 4-IBP28 (Supplementary Figures 1b-1e). Both haloperidol and NE-100 include a shared simple pharmacophore (Fig. 1a), and both adopt similar conformations in the ligand binding site (Fig. 1d and 1e). In each case, the ligand’s positively charged nitrogen forms an electrostatic interaction with E172, and the rest of the molecule adopts a linear pose that fits within the space not occluded by the many bulky hydrophobic residues that line the interior of the σ1 binding pocket (Fig. 1d and 1e). In general, the longer of the two hydrophobic regions occupies the region of the β-barrel that is proximal to the membrane, near the space between helices α4 and α5 (Fig. 1d, 1e, and Supplementary Figure 1b-e). In contrast, the shorter hydrophobic region occupies space near the bottom of the β-barrel, near D126 (Fig. 1d, 1e, and Supplementary Figure 1b-e).
Structure of the human σ1 receptor bound to an agonist
The structures above reveal the overall pose of ligands in the antagonist-bound σ1 receptor, confirming a highly conserved binding mode and receptor conformation even for chemically diverse antagonists. Next, we investigated the structure of the receptor bound to (+)-pentazocine at 3.1 Å resolution (Fig. 2, Table 1, Supplementary Figures 2d and 2e). In general, the agonist-bound receptor crystallized similarly to antagonist-bound σ1, and the overall conformation of the receptor did not change significantly (Fig. 2a). The exception is a movement of helix α4, which shifts roughly 1.8 Å away from helix α5 in the (+)-pentazocine bound structure relative to the PD 144418-bound structure (Fig. 2b). This movement appears to be a consequence of the pose adopted by (+)-pentazocine, which occupies a different portion of the receptor binding pocket than the other ligands examined thus far (Fig. 2b and 2c). This difference in helix α4 position is also consistently observed in MD simulations (Fig. 2d). In simulations of unliganded σ1, the helix adopts a similar position as when an antagonist is bound (Fig. 2d), also suggesting that the agonist is responsible for the conformational change. (+)-pentazocine engages in an electrostatic interaction with E172, and site 2 is positioned similarly to those of the antagonists, but its nonlinear shape forces site 1 to occupy space closer to helix α4 and further from α5 relative to the antagonists. In order to prevent a steric clash between the aromatic ring of (+)-pentazocine’s benzomorphan group and residue A185 in helix α4 (Fig. 2c), helix α4 must shift towards the membrane and away from the ligand (Supplementary Fig. 3a). This movement creates a slightly larger gap between helices α4 and α5 in the (+)-pentazocine-bound structure relative to the antagonist-bound structures. As a result, the distance between helix α4 and the adjacent protomer shrinks, and in the interface between chains A and C, an electrostatic bond between Q194 in chain C and the backbone of the α4 helix in chain A is broken (Supplementary Fig. 3b). In the two best-resolved protomers of the (+)-pentazocine-bound structure, chains A and C, two water molecules occupy the space normally occupied by a portion of the antagonist.
Fig. 2. | Crystal structure of the human σ1 receptor bound to the classical agonist (+)-pentazocine.
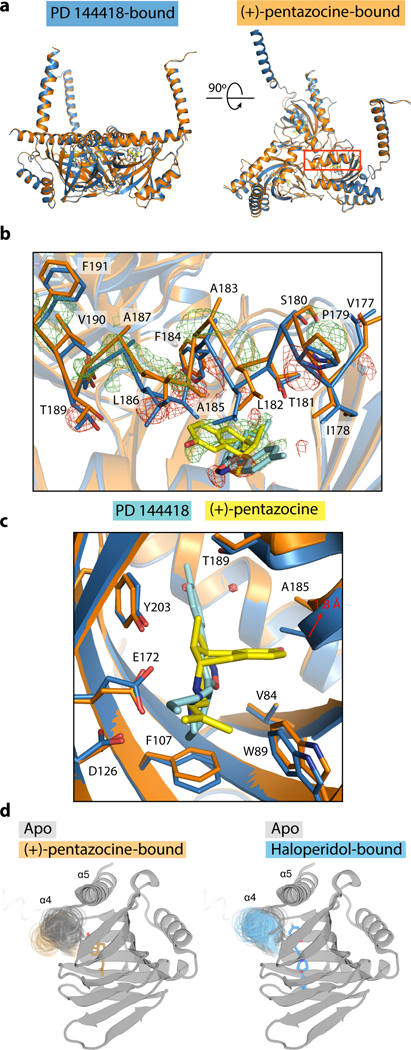
In all panels, the structure of human σ1 receptor bound to (+)-pentazocine (PDB ID: 6DK1) is shown in orange, and the structure of the human σ1 receptor bound to PD 144418 (PDB ID: 5HK1) is show in blue. The ligands (+)-pentazocine and PD 144418 are shown in yellow and cyan, respectively. Wire mesh represents Fo-Fo density, where green mesh corresponds to regions where there is more electron density in the (+)-pentazocine-bound structure relative to the PD 144418-bound structure, and red mesh is the opposite. a, An alignment of the overall structures of σ1 receptor bound to PD 144418 and (+)-pentazocine, with helix α4 highlighted by a red box. b, A close up of helix α4 alignment that was highlighted in a, shown in a stick representation with Fo-Fo density contoured at 2.5σ. c, An alignment of the (+)-pentazocine and PD 144418-bound structures in the binding pocket. d. Helix α4 position in simulation for unliganded (grey), (+)-pentazocine-bound (orange), and haloperidol-bound (blue) conditions. Multiple simulation frames comprising approximately 1 μs of simulation per condition are shown.
A defining feature of σ1 receptor pharmacology is high selectivity for (+)-benzomorphans over their (−) enantiomers. It is clear from this structure that (−)-pentazocine would be unable to bind the σ1 receptor in the same pose as (+)-pentazocine, as the benzomorphan ring would clash with both Y103 and the receptor backbone (Supplementary Fig. 3c). Therefore, (−)-pentazocine must adopt an alternative, presumably higher energy conformation to bind the σ1 receptor.
Though (+)-pentazocine and other (+) benzomorphans are classical σ1 agonists, other agonists are structurally unrelated. To see if other agonists may also put steric pressure on the α4 helix, we used the Schrödinger Glide package to dock the commonly used agonist PRE-084 into the (+)-pentazocine-bound σ1 receptor structure (Supplementary Fig. 3d). In its top-ranked pose, PRE-084 adopted a conformation similar to that seen in molecular modeling simulations performed by Yano et al.27, in which one of the carbon rings comes into close contact with the α4 helix. This pose would be sterically prohibited by the α4 helix in the antagonist-bound structure. Therefore, it is possible that this conformational change may be broadly important for agonist efficacy among σ1 receptor ligands.
