

Abstract
Chemical shift tensors (CSTs) are an exquisite probe of local geometric and electronic structure. 15N CST are very sensitive to hydrogen bonding, yet they have been reported for very few proteins to date. Here we present experimental results and statistical analysis of backbone amide 15N CSTs for 100 residues of four proteins, two E. coli thioredoxin reassemblies (1–73-(U-13C,15N)/74–108-(U-15N) and 1–73-(U-15N)/74–108-(U-13C,15N)), dynein light chain 8 LC8, and CAP-Gly domain of the mammalian dynactin. The 15N CSTs were measured by a symmetry-based CSA recoupling method, ROCSA. Our results show that the principal component δ11 is very sensitive to the presence of hydrogen bonding interactions due to its unique orientation in the molecular frame. The downfield chemical shift change of backbone amide nitrogen nuclei with increasing hydrogen bond strength is manifested in the negative correlation of the principal components with hydrogen bond distance for both α-helical and β-sheet secondary structure elements. Our findings highlight the potential for the use of 15N CSTs in protein structure refinement.
Keywords: magic-angle spinning NMR, chemical shift tensor, hydrogen bonding, proteins
1. INTRODUCTION
Hydrogen bond (HB) is one of the important noncovalent interactions stabilizing folded proteins [1]. NMR spectroscopy has long been used in the identification of hydrogen bonds in peptides and proteins through a number of NMR parameters. In particular, solution NMR signatures include chemical shift changes of the nuclei participating in HB interactions, reduced amide proton exchange rates, and indirect scalar or J-couplings mediated across hydrogen bonds to identify residues involved in hydrogen bonds. Among these, chemical shift perturbations are the easiest to measure directly using multidimensional homonuclear and heteronuclear correlation spectroscopy, as chemical shift parameters are highly sensitive to the local changes in the electronic environment due to HB interactions.
While solution NMR methods yield only the isotropic chemical shifts, in solid-state NMR (SSNMR) chemical shift tensors (CSTs) can be recorded. Chemical shift anisotropy (CSA) can be measured for multiple sites in a site-specific manner using magic angle spinning (MAS) NMR spectroscopy, where a chemical shift recoupling sequence is introduced into multidimensional experiments [2]. Yet, the literature on CSTs in proteins is sparse [3; 4] and the general rules governing the dependence of CST principal components on HB interactions remain to be established. For instance, the downfield change in the isotropic chemical shift due to the deshielding of amide protons involved in hydrogen bonds has been very well documented in the literature [5; 6; 7]. Recently our group reported site-specific amide proton CSA measurements for CAP-Gly domain of the mammalian dynactin, using the RN-symmetry based sequences [4]. The results revealed that indeed there is a high degree of correlation between the downfield shift of the principal components of the amide proton CST and the hydrogen bond length. Direct 1H–15N dipolar recoupling measurements have also been sometimes used in the literature to identify hydrogen bonds in terms of the elongation of N-H bond vector [8].
There have also been reports in the literature on the sensitivity of heteronuclear CS parameters to HB interactions in peptides and amino acids. In particular, the CSTs of both carbonyl carbon and amide nitrogen nuclei are as sensitive to HBs as amide protons. In an early SSNMR study of L-alanine containing short peptides, Asakawa et al. showed that as the hydrogen bond strength increases with decreasing distance between the nitrogen and oxygen atom (RN···O), the principal component δ 22 of the carbonyl carbon shows a large downfield shift accompanied by a similar trend in the isotropic chemical shift [9]. In a more detailed SSNMR study of CSA of carboxyl groups of amino acids, Gu et al. demonstrated the unique sensitivity of δ 11 and δ 22 to the protonation state and HB interaction of the CO group, respectively [10]. In a DFT study of model dipeptides, Walling et al. showed that, in addition to carbonyl atoms, backbone amide 15N CST parameters are also sensitive to hydrogen bonds [11]. In particular, they showed that both δ 11 and δ 33 exhibit downfield shift in both α-helical and β-sheet conformation, while δ 22 displays upfield shift in β-sheet conformation. This trend is consistent with 15N CST calculations performed on other model systems such as benzamide [12]. In contrast, a downfield shift of δ 22 with increasing RN···O and negligible sensitivity to HBs in both δ 11 and δ 33 was observed for the 15N nuclei of imidazole groups in histidines by Wei et al. [13], which is in agreement with results from 15N solution NMR studies of imidazoles [14]. Altough the magnitudes of the principal components of 15N CST are very sensitive to HB interactions, the orientation of the 15N CSTs in the molecular frame has been reported to be generally unperturbed on hydrogen bonding [15].
