

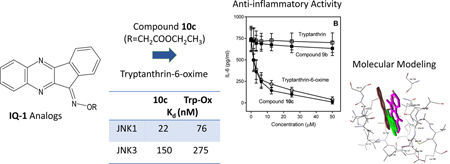
Abstract
c-Jun N-terminal kinases (JNKs) play a central role in many physiologic and pathologic processes. We synthesized novel 11H-indeno[1,2-b]quinoxalin-11-one oxime analogs and tryptanthrin-6-oxime (indolo(2,1-b)quinazoline-6,12-dion-6-oxime) and evaluated their effects on JNK activity. Several compounds exhibited sub-micromolar JNK binding affinity and were selective for JNK1/JNK3 versus JNK2.The most potent compounds were 10c (11H-indeno[1,2b]quinoxalin-11-one O-(O-ethylcarboxymethyl) oxime) and tryptanthrin-6-oxime, which had dissociation constants (Kd) for JNK1 and JNK3 of 22 and 76 nM and 150 and 275 nM, respectively. Molecular modeling suggested a mode of binding interaction at the JNK catalytic site and that the selected oxime derivatives were potentially competitive JNK inhibitors. JNK binding activity of the compounds correlated with their ability to inhibit lipopolysaccharide (LPS)-induced nuclear factor-κB/activating protein 1 (NF-κB/AP-1) activation in human monocytic THP-1Blue cells and interleukin-6 (IL-6) production by human MonoMac-6 cells. Thus, oximes with indenoquinoxaline and tryptanthrin nuclei can serve as specific smallmolecule modulators for mechanistic studies of JNK, as well as potential leads for the development of anti-inflammatory drugs.
Keywords: c-Jun N-terminal kinase; tryptanthrin; 11H-indeno[1,2-b]quinoxalin-11-one; oxime; tropomyosin‐related kinase; inflammation
Graphical abstract
1. Introduction
c-Jun N-terminal kinases (JNKs) belong to the family of mitogen-activated protein kinases (MAPK) that are activated in response to various stress stimuli, such as ultraviolet radiation, oxidative stress, heat and osmotic shock, and ischemia-reperfusion injury of the brain and heart [1–4]. In mammals, 10 highly similar isoforms are expressed by alternative splicing of three different genes: JNK1 (four isoforms), JNK2 (four isoforms), and JNK3 (two isoforms) [5, 6]. JNK1 and JNK2 are found in all cells and tissues of the body, while JNK3 is expressed mainly in the brain, heart, and testicles [2].
The JNKs have been shown to play an important role in regulation of the signaling pathways involved in apoptosis, necrosis, inflammation, and ischemia/reperfusion injury [7–10]. They are involved in a wide range of diseases, including rheumatoid arthritis, osteoarthritis, multiple sclerosis, inflammatory bowel disease, insulin resistance, tumorigenesis, stroke, renal ischemia, Alzheimer’s and Parkinson’s diseases [6, 11–17].Upstream kinases of the MAPK cascade (MKK4 and MKK7) phosphorylate and activate JNK [18], whereas transcription factors such as c-Jun, specificity protein 1 (Sp1), activating transcription factor 2 (ATF2), and nuclear factors of activated T-cells (NFATc2 and NFATc3) are substrates for phosphorylation-activated JNKs [5, 7, 9, 19]. There are also numerous non-nuclear substrates of JNK that participate in the degradation of proteins, signal transduction, and regulation of apoptotic cell death [3, 20]. For example, JNK1 phosphorylates insulin receptor substrate 1 (IRS-1), a key molecule in the insulin-sensing pathway, which down-regulates insulin signaling [21]. Recently Tudor-SN, a multifunctional protein that is implicated in a variety of cellular processes, was identified as a novel JNK target [22].
A significant amount of pharmacological and genetic evidence suggests that inhibition of JNK signaling may represent a promising therapeutic strategy [23], and numerous efforts have focused on the development of selective and nontoxic JNK inhibitors. For example, selective JNK1/3 inhibitors may have clinical benefit in treating neurodegenerative disorders [24]. However, it has been difficult to design selective JNK inhibitors because of the high sequence identity among JNK isoforms (from 73 to 75%) and, specifically, sequence identity of their ATP binding pockets (close to 98%) [25]. Recently, JNK2/3 inhibitors with an aminopyrazole scaffold that have >30-fold selectivity over JNK1 were identified [25].