Kinetic analysis of σ1 receptor ligand association and dissociation
As noted above, the ligand binding site of the σ1 receptor is sterically occluded, and so the receptor must undergo a conformational change to allow ligand entry and egress. Previous work has shown that (+)-pentazocine associates with the receptor slowly29,30, and rate constants have been determined for association and dissociation at a single concentration in guinea pig membranes30. In order to gain a better understanding of how ligands associate and dissociate with the σ1 receptor, we undertook an analysis of ligand binding kinetics using multiple concentrations of [3H](+)-pentazocine and membranes prepared from Sf9 cells expressing human σ1 receptor.
We began by measuring off rate at 37 °C and found it to follow a slow exponential decay with a half-life of over 200 minutes (Supplementary Fig. 4a). To obtain detailed association kinetics, we turned to a scintillation proximity assay (SPA), in which purified FLAG-tagged receptor was bound to YSi SPA beads coated with protein A and M1 αFLAG antibody. In this format, a single reaction can be monitored continuously at room temperature for an extended period (Supplementary Fig. 4b). The measured Kd in SPA experiments (Table 2) was indistinguishable from that measured in membrane binding experiments, suggesting that the receptor-ligand interaction is similar in both lipid membranes and in detergent (Supplementary Fig. 4c). Additionally, the values for these kinetic constants determined by SPA for [3H](+)-pentazocine association to and dissociation from human σ1 receptor were highly similar to the kobs (0.021 min−1) and koff (1.46 × 10−3 min−1) determined by Bowen and colleagues in guinea pig membranes30. Surprisingly, our experiments showed that the association of [3H](+)-pentazocine to the σ1 receptor was not monophasic, but could be well modeled by a two-step association model, in which a zero-order reaction, represented by the rate constant kslow, is followed by a concentration-dependent association step, modelled by the rate constant kfast (Fig. 3a, 3b, and 3c, Supplementary Fig. 4d). We also measured ligand dissociation in SPA format. In contrast to the association reaction, the dissociation data fit well to a simple monophasic dissociation curve (Fig. 3d and Supplementary Fig. 4e).
Table 2.
| Kinetic constants for σ1 receptor ligand binding. All data were obtained by scintillation proximity assay (SPA) at 23 °C. Errors represent standard deviation.
| Constant | [3H](+)-pentazocine | [3H]haloperidol |
|---|---|---|
| Kd (nM): | 22.5 ± 10.8 | N.D. |
| kslow (min−1)*: | (3.2 ± 0.6) × 10−3 | (4.4 ± 0.2) × 10−3 |
| kfast (min−1) † | ||
| at 1 nM: | (1.6 ± 0.6) × 10−2 | (2.8 ± 1.6) × 10−2 |
| 3 nM: | (2.3 ± 0.3) × 10−2 | (3.3 ± 0.2) × 10−2 |
| 10 nM: | (2.6 ± 0.4) × 10−2 | (4.3 ± 0.8) × 10−2 |
| 30 nM: | (3.0 ± 0.5) × 10−2 | N.D. |
| 100 nM: | (3.4 ± 0.4) × 10−2 | N.D. |
| 300 nM: | (3.4 ± 0.4) × 10−2 | N.D. |
| koff (min−1) ‡ | ||
| at 1 nM: | (9.4 ± 6.1) × 10−4 | (4.2 ± 1.6) × 10−4 |
| 3 nM: | (10.0 ± 7.0) × 10−4 | (5.1 ± 0.4) × 10−4 |
| 10 nM: | (8.0 ± 5.8) × 10−4 | (4.9 ± 0.6) × 10−4 |
| 30 nM: | (6.7 ± 4.8) × 10−4 | N.D. |
| 100 nM: | (5.1 ± 3.6) × 10−4 | N.D. |
| 300 nM: | (4.3 ± 2.9) × 10−4 | N.D. |
kslow is the observed rate constant for the slow step for ligand association to the σ1 receptor in the biphasic fit.
kfast is the observed rate constant for the fast step in ligand association to the σ1 receptor in the biphasic fit.
koff is the observed off rate for a given concentration in the SPA measurements, based on a monophasic fit.
Fig. 3. | kinetic analysis of ligand binding to the σ1 receptor.
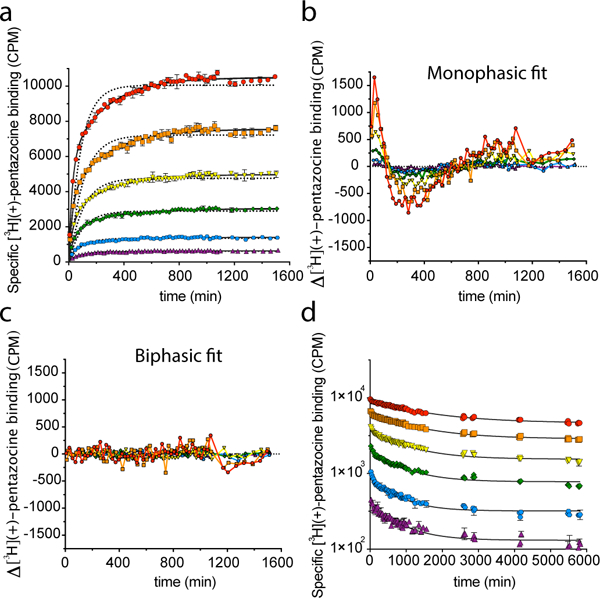
a, Association of [3H](+)-pentazocine with the σ1 receptor measured in SPA format at 23 °C. The six ligand concentrations assayed are 300 nM (red), 100 nM (orange), 30 nM (yellow), 10 nM (green), 3 nM (blue), and 1 nM (violet). The best-fit monophasic curve for each concentration is shown as a dotted black line, and the best-fit biphasic curve for each concentration is shown as solid black lines. Error bars represent SEM. Data shown are representative from three independent experiments performed in duplicate. b and c, Residual plots for the monophasic (b) and biphasic (c) curves and the individual data points shown in a. Colors differentiate concentration of [3H](+)-pentazocine used, and are the same as in a. d, Dissociation of [3H](+)-pentazocine from the σ1 receptor in SPA format at 23 °C. Colors represent initial [3H](+)-pentazocine concentrations as denoted in a. Solid black lines represent the best-fit monophasic exponential decay curve. Error bars represent SEM. Data shown are representative of two independent experiments performed in duplicate.
Interestingly, both the apparent kfast and the koff parameters for [3H](+)-pentazocine dissociation and association varied nonlinearly with concentration, indicative of cooperativity in ligand binding (Supplementary Fig. 4f and 4g, Table 2). Additionally, the dissociation curves were clearly nonlinear when plotted in a semi-log format, another indication of cooperative binding (Fig. 3d). However, the Hill coefficient for ligand binding in equilibrium experiments is indistinguishable from 1 (Supplementary Fig. 4h). Therefore, the binding of (+)-pentazocine to one σ1 monomer alters the rate of ligand binding to the next monomer, but must equally affect both on and off rates. Additionally, though the association curve for each individual concentration could be fit to a two-step exponential function, a simple two-state model is insufficient to account for the global data. This suggests that though there are at least two steps to ligand association with the σ1 receptor, there are probably additional steps or conformational states that are not accounted for with a simple two-step fit.