Despite the vast amount of theoretical and experimental-SSNMR studies of hydrogen bond effect on both 15N and 13C CST parameters on amino acids and model peptides, the corresponding experimental investigations in large systems, such as proteins are lacking. In this work, we present experimental SSNMR results and a statistical analysis of backbone amide 15N CST parameters for a total of 100 residues belonging to four proteins, two E. coli thioredoxin reassemblies ((1–73-(U-13C,15N)/74–108-(U-15N) and 1–73-(U-15N)/74–108-(U-13C,15N)), dynein light chain 8 LC8, and CAP-Gly domain of the mammalian dynactin. The 15N CST parameters measured by the ROCSA symmetry based recoupling are interpreted in terms of their sensitivity to HB interactions of the residues. Our results reveal unequivocal correlation between the principal components of the backbone 15N chemical shift tensors and hydrogen bonding interactions, underscoring the potential for use of 15N CSTs in protein structure refinement.
2. EXPERIMENTS AND METHODS
2.1. Protein samples preparation.
Preparation of solid-state NMR samples of E. coli thioredoxin reassemblies [1–73-(U-13C,15N)/74–108-(U-15N) and 1–73-(U-15N)/74–108-(U-13C,15N)], U-13C/15N DLC8, and U-13C/15N CAP-Gly domain of the mammalian dynactin by controlled precipitation was reported previously [16; 17; 18]. Approximately 10 mg of each sample was packed into a 3.2 mm Varian MAS rotor and sealed with an upper spacer and a top spinner.
2.2. SSNMR spectroscopy.
All SSNMR spectra presented in this work were acquired at 14.1 T on a narrow bore Varian InfinityPlus spectrometer operating at Larmor frequencies of 599.8 MHz for 1H, 150.8 MHz for 13C, and 60.8 MHz for 15N. The instrument was outfitted with a 3.2 mm T3 MAS probe. The MAS frequency was 10 kHz for all experiments, and was controlled to within ±1 Hz by a Varian MAS controller. The sample temperatures were kept in the range 0 oC to –15 oC. The temperature reported includes a MAS frequency-dependent correction determined experimentally by using PbNO3 as the temperature sensor [19]. 15N chemical shifts were referenced with respect to NH4Cl used as an external referencing standard following the standard protocol [20]. The 15N CSA recoupling was achieved by the symmetry-based ROCSA method [21]. The pulse sequence for the 3D NCA-ROCSA experiment is shown in Figure 3. The excitation frequencies were set at 122.4 ppm for 15N and at 55.5 ppm for 13C. Eighteen ROCSA points were acquired with the dwell time equal to one rotor period, and 64 15N t2 points with acquired with the dwell time of 120 μs. 128 scans were added to record each point in the indirect dimensions of the 3D spectra. 1H 90 pulse width was 2.78 μs. The contact time for the 1H-15N CP was 1.6 ms. The 1H radio frequency field strength was 51 kHz, the 15N field was linearly ramped 70–100% with the center of the ramp being 41 kHz. The ROCSA sequence was used with a window τa of 3.29 μs, and 42.8 kHz rf irradiation was employed. The Z-filter pulses were placed after the ROCSA block; the rf field strength was 50 kHz. The Z-filter delay was set equal to one rotor period. XY-16 decoupling [22] on the 13C channel was performed with the rf field strength of 50 kHz. 100 kHz CW decoupling was performed on the 1H channel during ROCSA. SPECIFIC-CP [23] for 15N-13CA transfer was utilized. The 13C field was tangentially ramped 90–100% with the center of the ramp being 35 kHz; the rf field strength for 15N was 25 kHz; the contact time was 6.2 ms. 100 kHz CW decoupling was applied during SPECIFIC-CP. The Z-filter pulses were incorporated on the 13C channel immediately after SPECIFIC-CP; the rf field strength was 67.9 kHz. 90 kHz TPPM decoupling [24] was applied during the t3 evolution; the TPPM pulse width was 5.4 μs.
Figure 3.
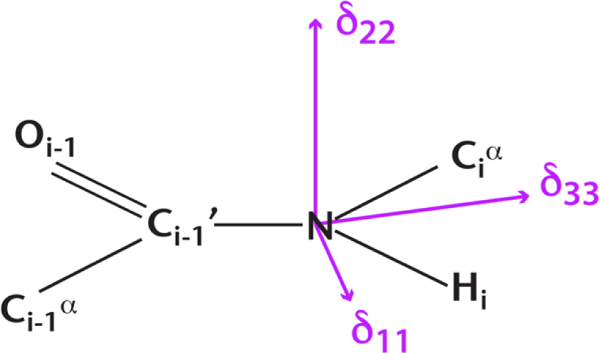
Illustration of the orientation of the backbone amide 15N CS tensor in the peptide plane (molecular frame). The bond lengths and bond angles are not to scale.
2.3. 15N ROCSA lineshape simulations.
Numerical simulations of the ROCSA lineshapes were performed on a single 15N spin using SIMPSON [25] with repulsion 320 powder angle set [26] and 36 γ-angles. An ideal 1H-15N cross polarization and proton decoupling during ROCSA evolution was assumed. The ROCSA lineshapes were fitted using MINUIT with the SIMPLEX method with three adjustable parameters: i) reduced anisotropy (δσ); ii) asymmetry parameter (ησ); and iii) exponential line-broadening parameter to model transverse relaxation during 15N ROCSA evolution. The uncertainties in the fitting parameters were determined using the Monte Carlo method as described in the literature [27]. The principal components of the 15N CS tensors in the standard convention (δ11 ≥ δ 22 ≥ δ 33) were determined using the relations, , , and .