Although no JNK inhibitors have been approved for use in humans, a few small molecule JNK inhibitors have entered clinical trials for various indications, including tanzisertib for the treatment of lupus erythematous and idiopathic pulmonary fibrosis, bentamapimod for the treatment of inflammatory endometriosis, and D-JNKi1 for the treatment of inflammation and stroke (for review [26–28]). Previously, we identified a new class of JNK inhibitors based on the 11H-indeno[1,2-b]quinoxalin-11-one scaffold [29]. Specifically, compound IQ-1 (11-Hindeno[1,2-b]quinoxalin-11-one oxime) and its oxime analogs (Fig. 1) inhibited JNK activity and, consequently, proinflammatory cytokine production by murine and human leukocytes [29]. We also found that IQ-1 reduced inflammation and cartilage loss associated with collageninduced arthritis (CIA) [30] and protected against cerebral ischemia–reperfusion injury in mice [31]. These JNK inhibitors contain oxime (IQ-1) or O-acyl-oxime (IQ-2 through IQ-4 and IQ-6) groups (Fig. 1) and exhibited some (~3.6–5.3 fold) selectivity for JNK3 versus JNK1/2 [29, 30]. Thus, we propose that modification of IQ-1 by introduction of various substituents could increase potency and/or a selectivity of the resulting analogs toward the JNK isoforms.
Figure 1.
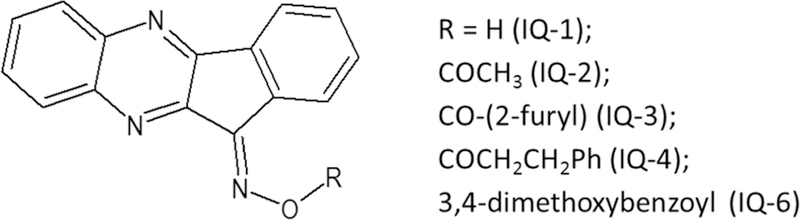
Chemical structure of previously identified JNK1–3 inhibitors with an 11H-indeno[1,2-b]quinoxalin-11one scaffold.
In the present studies, novel 11H-indeno[1,2-b]quinoxalin-11-one oxime analogs, such as aza-analogues, O-substituted derivatives, and analogs with different substituents in the indenoquinoxaline tetracyclic moiety, were synthesized and evaluated against JNK1–3. We conducted molecular modeling for selected compounds and estimated their anti-inflammatory potential using in vitro cell-based assays. We also report for the first time that tryptanthrin-6oxime, a derivative of the natural alkaloid tryptanthrin, has a relatively high binding affinity for JNK1–3 and tropomyosin‐related kinases (TRK) A and B.
2. Results and discussion
2.1. Chemistry
All new compounds were synthesized as reported in Scheme 1, and the structures were confirmed on the basis of analytical and spectral data. As reported previously, 11H-indeno[1,2b]quinoxalin-11-one (compound 1) was synthesized by the condensation of ninhydrin with ophenylenediamine [32, 33]. We synthesized oximes of known [34–36] and commercially available ketones 2a-i as described in Scheme 1A.
Scheme 1.

Reagents and conditions: (a) NH2OH·HSO4, NaOH in EtOH, 60 оC, 8 h, 8093%; (b,d) in EtOH, 60 оC, 10 h, 81%; (c,e) NH2OH·HCl in EtOH, 60 оC, 10 h, 80%; (f) NH2OH·HCl in pyridine, 60 оC, 2 h, 95%; (g) in EtOH, 78 oC, 9 h, 88–92%; (h) KOH in DMSO, r.t., 1 h (10a, 54%; 10c, Method A, 84%), or Na2CO3 in DMSO, r.t., 9–10 h (10b, 52%; 10c, Method B, 56%; 10d, 83%).