Since the rate-limiting step for [3H](+)-pentazocine association was not dependent on ligand concentration, we suspected that this step represented a conformational change from a ligand-inaccessible to a ligand-accessible state. To test this hypothesis, we repeated the experiment with [3H]haloperidol. The association of [3H]haloperidol to the σ1 receptor was also poorly modeled by a one-step reaction, but fit well to a two-step model (Supplementary Fig. 5a and 5b). Additionally, the rate of the slow step was very similar for both [3H]haloperidol and [3H](+)-pentazocine (Table 2). While a t-test indicated that the kslow parameter for each ligand was statistically different (p = 0.025), the 95% confidence intervals overlap and the difference in the rate constants is relatively small. Given how similar the two kslow estimates are, and the fact that oligomerization may cause cooperative effects which could influence the rate of conformational changes in unliganded protomers, we believe these data are consistent with the conclusion that the slow in ligand association step represents a conformational change intrinsic to the receptor that is mostly ligand-independent. As seen with [3H](+)-pentazocine, dissociation of [3H]haloperidol from σ1 receptor was slow and could be modeled by a monophasic exponential decay function (Supplementary Fig. 5c and 5d).
Ligand binding pathway via molecular dynamics simulation
To better characterize the pathway of ligand binding and dissociation, we performed MD simulations of σ1 with the goal of characterizing possible conformational rearrangements that could expose the binding pocket. To reduce the computational complexity of the system, we simulated the σ1 monomer and used accelerated molecular dynamics, which applies a boost to dihedral energy minima in order to speed up observation of conformational changes31.
The σ1 monomer from the crystal structure was inserted into a hydrated lipid bilayer, with (+)-pentazocine removed from the binding pocket and placed in the water. Using multiple rounds of simulation totaling over 110 μs, we were able to assemble a three-step binding pathway, with (+)-pentazocine reaching a bound state with an RMSD < 3 Å to the crystallographic pose (Fig. 4a).
Fig. 4. | Molecular dynamics simulation reveals a putative binding pathway for (+)-pentazocine.
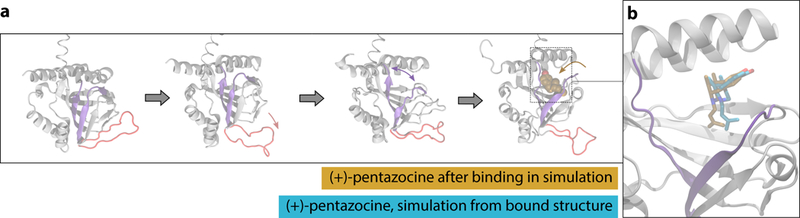
a. Binding pathway of (+)-pentazocine, with the “lid” region shown in red and the beta strands that separate shown in purple. From left to right: simulation begins with an unliganded receptor, where the “lid” region then opens. Next, the interior of the receptor opens, and the ligand enters and binds through this opening. b. A simulation frame after the ligand has entered the binding pocket (tan), compared to a frame initiated from the crystal structure with the ligand bound (blue). Protein backbone is shown for the binding simulation in grey. Helix α4 is located at the top of the rendering.
This binding pathway requires two major conformational rearrangements in order for the pocket to become accessible to the ligand. First, the “lid” of the receptor opens, breaking backbone hydrogen bonds between Trp136 and Ala161. Next, the beta-barrel structure in the interior of the receptor separates, breaking backbone hydrogen bonds between Glu123 and Arg175 and exposing the binding pocket. The ligand enters through this opening near the membrane, and assumes a near-crystallographic pose as the protein closes around it (Fig. 4b).
Each of these rare conformational changes or binding events was observed multiple times in simulation. Interestingly, the beta-barrel separation that exposes the binding site was only observed in simulations where the receptor “lid” had already opened, suggesting that two sequential conformational changes may be necessary before the ligand can bind. The lid opening may be a prerequisite for further conformational change as it may perturb the internal hydrogen-bond network of σ1 so that larger rearrangements may occur. These conformational changes occur spontaneously, and also spontaneously revert in some simulations, suggesting that the σ1 receptor exists in a conformational ensemble of open, ligand-accessible states and closed ones resembling the crystallographic pose.
Discussion
Antagonism or genetic ablation of σ1 receptor has analgesic effects in whole animals and humans14,15,32, and potentiates GPCR signaling in cells18,19. In contrast, σ1 receptor agonists are usually defined by their ability to oppose the effects of antagonists, and have been associated with cytoprotective effects33–35. Currently, the biochemical basis for agonism or antagonism at the σ1 receptor is largely unknown, which complicates the unambiguous assignment of efficacy class for σ1 ligands. The most well-documented biochemical difference between the two ligand classes is that antagonists increase the receptor’s oligomeric state, while agonists decrease the oligomeric state24–27. The structural data we present here show that these ligands occupy a different region of the binding pocket (Fig. 2c and 2d). Antagonists adopt a more linear pose, with the primary hydrophobic region of the molecule pointing towards the space between helices α4 and α5, while (+)-pentazocine’s primary hydrophobic site points towards helix α4 (figures 2c and 2d). Presumably, structurally similar agonists like (+)-SKF-10,047 adopt a similar pose, accounting for their shared biological activities. Indeed, computational docking (Supplemtary Fig. 3d and 3e) and modeling27 suggests that other agonists such as PRE-084 may also preferentially occupy space near helix α4 relative to antagonists. .
As a result of the steric constraints of agonists, most of helix α4 is forced to shift 1.1–1.8 Å away from helix α5 to accommodate the ligand. In our structure, this shift in α4 does not disrupt the oligomerization interface between individual protomers. However, if α4 were to move to a greater degree, it could disrupt the oligomerization interface. This is consistent with prior data, which suggest that σ1 receptor agonists bias the receptor towards lower molecular weight states, while antagonists bias it towards higher molecular weight states24–27. Additionally, molecular modeling by Yano et al. predicted that (+)-pentazocine and other multimer-impeding ligands would occupy this space differentially from haloperidol and other multimer-promoting ligands27, which is consistent with our structural results. Importantly, crystallographic studies by necessity favor conformationally stable, low-energy states, and so the structures shown here may not represent a fully activated state of the receptor. Indeed, studies of G protein-coupled receptors bound to agonists often show inactive-state structures in the absence of G proteins or antibody fragment stabilizers36.