2.4. Hydrogen bond analysis.
The hydrogen bond information of residues in the E. coli thioredoxin reassembly was inferred from its 1.8 Å crystal structure (PDB ID 2TRX) [28]. For LC8, a 2.8 Å resolution crystal structure (PDB ID 2PG1) was used [29]. MAS NMR structure was used for CAP-Gly domain of the mammalian dynactin (PDB ID 2M02) [30]. In all cases, an upper cutoff distance of 3.5 Å was set for RN···O. Residues exhibiting significant dynamic averaging of 15N CSA due to internal motions in the solid-state samples, such as G21, R73, and I75 of E. coli thioredoxin [31], were excluded from this analysis.
3. RESULTS AND DISCUSSION
3.1. Sensitivity of backbone amide 15N CST to presence of hydrogen bonds.
As shown in Figure 1B for E. coli thioredoxin reassembly (U-15N 1–73/13C,15N 74–108), the resolution of the NC chemical shift correlation plane of the 3D ROCSA-NCA spectrum is high permitting the extract the ROCSA lineshapes corresponding to the resolved peaks. In total, we have recorded ROCSA lineshapes for 100 residues in E. coli thioredoxin reassembly (U-13C/15N 1–73/15N 74–108, U-15N 1–73/13C,15N 74–108), dynein light chain 8 (LC8), and CAP-Gly domain of mammalian dynactin. The corresponding CST parameters are summarized in Table S1. Representative experimental and best-fit ROCSA lineshapes for E. coli thioredoxin reassembly (U-15N 1–73/13C,15N 74–108) and LC8 are shown in Figure 2.
Figure 1.
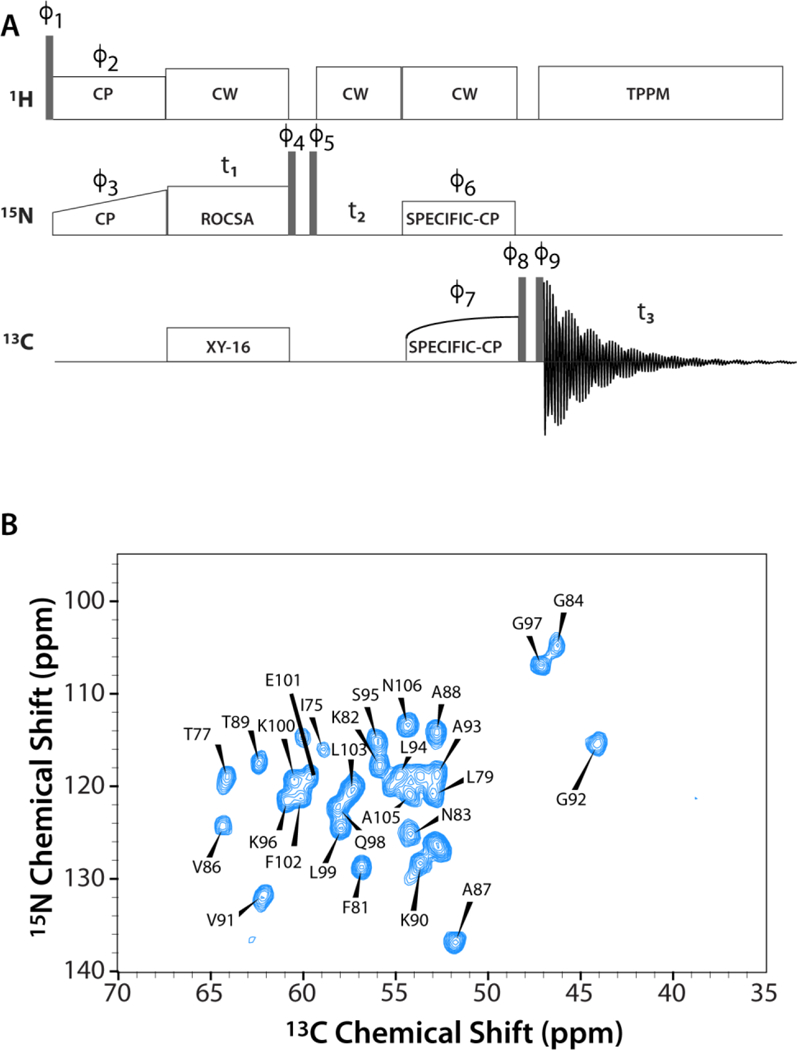
A) 3D 15N-13CA-ROCSA pulse sequence for site-specific measurement of the backbone amide 15N CSA lineshapes. B) The first 2D 15N-13CA plane of the 3D 15N-13CA-ROCSA spectrum of E. coli thioredoxin reassembly (U-15N 1–73/13C,15N 74–108).
Figure 2.