To synthesize indenoquinoxaline analogues 3a, 3b, and 5 containing an additional nitrogen atom in the tetracyclic nucleus, we used the reaction of 2,3-diaminopyridine, its 5chloro derivative, and 3,4-diaminopyridine, with ninhydrin in EtOH (Scheme 1B). It has been established that the use of H2O as a solvent instead of EtOH does not significantly affect yields of the products and selectivity of the process. The existence of two isomers is possible for each of the resulting aza-analogues. We determined the isomer ratios from integral intensities of the signals in 1H-NMR spectra of the products and found that 3,4-diaminopyridine reacts regiospecifically, with the sole formation of 5 (73% yield). To provide a rationale for regioselectivity of this reaction, we performed density functional theory (DFT) calculations (see the description and Supplementary Fig. S1). Previously, compound 5 was synthesized with comparable yield at higher temperature in boiling isobutyl alcohol [37]. From the reaction of 2,3diaminopyridine with ninhydrin, a mixture of isomers 3a and 4a (90:10%) was obtained, with a total yield of 82%, from which we isolated pure compound 3a. It should be noted that in boiling MeOH, an inseparable mixture of ketones 3a and 4a was obtained [36]. Reaction of 2,3-diamino6-methylpyridine leads to a mixture of compounds 3b and 4b (83:17%). From this mixture, we isolated pure isomer 3b by recrystallization from dimethylformamide.
Oximes were synthesized via ketone precursors through a reaction with hydroxylamine. Treatment of compounds 2a-i with hydroxylamine in hot EtOH in presence of NaOH led to the 11H-indeno[1,2-b]quinoxalin-11-one oximes (6a-i) (Scheme 1A). Likewise, oximes 7a, 7b, 8, and tryptanthrin-6-oxime were synthesized from ketones 3a, 3b, 5, and tryptanthrin, respectively, using hydroxylamine hydrochloride in EtOH or pyridine (Scheme 1B). For the synthesized oximes, the ratio of the Z- and E-isomers was determined from the integral intensities of the signals in 1H-NMR spectra. We found that compounds 6a-d, 6f-i, 8, and tryptanthrin-6-oxime were formed as individual isomers, while a mixture of Z- and E-isomers (90:10) was obtained for oxime 7a. The Z- and E-isomers of oximes 6e and 7a,b exist in dynamic equilibrium in solution and could not be isolated as individual forms. We speculate that the Z-isomer is predominant for the synthesized oximes, since it must be stabilized by an intramolecular H-bond between the OH group and the nitrogen atom of the pyrazine ring.
A convenient synthetic route to the O-substituted derivatives was synthesis from the corresponding ketone 1 by an oximation reaction with O-R-hydroxylamines (Scheme 1C). In the present work, we carried out the oximation of compound 1 using O-methyl, O-ethyl, O-benzyl, and O-allyl hydroxylamine hydrochlorides. According to the 1H-NMR spectra, products 9a-d synthesized according to Scheme 1C were isomerically pure individual compounds.
We also investigated the reactivity of IQ-1 towards alkylating reagents. IQ-1 has low solubility in most organic solvents, thus alkylation was evaluated in dimethylsulfoxide (DMSO); solubility of IQ-1 in this solvent is about 0.01 M at room temperature. KOH was used as a base. Being an aprotic solvent, DMSO easily solvates the potassium cations, while the OH-anions are solvated slightly, which leads to an extremely high basicity of the medium and activates the alkylation process in the DMSO-KOH system [38]. We also used Na2CO3 as a base. O-alkylation of IQ-1 in DMSO was carried out according to Scheme 1D at room temperature and vigorous stirring in the presence of a two-fold molar excess of a base (threefold on the synthesis of the carboxylic acid 10b).
Compound 10a in CDCl3 solution exists as a mixture of Z- and E-isomers with respect to the exocyclic C=N bond, as two sets of side-chain proton signals are observed in the 1H-NMR spectrum. The ratio of isomers is approximately 1:2, as determined from the integral intensities in each pair of signals. According to DFT calculations [B3LYP/6–31+G(d,p)] of the 10a isomers, the E-isomer is thermodynamically more stable. The effect of the solvent (chloroform) was taken into account within the polarizable continuum model (PCM). We determined that for the 10a(Z)10a(E) equilibrium, ∆Go298 is equal to 5.23 kJ/mol. The 1H-NMR results for compound 10c showed two sets of the side chain proton signals of the isomers with 1:3 integral intensity. Similarly to ⇌ 10a, indenoquinoxaline 10c in chloroform has a more stable E-isomer. Thus, for the 10c(Z)⇌10c(E) process, ∆Go298 evaluated by DFT is 10.38 kJ/mol. Obviously, the mixture of Z- and E-isomers is formed on synthesis under the reaction conditions.