We have also shown by kinetic analysis that (+)-pentazocine associates with the σ1 receptor in at least two steps, with MD simulation suggesting a three-step process requiring two substantial conformational changes to the receptor. Ligand association and dissociation at the σ1 receptor is very slow, and the rate-limiting step is independent of ligand concentration. Though many groups have analyzed the effects of σ1 receptor ligands in cells, there is no standard incubation time for observing σ1-dependent effects of σ ligands. A brief survey of the literature reveals that when using σ1 receptor ligands in cellular or biochemical assays, incubation times and temperatures vary from room temperature for 20 minutes37 to 37 °C incubation for up to 72 hours35. Ligand concentrations are sometimes nearly 10,000-fold over Kd38–40. Our data indicate that it can take 1.5 hours or longer to reach saturation at 37 °C (Supplementary Fig. 5f), and at room temperature it can take nearly a day (Fig. 3a). Furthermore, since the rate-limiting step is concentration-independent, high ligand concentrations cannot overcome the receptor’s slow binding kinetics. However, previous reports have shown that σ1 receptor-dependent effects can be observed in as little as 10 minutes with 100 nM (+)-pentazocine at 37 °C, though the effect sizes observed were quite small41,42. This is consistent with our data demonstrating that about 50% of the receptor population should be occupied under these conditions (Supplementary Figure 5f). Therefore, when ascribing the effects of σ1 receptor ligands to the σ1 receptor, one must ensure that sufficient time is allowed for the ligands to engage with the receptor, and signal-to-noise may be improved by longer incubation times. If effects are observed too quickly and are only observed at ligand concentrations that are vastly higher than Kd, then it is unlikely that the effects are σ1-mediated.
We have shown that the agonist (+)-pentazocine adopts a binding pose in the σ1 receptor binding pocket that is different from that of antagonists, which tend to bind similarly to one another despite their chemical diversity. We have also demonstrated that ligands associate with the σ1 receptor very slowly and in multiple steps. Our simulations suggest that ligands enter the binding pocket through a dynamic opening that would be challenging to predict based on a single crystal structure.
However, the precise details of σ1 signaling in cells have yet to be determined. While there are myriad proposed binding partners for the σ1 receptor19,21,24,34,43, the critical effectors of σ1 receptor signaling still need to be unambiguously established. Future work will need to focus on these functional questions in order to fully understand the function of the σ1 receptor and its potential as a therapeutic target.
Accession Codes
Refined coordinates and crystallographic structure factors have been deposited in the PDB under accession codes 6DJZ (σ1 receptor-haloperidol complex), 6DK0 (σ1 receptor-NE-100 complex), and 6DK1 (σ1 receptor-(+)-pentazocine complex).
Methods
Protein expression and purification
Human σ1 receptor and was expressed and purified in Sf9 cells in a manner similar to that described previously28. In brief, the receptor was cloned into pFastBac1 with an amino-terminal hemagglutinin signal sequence, followed by a FLAG epitope tag and a 3C protease cleavage site. The receptor was expressed in Sf9 cells (Expression Systems) using the FastBac baculovirus system (ThermoFisher). Cells were grown in a shaker at 27 °C and infected when they had reached a density of 4 × 106 cells/mL. After infection, the cells were allowed to grow for 48–52 h, at which point they were harvested for centrifugation. Pellets were stored at −80 °C until use.
For samples used in crystallography, 1 μM of either haloperidol, (+)-pentazocine, or NE-100 was added to all purification steps. Haloperidol and NE-100 were purchased from Tocris Biosciences, and (+)-pentazocine was kindly provided by Dr. Felix Kim. For samples used for SPA binding measurements, no ligand was added. Cell pellets were thawed and lysed by osmotic shock in 20 mM HEPES pH 7.5, 2 mM MgCl2, and 1:200,000 (v/v) benzonase nuclease (Sigma Aldrich). The lysate was then spun at 48,000 x g for 20 min at 4 °C. The supernatant was discarded, and the pellets were solubilized using a glass dounce tissue homogenizer in a buffer containing 1% (w/v) lauryl maltose neopentyl glycol (LMNG, Anatrace), 20 mM HEPES pH 7.5, 250 mM NaCl, and 20% glycerol. For samples used in the crystallization of σ1 receptor with haloperidol and (+)-pentazocine, the solubilization buffer also contained 0.1% (w/v) cholesterol hemisuccinate (CHS; Steraloids). However, samples used in the crystallization of σ1 receptor with NE-100 and in SPA experiments were not solubilized with CHS, as it was found to have no effect on protein quality or yield. Following homogenization, samples were stirred at 4 °C for 2 h, and then centrifuged again as before. The supernatant was filtered over glass microfiber filters, and 2 mM CaCl2 was added to the solution. The sample was then run over 4 mL of αFLAG affinity resin. Once the sample was loaded on the resin, it was washed once with 50 mL of buffer containing 0.1% LMNG, 20 mM HEPES pH 7.5, 150 mM NaCl, 2% glycerol, and 2 mM CaCl2, then again with a buffer containing 0.01% LMNG, 20 mM HEPES pH 7.5, 150 mM NaCl, 0.2% glycerol, and 2 mM CaCl2. Following these wash steps, the protein was eluted in an identical buffer that lacked CaCl2 but was supplemented with 0.2 mg/mL FLAG peptide and 5 mM EDTA. Samples used for crystallography were incubated with 3C protease at 4 °C overnight to remove the FLAG tag. Samples used for SPA experiments were also left at 4 °C overnight but were not digested.
The next day, samples were further purified by SEC on a Sephadex S200 column (GE Healthcare). The buffer for SEC contained 0.01% LMNG, 20 mM HEPES pH 7.5, and 150 mM NaCl. For samples used for crystallography, the buffer also contained 1 μM of the desired ligand. After SEC, samples intended for crystallization were concentrated to 25–35 mg/mL and flash frozen in liquid nitrogen in 8–9 μL aliquots. Samples intended for SPA experiments were concentrated to 300–400 μM and diluted to 200 μM in SEC buffer supplemented with 20% glycerol. The SPA samples were aliquoted into 6 μL aliquots and flash frozen in liquid nitrogen. All samples were stored at −80 °C and never frozen again after thawing.
Crystallography and data collection
Purified σ1 receptor was reconstituted into lipidic cubic phase as described previously28,44. The cubic phase was dispensed in 30 nL drops onto a hanging drop cover and overlaid with 600 nL of precipitant solution with the use of a Gryphon LCP robot (Art Robbins Instruments). The crystal that provided the haloperidol-bound structure was grown in 500 mM Li2SO4, 35% (v/v) PEG 300, 1% (v/v) hexanediol, and 100 mM MES pH 6.4. The crystal that provided the (+)-pentazocine-bound structure was grown in 240 mM Li2SO4, 42% (v/v) PEG 300, 1% (v/v) hexanediol, and 0.1 M MES pH 6.0. The crystals for the NE-100 bound dataset were grown in 400–500 mM Li2SO4, 30–40% PEG 300, 1% hexanediol, and 0.1 M MES pH 5.8–6.0. Crystals grew slowly over the course of one to three weeks, and were harvested with mesh loops (MiTeGen) and stored at −80 °C. For (+)-pentazocine-bound crystals, it was important to harvest within two weeks, or crystal quality would decline substantially.