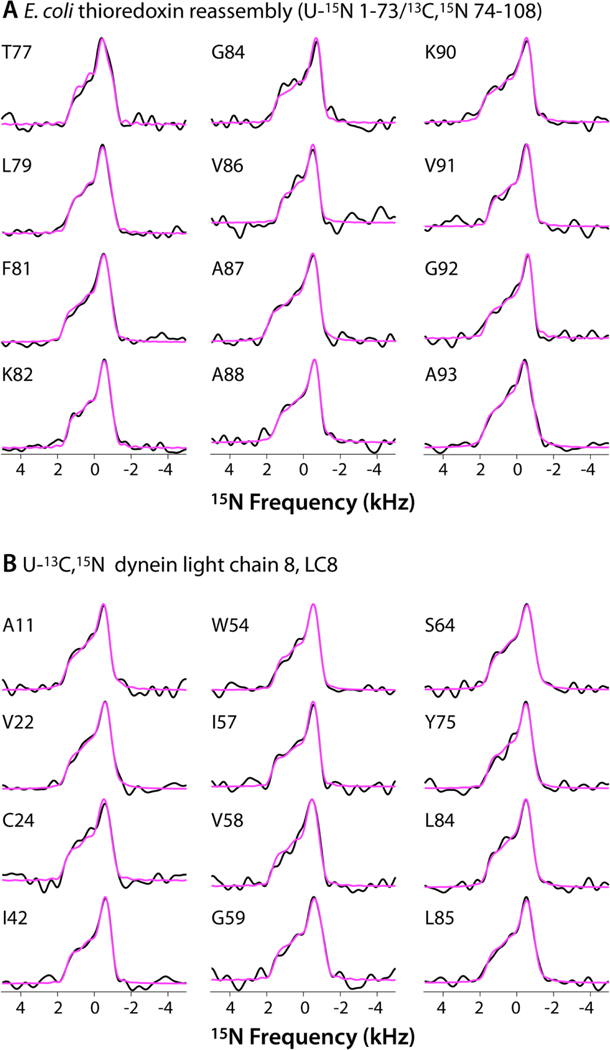
Representative backbone amide 15N ROCSA lineshapes of A) E. coli thioredoxin reassembly (U-15N 1–73/13C,15N 74–108) and B) dynein light chain 8, LC8.
Downfield shift of the isotropic component of the CS tensor of amide protons and amide nitrogens is known to be a characteristic marker of HB interactions [5; 6; 7]. This trend is confirmed in our results by the comparison of the statistical average value of the isotropic chemical shifts of the hydrogen-bonded 15N nuclei versus those that do not participate in hydrogen bonds. As shown in Table 1, the average isotropic chemical shift δ iso, of the hydrogen bonded 15N nuclei is around 120.6 ppm while that of the non-hydrogen-bonded 15N is 118.9 ppm. The difference of 1.7 ppm is significant enough for the direct detection of the nuclei participating in H-bonds from the 1D 15N CPMAS spectra or 2D correlation spectra, such as NCA or NCO.
Table 1.
Statistical Averages of the Principal Components of 15NH Chemical Shift Tensors for Hydrogen-Bonded and Non-Hydrogen-Bonded Residues of E. coli Thioredoxin Reassemblies, LC8, and CAP-Gly.
| Type | δiso (ppm) |
δσ (ppm) |
ησ | δ11
(ppm) |
δ22 (ppm) |
δ33 (ppm) |
|---|---|---|---|---|---|---|
| HB | 120.6 ± 6.6 | 95.8 ± 5.1 | 0.23 ± 0.08 | 216.3 ± 8.9 | 83.8 ± 8.6 | 61.5 ± 6.6 |
| NHB | 118.9 ± 7.1 | 93.6 ± 7.0 | 0.24 ± 0.09 | 212.5 ± 8.8 | 83.3 ± 10.6 | 61.0 ± 8.4 |
| Difference | 1.7 | 2.2 | –0.01 | 3.8 | 0.5 | 0.5 |
HB – hydrogen-bonded residues, NHB – non-hydrogen-bonded residues. The values presented here are the statistical average ± standard deviations.
The hydrogen bond interaction increases the width of 15N CSA lineshapes, which can easily be measured from the width of the ROCSA patterns. The reduced anisotropy, δσ, of the 15N CST exhibits the same trend as the isotropic chemical shifts. Specifically, the average δσ value of hydrogen bonded residues is 95.8 ppm while δσ of residues not forming hydrogen bonds is 93.6 ppm. Although the difference of 2.2 ppm is relatively small, it is statistically significant and exceeds the typical experimental errors associated with modern CSA recoupling sequences designed for MAS frequencies exceeding 10 kHz, such as ROCSA and RNCSA [3]. Unlike δiso and δσ, the asymmetry parameter of the 15N CSA tensor (ησ) is not sensitive to hydrogen bond interaction. The average ησ values of both hydrogen-bonded and non-hydrogen-bonded 15N nuclei are essentially the same 0.2.