It should be noted that the use of DMSO-Na2CO3 instead of DMSO-KOH (Scheme 1D) led to a longer reaction time: complete alkylation of IQ-1 was attained in 9–10 hours. However, on the alkylation by ethyl chloroacetate, isomerically pure 10c was formed with only traces of the minor isomer present, in contrast to the method using the superbasic medium DMSO-KOH. Comparison of the 1H-NMR spectra of the isomer mixture and the individual isomer 10c synthesized in DMSO-KOH and DMSO-Na2CO3 systems, respectively, showed that in the latter case, the product consisted of the isomer that was predominant when DMSO-KOH medium was used. According to results of our DFT calculations presented above, this isomer has an E-configuration, and this product was used for further biological evaluation.
2.2. Structure–activity relationship (SAR) analysis for JNK1–3 binding affinity
All compounds were evaluated for their ability to bind to the three JNK isoforms in comparison with IQ-1, and the results presented in Table 1 demonstrated that the 11H-indeno[1,2-b]quinoxalin-11-one nucleus is an appropriate scaffold for JNK inhibitor development. Indenoquinoxalines 6e, 7a, and 10c exhibited Kd values in the nanomolar range for all three JNKs, with the most potent being 10c, which had even lower Kd values for JNK1 and JNK3 compared to IQ-1 [29]. Moreover, 10c had much higher specificity toward JNK1 and JNK3 (Kd values of 22 nM and 76 nM, respectively) versus JNK2 (Kd = 735 nM). To further evaluate the relative potency of 10c, we compared its binding affinity with that of a commercially available JNK inhibitor, SP600125. As shown in Table 1, the Kd of 10c toward JNK1 was even lower to that of SP600125.
Table 1.
Chemical structures of synthesized oxime derivatives, their binding affinity, and effect on LPS-induced NF-κB/AP-1 transcriptional activity and interleukin-6 production.
 |
 |
 |
 |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Compd | R | R1 | R2 | R3 | JNK1 | JNK2 | JNK3 | NF-κB/ AP-1 |
IL-6 |
| Kd (µM) | IC50 (µM) | ||||||||
| SP600125a | 0.10 ± 0.043 | 0.084 ± 0.023 | 0.022 ± 0.009 | 3.4 ± 0.5 | 5.2 ± 1.2 | ||||
| IQ-1 | H | H | H | H | 0.24 ± 0.11 | 0.36 ± 0.054 | 0.10 ± 0.036 | 2.3 ± 0.4 | 3.8 ± 0.8 |
| 6a | H | H | H | CH3 | 3.3 ± 0.9 | 2.5 ± 0.7 | 6.0 ± 1.1 | 3.0 ± 0.9 | 0.5 ± 0.2 |
| 6b | H | Cl | H | Cl | N.A. | N.A. | N.A. | N.A. | N.A. |
| 6c | H | CH3 | CH3 | H | 2.7 ± 0.4 | 2.5 ± 0.6 | 1.3 ± 0.2 | 2.9 ± 0.8 | 0.6 ± 0.2 |
| 6d | H | COOH | H | H | 2.1 ± 0.3 | 6.0 ± 0.2 | 3.9 ± 0.6 | 24.6 ± 5.7 | 11.1 ± 2.3 |
| 6e | H | H | CH3 | H | 0.58 ± 0.035 | 0.84 ± 0.17 | 0.34 ± 0.014 | 3.9 ± 1.2 | 0.9 ± 0.3 |
| 6f | H | H | OC2H5 | H | 2.3 ± 0.57 | 2.0 ± 0.14 | 0.91 ± 0.13 | 5.9 ± 1.7 | 6.7 ± 2.2 |
| 6g | H | H | NO2 | H | 6.0 ± 0.21 | 12.0 ± 1.41 | 4.3 ± 0.28 | 2.7 ± 0.7 | 3.0 ± 1.2 |
| 6h | H | H | CF3 | H | N.A. | N.A. | N.A. | N.A. | N.A. |
| 6i | H | Cl | H | H | 1.1 ± 0.2 | 21.5 ± 7.8 | 0.71 ± 0.11 | 5.5 ± 1.6 | 1.2 ± 0.4 |