Data collection was performed at Advanced Photon Source GM/CA beamlines 23ID-B (NE-100 and (+)-pentazocine-bound structures) and 23ID-D (haloperidol-bound structure). Data collection was performed as described previously, using a 1.033 Å x-ray wavelength28. The datasets for haloperidol-bound and (+)-pentazocine-bound σ1 receptor were obtained from single crystals, while the dataset for NE-100-bound σ1 receptor was obtained by merging partial datasets from seven crystals.
Data processing, structure refinement, and model building
Data were processed using XDS45. For the haloperidol and NE-100 bound complexes, scaling was done with XSCALE45. For the (+)-pentazocine-bound structure, scaling was done with Aimless46. Phases for all three structures were solved via molecular replacement, using PD144418-bound σ1 receptor (PDB ID: 5HK1) as a search model. Model building was done with Coot47, and refinement was performed in phenix.refine48. Following refinement, structures were evaluated with MolProbity49, and figures were prepared with PyMOL50. In the haloperidol and NE-100-bound structures, 98.3% of the bond angles fall within favorable regions of Ramachandran space, and 1.7% are within the allowed region. For the (+)-pentazocine-bound structure, 97.7% of the bond angles are within favorable regions of Ramachandran space, and 2.3% are in allowed regions. None of the structures have peptide bonds that are not allowed by Ramachandran statistics. The SBGrid Consortium supported all crystallographic data processing, refinement, and analysis software51.
Preparation of membranes for radioligand binding
Membranes were prepared as described previously52, using a protocol adapted from that of Vilner et al.53. In brief, Sf9 cells expressing σ1 receptor were harvested by centrifugation and lysed by osmotic shock in a buffer containing 20 mM HEPES pH 7.5, 2 mM MgCl2,1:200,000 (v/v) benzonase nuclease (Sigma Aldrich), and cOmplete Mini EDTA-free protease inhibitor tablets (Sigma Aldrich). The lysates were homogenized using a glass dounce tissue homogenizer and then centrifuged at 48,000 x g for 20 min. Following centrifugation, the membranes were resuspended in buffer containing 50 mM Tris pH 8.0 and cOmplete Mini EDTA-free protease inhibitor tablets (Sigma Aldrich). The samples were spun down as before and resuspended in the same buffer. Next, the samples were homogenized using a needle and syringe. Protein content was determined using the DC protein assay (Bio Rad). Samples were aliquoted into 100 μL aliquots at protein concentrations of 10–20 mg/mL and flash frozen in liquid nitrogen. All samples were stored at −80 °C until use.
Saturation binding in Sf9 membranes
Saturation binding was performed as described previously52, using a method similar to that of Chu and Ruoho54. Briefly, membrane samples from Sf9 cells expressing wild-type or mutant σ1 receptor prepared as described above were thawed, homogenized with a syringe, and diluted in 50 mM Tris pH 8.0. Each reaction was 100 μL, with a final concentration of 0.025 mg/mL protein and the indicated concentration of [3H](+)-pentazocine. To assay nonspecific binding, equivalent reactions containing 2 μM haloperidol were performed in parallel. Samples were shaken at 37 °C for 90 minutes. Afterwards, the reaction was terminated by massive dilution and filtration over a glass microfiber filter using a Brandel harvester. Filters were soaked with 0.3% polyethyleneimine (PEI) for at least 30 minutes before use. Radioactivity was measured by liquid scintillation counting.
Measurement of ligand dissociation in Sf9 membranes
Membrane samples prepared as described above were thawed, syringe homogenized, and diluted in 50 mM Tris pH 8.0 to a final concentration of 0.05 mg/mL in a 96-well plate with a final volume of 100 μL per well. For association experiments, samples were incubated with 1 nM, 10 nM, or 100 nM [3H](+)-pentazocine for 5 min to 6 h, with all points performed in triplicate. For dissociation experiments, samples were first incubated with 10 nM [3H](+)-pentazocine (Perkin Elmer) for 90 minutes at 37 °C to reach equilibrium. After equilibration, 1 μL 500 μM haloperidol (Tocris Biosciences) was added to a set of wells in triplicate for a particular time point. This was repeated for a total of eight time points over the course of twenty-four hours. Upon completion of the time course in either the association or dissociation experiment, the reaction was terminated by massive dilution in ice-cold water and filtration over a glass microfiber filter using a Brandel harvester. Filters were soaked in 0.3% PEI for at least 30 minutes prior to use. Radioactivity was quantified by liquid scintillation counting. Data analysis was performed using GraphPad Prism.
Scintillation proximity assay
All scintillation proximity experiments were performed using protein-A coated YSi scintillation proximity beads (PerkinElmer, RPN143). Beads coupled with M1 αFLAG antibody and stored in HBS at 4 °C until use in 5 mg aliquots. Upon use, 4–6 mg of beads were spun down twice in a cold centrifuge and resuspended each time in HBS with 0.01% LMNG and 2 mM CaCl2. Then, the beads were incubated with 50 nM σ1 receptor purified as described above for 30 min at 4 °C. Following coupling of the receptor, the beads were again centrifuged and resuspended twice in HBS with 0.01% LMNG and 2 mM CaCl2. To start the reaction, 40 μL containing 0.2 mg of receptor-linked beads was added to a solution containing the desired concentration of radioligand in a total volume of 360 μL, for a total reaction volume of 400 μL. To assay nonspecific binding, equivalent reactions were prepared that also contained either nonradioactive haloperidol ([3H](+)-pentazocine binding) or nonradioactive NE-100 ([3H]haloperidol binding) at a concentration of 5 μM. Once association measurements were completed, 5 μM haloperidol ([3H](+)-pentazocine binding) or NE-100 ([3H]haloperidol binding) was added to each vial to begin the dissociation measurements. Samples were measured in duplicate at room temperature using a Beckman Coulter LS 6500 multi-purpose scintillation counter.
In order to average duplicate points, both the signal in CPM and the time at which the two different vials were measured were averaged. The values for each kinetic constant were first determined for each independent experiment using averaged duplicate time points. Thereafter, the constants from each independent experiment were averaged to obtain a final value with associated errors. Standard deviation and 95% confidence intervals were determined using only the differences in constants between independent experiments, and did not include error estimates from technical replicates, as the duplicate measures were averaged into single points prior to calculation of each constant. For all SPA association experiments, the final fits were obtained with kslow constrained to be identical among all concentrations of ligand tested in a given independent experiment. This was only done after confirming that all kslow values for a given ligand were statistically indistinguishable with an ANOVA test, as it improved the quality of the fits. Data were analyzed using GraphPad Prism.