More insight into the sensitivity of the individual components of the CSA tensor to the hydrogen bonding interactions can be gained from the analysis of the principal components using the standard convention, (δ11, δ22, δ33). It is evident from Table 1 that the most downfield shifted component δ11 is the most sensitive to the presence of hydrogen bonds. The average δ 11 value for hydrogen-bonded 15N nuclei is 216.3 ppm, while it is 212.5 ppm for non-hydrogen-bonded residues. The component with intermediate shielding, δ22, and the most shielded component δ33 of the 15N CSA tensor show relatively minor downfield shift due to the presence of HB interactions. The average value of δ22 for hydrogen-bonded residues is 83.8 ppm and 83.3 ppm for the non-hydrogen-bonded residues. The corresponding values for δ33 are 61.5 and 61.0 ppm.
The result that the δ 11 component of the 15N CSA tensor has the highest sensitivity to hydrogen bonding is in perfect agreement with theoretical studies of 15N shielding tensors on hydrogen bonded amino acids and peptides [15; 32]. In these studies, it has been shown that the orientation of the 15N CS tensor relative to the molecular frame is such that the δ11 component lies in the peptide plane with the angle between the N–H bond vector and the δ11 component being around ca. 10–20 degrees (Figure 3). The almost parallel orientation of δ11 component with respect to the N–H bond vector gives it the highest perturbation due to hydrogen bond interaction. The δ22 component which shows weak sensitivity to hydrogen bond interactions lies almost perpendicular to the peptide plane. The equally weakly sensitive δ33 component lies in the peptide plane with almost perpendicular orientation to the N–H bond vector.
It is important to mention that the backbone amide 15N CS tensors are not only sensitive to hydrogen bonds, but are also dependent on secondary structure, nature of the amino acid, and internal dynamics of backbone. This fact is evident in the large standard deviations associated with each of the three principal components for both hydrogen-bonded and non-hydrogen-bonded residues as presented in Table 1. Nevertheless, despite the limited number of 15N CSA lineshapes available so far and analyzed in this work, we do observe a clear downfield shift in δ11 due to hydrogen bond interactions.
3.2. Correlation of backbone amide 15N CST components with isotropic chemical shift.
In the case of amide proton CSA parameters, Tjandra et al. reported the correlation of the three principal components with the isotropic chemical shift as an essential metric for the sensitivity of amide proton CSA parameters to HB interactions [5]. Recently, in our work on MAS NMR measurements of backbone amide proton CSTs of CAP-Gly domain of the mammalian dynactin, the correlation of the three principal components with the isotropic chemical of the backbone amide protons was verified to be correct [4]. In order to examine whether the same trend is observed for the backbone amide 15N CST parameters, in Figure 4 we have plotted the three 15N principal components versus the isotropic chemical shift. Similar to the findings for the backbone amide proton CSAs, 15N CS principal components also follow a linear relationship with the isotropic chemical shift, with a strong positive correlation indicated by positive values of Pearson correlation coefficients of 0.73, 0.84, and 0.79 for δ11, δ22, and δ33 respectively. These results suggest that that one can expect to see a similar positive correlation of the three amide 15N CSAs with hydrogen bond strength or a negative correlation with hydrogen bond distance. This assertion is corroborated in the following section.
Figure 4.
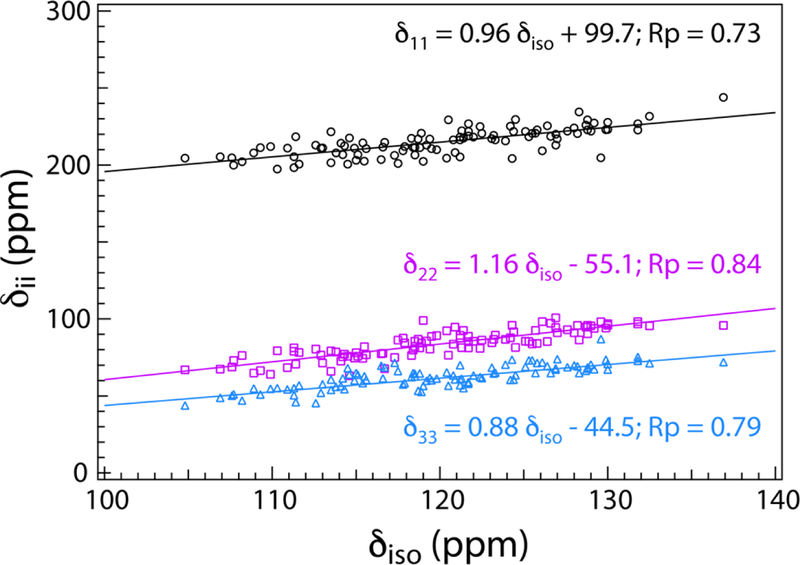
Correlation of the backbone amide 15N CST principal components with isotropic chemical shift for the four proteins under investigation, E. coli thioredoxin reassemblies, LC8, and CAP-Gly. The correlation is determined the Pearson correlation coefficients (RP) with values greater than 0.70 indicating a strong positive correlation of the three principal components with 15N isotropic chemical shift.