| 7a | 0.24 ± 0.057 | 0.36 ± 0.092 | 0.39 ± 0.014 | 4.3 ± 1.1 | 3.5 ± 1.3 | ||||
| 7b | 5.3 ± 1.3 | 6.4 ± 0.1 | 2.9 ± 0.1 | 3.3 ± 1.1 | 6.7 ± 2.2 | ||||
| 8 | 0.95 ± 0.071 | 1.4 ± 0.14 | 0.28 ± 0.042 | 5.4 ± 0.4 | 2.1 ± 0.8 | ||||
| 9a | CH3 | H | H | H | N.A. | N.A. | N.A. | N.A. | N.A. |
| 9b | CH2CH3 | H | H | H | N.A. | N.A. | N.A. | N.A. | N.A. |
| 9c | CH2Ph | H | H | H | N.A. | N.A. | N.A. | N.A. | N.A. |
| 9d | CH2CH=CH2 | H | H | H | N.A. | N.A. | N.A. | N.A. | N.A. |
| 10a | i-Bu | H | H | H | N.A. | N.A. | N.A. | N.A. | N.A. |
| 10b | CH2COOH | H | H | H | 23.5 ± 12.0 | 16.5 ± 0.7 | 28.5 ± 0.7 | N.A. | N.A. |
| 10c | CH2COOCH2CH3 | H | H | H | 0.022 ± 0.006 | 0.735 ± 0.19 | 0.076 ± 0.006 | 2.5 ± 0.8 | 3.3 ± 0.7 |
| 10d | (CH2)2OH | H | H | H | 1.15 ± 0.071 | 1.25 ± 0.071 | 0.31 ± 0.042 | 4.5 ± 1.8 | 5.9 ± 1.8 |
| Trp | 23.0 ± 1.4 | N.A. | N.A. | N.A. | N.A. | ||||
| Trp-Ox | 0.15 ± 0.081 | 1.0 ± 0.14 | 0.275 ± 0.21 | 3.8 ± 1.1 | 3.2 ± 1.2 | ||||
JNK binding data for known JNK inhibitors SP600125 (anthra[1–9-cd]pyrazol-6(2H)-one) and IQ-1 are taken from [29]. N.A., no inhibition at concentrations <40 µM; N.T., nontoxic at concentrations <40 µM. Trp, tryptanthrin; Trp-Ox, tryptanthrin-6-oxime. For biological experiments, oximes 6b and 6h were used in the form of sodium salts.
As reported previously [29], we found that the side chain oxime R substituent was critical for JNK binding and biological activities. The observation that oxime derivatives 9a-d and 10a, which have hydrocarbon side chains, were inactive in the competition binding assay suggests that the R oxime substituent is involved in H-bond donor/acceptor interactions with JNK. These interactions occur possibly due to the presence of additional oxygen atoms in the carboxyl, ester, or OH groups of molecules 10b-d, which can be anchored in the binding site in a favorable conformation, as shown below for 10c. Although oxime groups may contribute important interactions in the JNK binding site, the tetracyclic nucleus seems to be responsible for proper ligand positioning. Indeed, substitution of an aromatic carbon atom at position 6 with nitrogen led to less active compound 8 with all JNK isoforms. On the other hand, substitution of a carbon atom at position 8 with nitrogen (7a) or introduction of a CH3 group as the R2 substituent (6f) had little effect on binding affinity with all three JNK isoforms. Other modifications of the tetracyclic nucleus, including introduction of CH3 at R3 (6a), OCH2CH5 or NO2 at R2 (6f and 6g, respectively), COOH at R1 (6d), and two CH3 groups at R1 and R2 (6d), led to compounds with relatively low JNK binding affinity. Furthermore, 6h containing a CF3 group at R2 and 6b with two Cl atoms at R1/R3 were completely inactive. The most interesting modification of the tetracyclic nucleus was the introduction of Cl at position R1 (6i), as it led to an increase in relative specificity toward JNK1/JNK3 versus JNK2.