Glide docking
Molecular docking into the σ1 receptor was performed in the manner of previous work55 using Glide 5.5 extra precision (XP) Maestro 11 Schrodinger release 2016–356. PRE-084 was docked into either the structure of the σ1 receptor bound to (+)-pentazocine (PDB ID: 6DK1). Since the structure has three protomers in the asymmetric unit, only chain C was used for docking studies. Lipids, ions, and waters were removed prior to protein preparation, leaving only the protein and ligand. Hydrogen atoms were added, and the protein was further refined by assigning H-bonds and minimizing energy for the OPLS3 force field. The grid used for docking was centered on the location of the co-crystallized ligand, and was 20 Å in the x, y, and z dimensions. Poses were ranked by glide score.
Statistics
To compare the kslow values from SPA association measurements of [3H](+)-pentazocine and [3H]haloperidol association, we used a two-tailed t-test. This t-test had four degrees of freedom and a t-value of 3.502, and gave a P value of P = 0.0249.
To test for potential kslow differences between concentrations of [3H](+)-pentazocine we performed a one-way ANOVA test, which had an F value of 6.535 and 17 degrees of freedom. The P value was 0.1078, indicating that the kslow values were not significantly different.
Molecular dynamics simulations
MD simulations setup
Simulations of the σ1 receptor were based on either the (+)-pentazocine or haloperidol-bound crystal structures described in this manuscript. The receptor was simulated in four distinct conditions (Supplementary Table 1): (A) the (+)-pentazocine bound structure, (B) the haloperidol bound structure, (C) the (+)-pentazocine bound structure with ligand removed, and (D) the (+)-pentazocine bound structure with ligand removed and placed in solvent. All simulations were of a σ1 monomer.
Coordinates were prepared by first removing chains B and C to obtain a monomer, and removing crystallographic ligands. For conditions C and D, the ligand was also removed from the binding pocket and for condition D it was replaced at least 10 Å from the protein. Prime (Schrodinger, Inc) was used to model in missing side chains, add hydrogens, and cap the protein chain termini with the neutral groups acetyl and methylamide.
As suggested previously28, Glu172 should be charged to interact with the ligand, and Asp126 should be protonated to hydrogen bond with Glu172; the protonation states of these two residues were set accordingly. All other residues were left in their dominant protonation state at pH 7.0.
The prepared protein was aligned to the Orientation of Proteins in Membranes57 (OPM) structure of PDB 5HK1, and internal waters added with Dowser58. The structures were then inserted into a pre-equilibrated palmitoyl-oleoyl-phosphatidylcholine (POPC) bilayer, and solvated with 0.15 M NaCl in explicitly represented water, then neutralized by removing sodium ions using the program Dabble59. Final system dimensions were about 63 × 63 × 140 Å3, including about 140 lipids, 12000 water molecules, 34 sodium ions, and 27 chloride ions.
MD simulation force field parameters
We used the CHARMM36m parameter set for protein molecules, the CHARMM36 parameter set for lipid molecules and salt ions, and the CHARMM TIP3P water model60–63. Parameters for (+)-pentazocine and haloperidol were generated using the CHARMM General Force Field (CGenFF) with the ParamChem server, version 1.0.064–66. Full parameter sets are available upon request.
MD simulation protocol
Simulations were performed on GPUs using the CUDA version of PMEMD (Particle Mesh Ewald Molecular Dynamics) on Amber1667. Prepared systems were minimized, then equilibrated as follows: the system was heated using the Langevin thermostat from 0 to 100 K in the NVT ensemble over 12.5 ps with harmonic restraints of 10.0 kcal∙mol−1 ∙Å−2 on the non-hydrogen atoms of lipid, protein and ligand, with initial velocities sampled from the Boltzmann distribution. The system was then heated to 310 K over 125 ps in the NPT ensemble with semi-isotropic pressure coupling and a pressure of 1 bar. Further equilibration was performed at 310 K with harmonic restraints on the protein and ligand starting at 5.0 kcal∙mol−1 ∙Å−2 and reduced by 1.0 kcal∙mol−1 ∙Å−2 in a stepwise fashion every 2 ns, for a total of 10 ns of additional restrained equilibration.
Five independent simulations were initialized from the final snapshot of the restrained equilibration for each condition (Supplementary Table 1). These simulations were conducted in the NPT ensemble at 310 K and 1 bar, using a Langevin thermostat and Monte Carlo barostat. In each of these simulations we performed 5 ns of unrestrained equilibration followed by 0.8 – 6.7 μs production run. Simulations used periodic boundary conditions and a time step of 4.0 fs with hydrogen mass repartitioning. Bond lengths to hydrogen atoms were constrained using SHAKE. Non-bonded interactions were cut off at 9.0 Å, and long-range electrostatic interactions were computed using the particle mesh Ewald (PME) method with an Ewald coefficient β of approximately 0.31 Å and B-spline interpolation of order 4. The FFT grid size was chosen such that the width of a grid cell was approximately 1 Å. Accelerated molecular dynamics (aMD) was used to boost dihedral potential energies, with parameters ED=10427 and αD=170.
For condition C, the ensemble of simulations was periodically visualized for major conformational change in the protein, and new sets of 5–10 simulation replicates initialized from restart files corresponding to rare events, with velocities either retained or equilibration being performed once more (Supplementary Table 1 )
MD simulation analysis protocols
Trajectory snapshots were saved every 200 ps during production simulations. Trajectory analysis was performed using VMD and CPPTRAJ, and visualization performed using VMD.
Supplementary Material
Acknowledgments
We thank Dr. Brendan Kelly for performing preliminary simulations of the σ1 receptor trimer, and Advanced Photon Source GM/CA beamline staff for excellent technical support of crystallographic data collection. We also thank Dr. Felix Kim (Drexel University) for generously providing (+)-pentazocine for crystallographic studies. This work was supported by a Klingenstein-Simons Fellowship in Neuroscience (A.C.K.), National Institutes of Health grant 1R01GM119185 (A.C.K.), the Winthrop Fund/Harvard Brain Science Initiative (A.C.K.) and National Science Foundation Graduate Research Fellowship award numbers DGE1745303 (H.R.S.) and DGE1656518 (R.M.B.).
Footnotes
Data Availability Statement
Refined coordinates and crystallographic structure factors have been deposited in the PDB under accession codes 6DJZ (σ1 receptor-haloperidol complex), 6DK0 (σ1 receptor-NE-100 complex), and 6DK1 (σ1 receptor-(+)-pentazocine complex). All other data that support the findings of this study are available from the corresponding author upon request.
Competing Interests Statement
The authors declare no competing interests.