3.3. Correlation of backbone amide 15N CST with hydrogen bond length.
Ab initio theoretical and experimental studies have shown that CS tensor values of nuclei involved in hydrogen bond correlate well with hydrogen bond length values [7; 12]. In particular, the downfield shift of nuclei of the donor group with increasing hydrogen bond strength or decreasing hydrogen bond length (RN…O or RH…O) due to increased deshielding has been well investigated [6; 7]. In this section, we present the statistical correlation of backbone amide 15N CS principal components with the distance between the amide 15N atom in the donor group and the oxygen atom in the acceptor group. As mentioned in the previous section, in addition to HB interactions backbone amide 15N CST values are dependent on several other parameters such as secondary structure, local conformation and internal dynamics. In order to decouple the sensitivity of amide 15N CST on secondary structure, we have performed two separate correlation analyses, one for α-helical residues, and the other for β-strand residues. The results are shown in Figure 5. Out of the 100 residues we have analyzed from the four proteins, 65 are hydrogen bonded and 35 are non-hydrogen bonded. Among the hydrogen-bonded, 21 are α-helical residues and 32 are located in β -strands.
Figure 5.
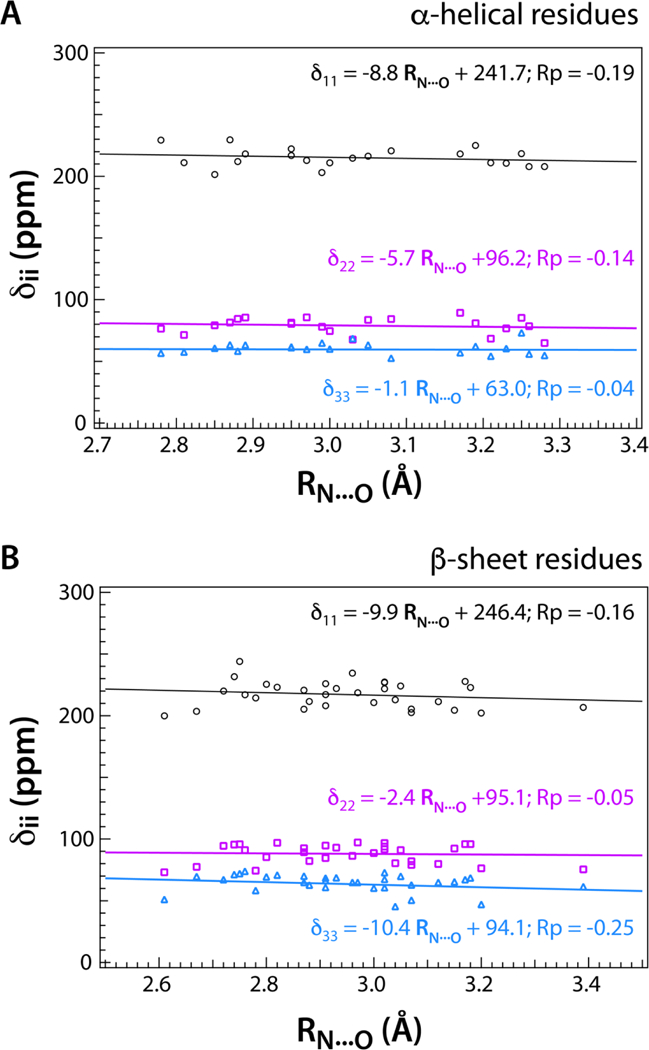
Correlation of the hydrogen-bonded backbone amide 15N CST principal components with hydrogen bond distance RN…O for A) α-helical and B) β-sheet structure, for the four proteins under investigation, E. coli thioredoxin reassemblies, LC8, and CAP-Gly. The downfield shift of all three 15N CST principal components, notably in δ 11, with decreasing RN…O is indicated by the negative Pearson correlation coefficients (RP) for both α-helical and β-sheet secondary structures.
In Figure 5, the correlation of the principal components of backbone amide 15N CST is plotted against RN…O for both α-helical and β -strand residues. First, a negative correlation as determined by the Pearson correlation coefficient is evident in all the graphs. This statistical observation is consistent with the fact that the downfield chemical shift change of backbone amide 15N CST values correlate with decreasing hydrogen bond lengths as reported in literature [13]. Among the two secondary structure types, for α-helices, the δ11 component exhibits the highest sensitivity to the hydrogen bonding distance with RP of –0.19. The δ22 component is somewhat less sensitive with RP of –0.14, and δ33 has the smallest or almost no correlation (RP equal to –0.04). This trend is in very good agreement with the assertion that the least-shielded component δ11 acquires maximum sensitivity due to its collinear orientation with the N-H bond vector.
Interestingly, for β-strands, the δ33 component, which lies in the peptide plane with almost perpendicular orientation to the N-H bond vector, has the maximum negative correlation with RP of –0.25 while the δ11 component is somewhat less sensitive with RP equal to –0.16. The δ22 component shows the weakest correlation with RP equal to –0.05. The less negative correlation of δ22 component with RN…O for β-sheets is consistent with earlier reports that δ22 component is shifted upfield or shows positive correlation with increasing hydrogen bond strength or decreasing RN…O [11]. However, the decreased sensitivity of δ11 and the increased sensitivity of δ33 to hydrogen bond length in β -strands is somewhat surprising. It would be interesting to explore this further through quantum chemical calculations of the hydrogen-bonded backbone amide 15N chemical shift tensors as a function of the secondary structure type.