The natural alkaloid tryptanthrin has an indolo(2,1-b)quinazoline-6,12-dion nucleus, which is analogous to the 11H-indeno[1,2-b]quinoxalin-11-one scaffold. Indeed, charge distributions in IQ-1 and tryptanthrin-6-oxime molecules are very similar (Fig. 2), although the latter has a very polar carbonyl group, which results in lower hydrophobicity (LogP values are 4.04 and 2.92 for IQ-1 and tryptanthrin oxime, respectively). Thus, we also evaluated JNK binding activity of this IQ-1 analog. Although tryptanthrin was inactive for JNK2/JNK3 and had a very low binding affinity for JNK1 (Kd ~23.0 µM), tryptanthrin-6-oxime exhibited high binding affinity for JNK1 and JNK3 (Table 1).
Figure 2.

Electrostatic potential maps for IQ-1 (11H-indeno[1,2b] quinoxalin-11- one oxime) (left) and tryptanthrin-6-oxime (right). Different colors correspond to the potential values between −0.020 (red) and +0.002 (blue).
2.3. Kinase inhibition profile of tryptanthrin-6-oxime
Since tryptanthrin-6-oxime demonstratedd high affinity for JNKs, we evaluated its specificity for various other kinases to evaluate its specificity compared to IQ-1. Specifically, it was profiled in a competition binding assay for its ability to compete with an active-site directed ligand for 97 different kinases (KINOMEscan, Eurofins Pharma Discovery, San Diego), representing all known kinase families. The panel included 10 kinases that were reported previously to be targets of SP600125 with similar or greater potency than the JNKs [39]. Tryptanthrin-6-oxime was screened at 10 µM, and the kinases for which >90% inhibition of ligand binding and kinase activity was observed were designated as “kinase targets of the compound.” Five such kinase targets were identified, including casein kinase 1 σ (CK1σ, gene symbol CSNK1D), tropomyosin‐related kinase A (TRK-A, gene symbol NTRK1), JNK1, JNK2, and JNK3 (Fig. 3). Thus, similar to IQ-1 [30], tryptanthrin-6oxime had high specificity for inhibition of human JNK isoforms. Note however, that IQ-1 and IQ-3 (both potent JNK inhibitors with an indenoquinoxaline scaffold) did not bind TRK-A [29, 30]. Because TRKA-C are important targets for treatment of several tumors [40–42], the parent tryptanthrin and tryptanthrin-6-oxime were evaluated for their binding affinities (Kd) to these 3 kinases. We found that tryptanthrin-6-oxime had higher affinity toward TRKA-C in comparison with the parent alkaloid (Table 2).
Figure 3.

Kinase profile of tryptanthrin-6-oxime. Shown is the percentage inhibition of binding to an active-site directed ligand for each of the indicated kinases after treatment with 10 µM tryptanthrin-6oxime.
Table 2.
Binding affinity of tryptanthrin and tryptanthrin-6oxime toward TRKA-C isoforms.
| Compound | Kd (µM) | ||
|---|---|---|---|
| TRKA | TRKB | TRKC | |
| Tryptanthrin | 9.2 ± 1.1 | 7.2 ± 2.6 | N.A. |
| Tryptanthrin-6-oxime | 1.1 ± 0.2 | 3.8 ± 0.2 | 13.0 ± 2.1 |
Activity of TRK-family proteins (TRKA-C) is associated with poor survival in many types of cancer [43]. For example, TRK-A, a high affinity receptor for nerve growth factor (NGF) has been associated with the development of epithelial ovarian cancer [44]. Brain-derived neurotrophic factor (BDNF) is a potent neurotrophic factor that has been shown to stimulate breast cancer cell growth and metastasis via TRK-A and TRK-B [45]. Several compounds, including crizotinib and entrectinib, have been shown to inhibit the growth of tumor cells that express TRK-family fusion proteins and have demonstrated remarkable clinical response in patients with TRK-A fusion-positive tumors [46–48]. Sharma et al. [49] reported that some oxime derivatives of tryptanthrin exhibited anticancer activity in vitro against a panel of human cancer cell lines, but mechanisms of this activity are still non-identified. To our knowledge, this is first report demonstrating co-activity of a kinase inhibitor toward TRK and JNK isoforms. Using a selectivity score S(10), based on >90% inhibition of ligand binding at a single 10 µM screen concentration [50], we found that the S(10) for tryptanthrin-6-oxime was much lower (0.015 = 5/99) compared with the S(10) for SP600125 (0.328 = 39/119) [51], indicating much higher target kinase selectivity for tryptanthrin-6-oxime.