References
- 1.Martin WR, Eades CG, Thompson JA, Huppler RE & Gilbert PE The effects of morphine- and nalorphine- like drugs in the nondependent and morphine-dependent chronic spinal dog. J Pharmacol Exp Ther 197, 517–532 (1976). [PubMed] [Google Scholar]
- 2.Tam SW & Cook L Sigma opiates and certain antipsychotic drugs mutually inhibit (+)-[3H] SKF 10,047 and [3H]haloperidol binding in guinea pig brain membranes. Proc Natl Acad Sci U S A 81, 5618–5621 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Su TP Evidence for sigma opioid receptor: binding of [3H]SKF-10047 to etorphine-inaccessible sites in guinea-pig brain. J Pharmacol Exp Ther 223, 284–290 (1982). [PubMed] [Google Scholar]
- 4.Hellewell SB & Bowen WD A sigma-like binding site in rat pheochromocytoma (PC12) cells: decreased affinity for (+)-benzomorphans and lower molecular weight suggest a different sigma receptor form from that of guinea pig brain. Brain Res 527, 244–253 (1990). [DOI] [PubMed] [Google Scholar]
- 5.Hanner M et al. Purification, molecular cloning, and expression of the mammalian sigma1-binding site. Proc Natl Acad Sci U S A 93, 8072–8077 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alon A et al. Identification of the gene that codes for the sigma2 receptor. Proc Natl Acad Sci U S A 114, 7160–7165, doi:10.1073/pnas.1705154114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walker JM et al. Sigma receptors: biology and function. Pharmacol Rev 42, 355–402 (1990). [PubMed] [Google Scholar]
- 8.Glennon RA et al. Structural features important for sigma 1 receptor binding. J Med Chem 37, 1214–1219 (1994). [DOI] [PubMed] [Google Scholar]
- 9.Ullah MI et al. In silico analysis of SIGMAR1 variant (rs4879809) segregating in a consanguineous Pakistani family showing amyotrophic lateral sclerosis without frontotemporal lobar dementia. Neurogenetics 16, 299–306, doi:10.1007/s10048-015-0453-1 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Wong AY et al. Aberrant Subcellular Dynamics of Sigma-1 Receptor Mutants Underlying Neuromuscular Diseases. Mol Pharmacol 90, 238–253, doi:10.1124/mol.116.104018 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Gregianin E et al. Loss-of-function mutations in the SIGMAR1 gene cause distal hereditary motor neuropathy by impairing ER-mitochondria tethering and Ca2+ signalling. Hum Mol Genet, doi:10.1093/hmg/ddw220 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Hong J et al. Sigma-1 receptor deficiency reduces MPTP-induced parkinsonism and death of dopaminergic neurons. Cell Death Dis 6, e1832, doi:10.1038/cddis.2015.194 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Katz JL, Hong WC, Hiranita T & Su TP A role for sigma receptors in stimulant self-administration and addiction. Behav Pharmacol, doi:10.1097/fbp.0000000000000209 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Castany S, Gris G, Vela JM, Verdu E & Boadas-Vaello P Critical role of sigma-1 receptors in central neuropathic pain-related behaviours after mild spinal cord injury in mice. Sci Rep 8, 3873, doi:10.1038/s41598-018-22217-9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bruna J et al. Efficacy of a Novel Sigma-1 Receptor Antagonist for Oxaliplatin-Induced Neuropathy: A Randomized, Double-Blind, Placebo-Controlled Phase IIa Clinical Trial. Neurotherapeutics 15, 178–189, doi:10.1007/s13311-017-0572-5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.An Extension Study of ANAVEX2–73 in Patients With Mild to Moderate Alzheimer’s Disease. Report No. NCT02756858, (Anavex Life Sciences Corp., ClincalTrials.gov, 2018).
- 17.Urfer R et al. Phase II trial of the Sigma-1 receptor agonist cutamesine (SA4503) for recovery enhancement after acute ischemic stroke. Stroke 45, 3304–3310, doi:10.1161/strokeaha.114.005835 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Kim FJ et al. Sigma 1 receptor modulation of G-protein-coupled receptor signaling: potentiation of opioid transduction independent from receptor binding. Mol Pharmacol 77, 695–703, doi:10.1124/mol.109.057083 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Navarro G et al. Direct involvement of sigma-1 receptors in the dopamine D1 receptor-mediated effects of cocaine. Proc Natl Acad Sci U S A 107, 18676–18681, doi:10.1073/pnas.1008911107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maurice T & Su TP The pharmacology of sigma-1 receptors. Pharmacol Ther 124, 195–206, doi:10.1016/j.pharmthera.2009.07.001 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hayashi T & Su TP Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 131, 596–610, doi:10.1016/j.cell.2007.08.036 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Aydar E, Palmer CP, Klyachko VA & Jackson MB The sigma receptor as a ligand-regulated auxiliary potassium channel subunit. Neuron 34, 399–410 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Kourrich S et al. Dynamic interaction between sigma-1 receptor and Kv1.2 shapes neuronal and behavioral responses to cocaine. Cell 152, 236–247, doi:10.1016/j.cell.2012.12.004 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hong WC et al. The sigma-1 receptor modulates dopamine transporter conformation and cocaine binding and may thereby potentiate cocaine self-administration in rats. J Biol Chem 292, 11250–11261, doi:10.1074/jbc.M116.774075 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mishra AK et al. The sigma-1 receptors are present in monomeric and oligomeric forms in living cells in the presence and absence of ligands. Biochem J 466, 263–271, doi:10.1042/bj20141321 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gromek KA et al. The oligomeric states of the purified sigma-1 receptor are stabilized by ligands. J Biol Chem 289, 20333–20344, doi:10.1074/jbc.M113.537993 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yano H et al. Pharmacological profiling of sigma 1 receptor ligands by novel receptor homomer assays. Neuropharmacology 133, 264–275, doi:10.1016/j.neuropharm.2018.01.042 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmidt HR et al. Crystal structure of the human sigma1 receptor. Nature 532, 527–530, doi:10.1038/nature17391 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kovacs KJ & Larson AA Discrepancies in characterization of sigma sites in the mouse central nervous system. Eur J Pharmacol 285, 127–134 (1995). [DOI] [PubMed] [Google Scholar]
- 30.Bowen WD, de Costa BR, Hellewell SB, Walker JM, and Rice KC [3H](+)-Pentazocine: A potent and highly selective benzomorphan-based probe for sigma-1 receptors. Mol. Neuropharmacol. 3, 117–126 (1993). [Google Scholar]