As shown in Figure 5, the magnitudes of the Pearson correlation coefficients are in the range of 0.04–0.19, which indicate only a modest correlation of backbone amide 15N CST with hydrogen bond length. Despite these modest values of RP, a clear trend of δii (ii = 11, 22, 33) linearly decreasing with RN…O is evident for both α-helical and β-strand secondary structure types.
4. CONCLUSIONS
We have presented experimental SSNMR data and statistical analysis of site-specific backbone amide 15N CS tensor components of 100 residues of four different proteins, E. coli thioredoxin, DLC8 and CAP-Gly domain of the mammalian dynactin. Our results reveal that the least-shielded component δ 11 of the backbone amide 15N is very sensitive to hydrogen bond interaction, displaying a significant downfield shift. The downfield chemical shift change of backbone amide nitrogen nuclei with increasing hydrogen bond strength is manifested in the negative correlation of the principal components with hydrogen bond distance for both secondary structure elements, α-helices and β -sheets. Our results are consistent with the theoretical studies of 15N shielding tensors on model compounds and short peptides. In order to improve this analysis further, one needs to record a larger database of 15N chemical shift tensors, which would encompass multiple proteins and different secondary structure types. These studies are currently ongoing in our laboratory.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (NIH Grant P50 GM082251, Technology Development Project on MAS NMR). We acknowledge the support of the National Institutes of Health (NIH Grants P30GM103519 and P30GM110758) for the support of core instrumentation infrastructure at the University of Delaware.
REFERENCES
- [1].Baker EN, and Hubbard RE, Hydrogen bonding in globular proteins. Prog. Biophys. Mol. Biol. 44 (1984) 97–179. [DOI] [PubMed] [Google Scholar]
- [2].Saitô H, Ando I, and Ramamoorthy A, Chemical Shift Tensor – the Heart of NMR: Insights into Biological Aspects of Proteins. Prog. Nucl. Magn. Reson. Spectrosc. 57 (2010) 181–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hou G, Byeon IJ, Ahn J, Gronenborn AM, and Polenova T, Recoupling of chemical shift anisotropy by R-symmetry sequences in magic angle spinning NMR spectroscopy. J. Chem. Phys. 137 (2012) 134201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hou G, Paramasivam S, Yan S, Polenova T, and Vega AJ, Multidimensional magic angle spinning NMR spectroscopy for site-resolved measurement of proton chemical shift anisotropy in biological solids. J. Am. Chem. Soc. 135 (2013) 1358–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Tjandra N, and Bax A, Solution NMR Measurement of Amide Proton Chemical Shift Anisotropy in 15N-Enriched Proteins. Correlation with Hydrogen Bond Length. Journal of the American Chemical Society 119 (1997) 8076–8082. [Google Scholar]
- [6].Yao L, Grishaev A, Cornilescu G, and Bax A, The Impact of Hydrogen Bonding on Amide (1)H Chemical Shift Anisotropy Studied by Cross-Correlated Relaxation and Liquid Crystal NMR Spectroscopy. J. Am. Chem. Soc. 132 (2010) 10866–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sharma Y, Kwon OY, Brooks B, and Tjandra N, An ab initio study of amide proton shift tensor dependence on local protein structure. J. Am. Chem. Soc. 124 (2002) 327–35. [DOI] [PubMed] [Google Scholar]
- [8].Zhao X, Sudmeier JL, Bachovchin WW, and Levitt MH, Measurement of NH bond lengths by fast magic-angle spinning solid-state NMR spectroscopy: a new method for the quantification of hydrogen bonds. J. Am. Chem. Soc. 123 (2001) 11097–8. [DOI] [PubMed] [Google Scholar]
- [9].Asakawa N, Kuroki S, Kurosu H, Ando I, Shoji A, and Ozaki T, Hydrogen-bonding effect on carbon-13 NMR chemical shifts of L-alanine residue carbonyl carbons of peptides in the solid state. Journal of the American Chemical Society 114 (1992) 3261–3265. [Google Scholar]
- [10].Wei Y, de Dios AC, and McDermott AE, Solid-State 15N NMR Chemical Shift Anisotropy of Histidines: Experimental and Theoretical Studies of Hydrogen Bonding. Journal of the American Chemical Society 121 (1999) 10389–10394. [Google Scholar]
- [11].Walling AE, Pargas RE, and de Dios AC, Chemical Shift Tensors in Peptides: A Quantum Mechanical Study. J. Phys. Chem. A 101 (1997) 7299–7303. [Google Scholar]
- [12].Facelli JC, Pugmire RJ, and Grant DM, Effects of Hydrogen Bonding in the Calculation of 15N Chemical Shift Tensors: Benzamide. Journal of the American Chemical Society 118 (1996) 5488–5489. [Google Scholar]