2.4. Molecular modelling
To further characterize our most active analogs, we performed docking studies of 10c and tryptanthrin-oxime into the binding sites of the three JNK isoforms. Since tryptanthrin was inactive, we were also able to directly compare binding of the inactive parent and active oxime derivative. According to our modelling, tryptanthrin formed a weak H-bond with Asn114 on binding with JNK1. At the same time, the highest partial interaction energy of this molecule was observed with Met111, which was due to van der Waals forces. The docking pose of tryptanthrin-6-oxime (Fig. 4) was characterized by strong H-bonding between the oxygen atom of the amide group and Met111. It should be noted that Met111 is considered as an important residue for small molecule interactions with JNK [52, 53]. The calculated docking score for tryptanthrin-6-oxime was about 15 kcal/mol more negative than for tryptanthrin, which may explain the higher binding affinity of the oxime derivative. Docking studies of these compounds to JNK2 showed that the parent alkaloid did not form H-bonds with any of the residues of this kinase, retaining in the binding center only by non-valent interactions. The highest attraction with a score of 14 kcal/mol was obtained for His149. In contrast, tryptanthrin-6-oxime was Hbonded with JNK2 through its oxime group with Gly171 (Fig. 4). Docking scores for tryptanthrin and its oxime derivative differed by 16 kcal/mol in favor of the oxime. According to the docking results obtained for JNK3, the low-energy pose for tryptanthrin formed a weak Hbond through its amide oxygen with Asn152 and was fixed in the binding site mainly by van der Waals interactions. On the other hand, tryptanthrin-6-oxime was anchored in the kinase cavity through H-bonding of the oxime group with Asp207 (Fig. 4). The docking score of the oxime in JNK3 was ~ 30 kcal/mol more negative than that of tryptanthrin. Thus, it can be assumed that, at least for JNK2 and JNK3, the introduction of an oxime moiety into the molecule of tryptanthrin caused the formation of a new H-bond with the kinase through participation of this moiety.
Figure 4.
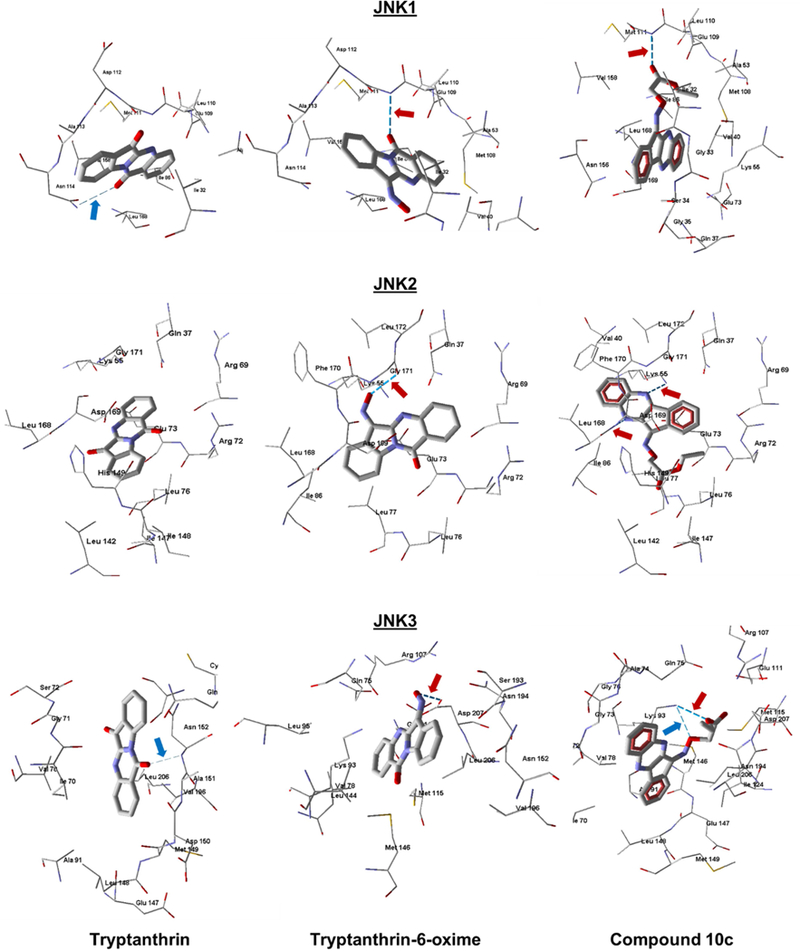
Docking poses of tryptanthrin (left), tryptanthrin-6-oxime (middle), and 10c (right) in JNK1 (PDB code 1UKI), JNK2 (PDB code 3NPC), and JNK3 (PDB code 1PMV). Strong H-bonds are shown as darker dashed lines and indicated by red arrows. Weak H-bonds are shown as light dashed lines and indicated by blue arrows. Residues within 4 Å from each pose are shown.