- 31.Pierce LC, Salomon-Ferrer R, Augusto F.d. O. C., McCammon JA & Walker RC Routine Access to Millisecond Time Scale Events with Accelerated Molecular Dynamics. J Chem Theory Comput 8, 2997–3002, doi:10.1021/ct300284c (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Romero L, Merlos M & Vela JM Antinociception by Sigma-1 Receptor Antagonists: Central and Peripheral Effects. Adv Pharmacol 75, 179–215, doi:10.1016/bs.apha.2015.11.003 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Mancuso R et al. Sigma-1R agonist improves motor function and motoneuron survival in ALS mice. Neurotherapeutics 9, 814–826, doi:10.1007/s13311-012-0140-y (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maher CM et al. Small-Molecule Sigma1 Modulator Induces Autophagic Degradation of PD-L1. Mol Cancer Res 16, 243–255, doi:10.1158/1541-7786.MCR-17-0166 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Li D et al. Sigma-1 receptor agonist increases axon outgrowth of hippocampal neurons via voltage-gated calcium ions channels. CNS Neurosci Ther 23, 930–939, doi:10.1111/cns.12768 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rosenbaum DM et al. Structure and function of an irreversible agonist-beta(2) adrenoceptor complex. Nature 469, 236–240, doi:10.1038/nature09665 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goguadze N, Zhuravliova E, Morin D, Mikeladze D & Maurice T Sigma-1 Receptor Agonists Induce Oxidative Stress in Mitochondria and Enhance Complex I Activity in Physiological Condition but Protect Against Pathological Oxidative Stress. Neurotox Res, doi:10.1007/s12640-017-9838-2 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Zhu J et al. Involvement of the delayed rectifier outward potassium channel Kv2.1 in methamphetamine-induced neuronal apoptosis via the p38 mitogen-activated protein kinase signaling pathway. J Appl Toxicol 38, 696–704, doi:10.1002/jat.3576 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Ortiz-Renteria M et al. TRPV1 channels and the progesterone receptor Sig-1R interact to regulate pain. Proc Natl Acad Sci U S A 115, E1657–E1666, doi:10.1073/pnas.1715972115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim FJ, Schrock JM, Spino CM, Marino JC & Pasternak GW Inhibition of tumor cell growth by Sigma1 ligand mediated translational repression. Biochem Biophys Res Commun 426, 177–182, doi:10.1016/j.bbrc.2012.08.052 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hayashi T, Maurice T & Su TP Ca(2+) signaling via sigma(1)-receptors: novel regulatory mechanism affecting intracellular Ca(2+) concentration. J Pharmacol Exp Ther 293, 788–798 (2000). [PubMed] [Google Scholar]
- 42.Hayashi T & Su TP Regulating ankyrin dynamics: Roles of sigma-1 receptors. Proc Natl Acad Sci U S A 98, 491–496, doi:10.1073/pnas.021413698 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kourrich S, Su TP, Fujimoto M & Bonci A The sigma-1 receptor: roles in neuronal plasticity and disease. Trends Neurosci 35, 762–771, doi:10.1016/j.tins.2012.09.007 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only References
- 44.Caffrey M & Cherezov V Crystallizing membrane proteins using lipidic mesophases. Nat. Protocols 4, 706–731 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kabsch W Xds. Acta Crystallogr D Biol Crystallogr 66, 125–132, doi:10.1107/S0907444909047337 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Evans PR & Murshudov GN How good are my data and what is the resolution? Acta Crystallogr D Biol Crystallogr 69, 1204–1214, doi:10.1107/S0907444913000061 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Emsley P & Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132, doi:10.1107/s0907444904019158 (2004). [DOI] [PubMed] [Google Scholar]
- 48.Afonine PV et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr 68, 352–367, doi:10.1107/s0907444912001308 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen VB et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66, 12–21, doi:10.1107/s0907444909042073 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schrodinger LLC. The PyMOL Molecular Graphics System, Version 1.3r1 (2010). [Google Scholar]
- 51.Morin A et al. Collaboration gets the most out of software. eLife 2, doi:10.7554/eLife.01456 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sguazzini E, Schmidt HR, Iyer KA, Kruse AC & Dukat M Reevaluation of fenpropimorph as a sigma receptor ligand: Structure-affinity relationship studies at human sigma1 receptors. Bioorg Med Chem Lett 27, 2912–2919, doi:10.1016/j.bmcl.2017.04.088 (2017). [DOI] [PubMed] [Google Scholar]
- 53.Vilner BJ, John CS & Bowen WD Sigma-1 and sigma-2 receptors are expressed in a wide variety of human and rodent tumor cell lines. Cancer Res 55, 408–413 (1995). [PubMed] [Google Scholar]
- 54.Chu UB & Ruoho AE Biochemical Pharmacology of the Sigma-1 Receptor. Mol Pharmacol 89, 142–153, doi:10.1124/mol.115.101170 (2016). [DOI] [PubMed] [Google Scholar]
- 55.Linkens K, Schmidt HR, Sahn JJ, Kruse AC & Martin SF Investigating isoindoline, tetrahydroisoquinoline, and tetrahydrobenzazepine scaffolds for their sigma receptor binding properties. Eur J Med Chem 151, 557–567, doi:10.1016/j.ejmech.2018.02.024 (2018). [DOI] [PubMed] [Google Scholar]
- 56.Friesner RA et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem 49, 6177–6196, doi:10.1021/jm051256o (2006). [DOI] [PubMed] [Google Scholar]
- 57.Lomize MA, Lomize AL, Pogozheva ID & Mosberg HI OPM: orientations of proteins in membranes database. Bioinformatics 22, 623–625, doi:10.1093/bioinformatics/btk023 (2006). [DOI] [PubMed] [Google Scholar]
- 58.DOWSER program (Department of Biochemistry and Biophysics, School of Medicine, University of North Carolina, Chapel Hill, NC, 27599–7260., 2003). [Google Scholar]
- 59.Dabble (version v2.6.3) (Zenodo, 2017).
- 60.Huang J & MacKerell AD Jr. CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J Comput Chem 34, 2135–2145, doi:10.1002/jcc.23354 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Best RB, Mittal J, Feig M & MacKerell AD Jr. Inclusion of many-body effects in the additive CHARMM protein CMAP potential results in enhanced cooperativity of alpha-helix and beta-hairpin formation. Biophys J 103, 1045–1051, doi:10.1016/j.bpj.2012.07.042 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Best RB et al. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles. J Chem Theory Comput 8, 3257–3273, doi:10.1021/ct300400x (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Klauda JB et al. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J Phys Chem B 114, 7830–7843, doi:10.1021/jp101759q (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vanommeslaeghe K et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem 31, 671–690, doi:10.1002/jcc.21367 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vanommeslaeghe K & MacKerell AD Jr. Automation of the CHARMM General Force Field (CGenFF) I: bond perception and atom typing. J Chem Inf Model 52, 3144–3154, doi:10.1021/ci300363c (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vanommeslaeghe K, Raman EP & MacKerell AD Jr. Automation of the CHARMM General Force Field (CGenFF) II: assignment of bonded parameters and partial atomic charges. J Chem Inf Model 52, 3155–3168, doi:10.1021/ci3003649 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.AMBER 2015 (University of California, San Francisco, 2015). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.