- [13].Wei Y, De Dios AC, and McDermott AE, Solid-state 15N NMR chemical shift anisotropy of histidines: Experimental and theoretical studies of hydrogen bonding. Journal of the American Chemical Society 121 (1999) 10389–10394. [Google Scholar]
- [14].Schuster II, and Roberts JD, Nitrogen-15 nuclear magnetic resonance spectroscopy. Effects of hydrogen bonding and protonation on nitrogen chemical shifts in imidazoles. J. Org. Chem. 44 (1979) 3864–3867. [Google Scholar]
- [15].Brender JR, Taylor DM, and Ramamoorthy A, Orientation of Amide-Nitrogen-15 Chemical Shift Tensors in Peptides: A Quantum Chemical Study. Journal of the American Chemical Society 123 (2001) 914–922. [DOI] [PubMed] [Google Scholar]
- [16].Marulanda D, Tasayco ML, McDermott A, Cataldi M, Arriaran V, and Polenova T, Magic angle spinning solid-state NMR spectroscopy for structural studies of protein interfaces. resonance assignments of differentially enriched Escherichia coli thioredoxin reassembled by fragment complementation. J. Am. Chem. Soc. 126 (2004) 16608–20. [DOI] [PubMed] [Google Scholar]
- [17].Yang J, Paramasivam S, Marulanda D, Cataldi M, Tasayco ML, and Polenova T, Magic angle spinning NMR spectroscopy of thioredoxin reassemblies. Magn. Reson. Chem. 45 Suppl 1 (2007) S73–83. [DOI] [PubMed] [Google Scholar]
- [18].Sun S, Butterworth AH, Paramasivam S, Yan S, Lightcap CM, Williams JC, and Polenova T, Resonance Assignments and Secondary Structure Analysis of Dynein Light Chain 8 by Magic Angle Spinning NMR Spectroscopy. Can. J. Chem. 89 (2011) 909–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Neue G, and Dybowski C, Determining temperature in a magic-angle spinning probe using the temperature dependence of the isotropic chemical shift of lead nitrate. Solid State Nucl. Magn. Reson. 7 (1997) 333–6. [DOI] [PubMed] [Google Scholar]
- [20].Morcombe CR, and Zilm KW, Chemical shift referencing in MAS solid state NMR. J. Magn. Reson. 162 (2003) 479–86. [DOI] [PubMed] [Google Scholar]
- [21].Chan JCC, Recoupling of chemical shift anisotropies in solid-state NMR under high-speed magic-angle spinning and in uniformly 13C-labeled systems. The Journal of chemical physics 118 8378–8389. [Google Scholar]
- [22].Gullion T, Baker DB, and Conradi MS, New, compensated Carr-Purcell sequences. J. Magn. Reson. 89 (1990) 479–484. [Google Scholar]
- [23].Baldus M, Cross polarization in the tilted frame: assignment and spectral simplification in heteronuclear spin systems. Mol. Phys. 95 1197–1207. [Google Scholar]
- [24].Bennett AE, Heteronuclear decoupling in rotating solids. The Journal of chemical physics 103 6951–6958. [Google Scholar]
- [25].Bak M, Rasmussen JT, and Nielsen NC, SIMPSON: A General Simulation Program for Solid-State NMR Spectroscopy. J. Magn. Reson. 147 (2000) 296–330. [DOI] [PubMed] [Google Scholar]
- [26].Bak M, and Nielsen NC, REPULSION, A Novel Approach to Efficient Powder Averaging in Solid-State NMR. J. Magn. Reson. 125 (1997) 132–139. [DOI] [PubMed] [Google Scholar]
- [27].Wylie BJ, Franks WT, and Rienstra CM, Determinations of 15N Chemical Shift Anisotropy Magnitudes in a Uniformly 15N,13C-Labeled Microcrystalline Protein by Three-Dimensional Magic-Angle Spinning Nuclear Magnetic Resonance Spectroscopy. J. Phys. Chem. B 110 (2006) 10926–10936. [DOI] [PubMed] [Google Scholar]
- [28].Katti SK, Crystal structure of thioredoxin from Escherichia coli at 1.68 Å resolution. J. Mol. Biol. 212 167–184. [DOI] [PubMed] [Google Scholar]
- [29].Williams JC, Roulhac PL, Roy AG, Vallee RB, Fitzgerald MC, and Hendrickson WA, Structural and thermodynamic characterization of a cytoplasmic dynein light chain-intermediate chain complex. Proc. Natl. Acad. Sci. USA 104 (2007) 10028–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yan S, Hou G, Schwieters CD, Ahmed S, Williams JC, and Polenova T, Three-dimensional structure of CAP-gly domain of mammalian dynactin determined by magic angle spinning NMR spectroscopy: conformational plasticity and interactions with end-binding protein EB1. J Mol Biol 425 (2013) 4249–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Yang J, Tasayco ML, and Polenova T, Dynamics of reassembled thioredoxin studied by magic angle spinning NMR: snapshots from different time scales. J. Am. Chem. Soc. 131 (2009) 13690–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Poon A, Birn J, and Ramamoorthy A, How Does an Amide-N Chemical Shift Tensor Vary in Peptides? J. Phys. Chem. B 108 (2004) 16577–16585. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.