Docking of the highly active compound 10c in JNK1 gave a pose similar to tryptanthrin6-oxime, meaning that the molecule formed a strong H-bond with Met111 via the ester group of the ligand (Fig. 4). It is important that such an arrangement of the ester group is achieved for the Z-isomer of 10c. We also performed docking of the E-isomer, but another pose with a markedly worse docking score was obtained in this case. In its unbound form, the E-isomer of 10c is more stable; however, our DFT calculations show that the Z-isomer in solution is only slightly higher in energy than the E-isomer of the substituted oxime 10c. Obviously, when interacting with the kinase, 10c adopts the Z-configuration, which binds more effectively to the JNK1 active site, and in general, a gain in energy is achieved. When docking compound 10c in the JNK2 binding site, a more energy-efficient pose was obtained for the E-isomer (by 20.4 kcal/mol better according to the docking score) than for the Z-isomer. Compound 10c forms two strong H-bonds with Lys55 and Leu168 of JNK2 with participation of two nitrogen atoms in the heterocycle. It should be noted that the oxygen of the oxime group is located near one of these nitrogen atoms and forms an H-bond with Gly171 located in the vicinity of Lys55 for the pose of tryptanthrin-6-oxime (Fig. 4). We determined that 10c binds JNK3 in the form of the Z-isomer (by 60.2 kcal/mol lower in docking score than the corresponding E-isomer), forming two H-bonds to Lys93 with participation of the oxime and ethoxy oxygen atoms. In the pose of tryptanthrin-6-oxime, the oxygen atom, although located in the same region of space, forms an H-bond with Asp207 (Fig. 4). Consequently, there is a similarity in the location of the most active compounds (10c and tryptanthrin-6-oxime) by their location within the binding sites of the three JNK isoforms. Note that these molecules occupy the same region of space where co-crystallized ligand SP600125 is located. As shown in Supplementary Fig. 2S, the tetracyclic moieties of all three compounds are approximately parallel within a narrow binding site of JNK3.
We also performed docking studies of tryptanthrin and tryptanthrin-6-oxime into the TRK-A binding site. The major difference between their docking poses was the presence of Hbonding between the oxime moiety of tryptanthrin-6-oxime and the kinase (Fig. 5). Specifically, the oxygen atom of the oxime group is strongly H-bonded with Asp596 and Arg599. Additionally, a weaker H-bonding interaction is possible between a nitrogen atom in the tetracyclic alkaloid derivative and Arg599. In contrast, tryptanthrin interacted with the kinase via van der Waals forces only, although a strong attraction of the ligand to Asp596 exists according to our calculations. The dissimilarity in docking modes of tryptanthrin and tryptanthrin-6-oxime is likely responsible for the difference in their binding affinities to TRPA-C (Table 2).
Figure 5.

Docking poses of tryptanthrin (left) and tryptanthrin-6-oxime (right) in the binding site of TRK-A (PDB code 4AOJ). Strong H-bonds are shown as darker dashed lines and indicated by red arrows. Weak H-bonds are shown as light dashed lines and indicated by blue arrows. Residues within 4 Å from each pose are shown.
2.5. Evaluation of compound biological activity
All compounds were evaluated for their ability to inhibit LPS-induced NF-κB/AP-1 reporter activity and interleukin (IL)-6 production in human monocytic THP-1Blue and MonoMac-6 cells, respectively. As shown in Table 1, the 13 oxime compounds inhibited LPSinduced NF-κB/AP-1 activity and IL-6 production. As examples, the dose-dependent inhibitory effects of 10c and tryptanthrin-6-oxime on NF-κB/AP-1 activity and IL-6 production are shown in Fig. 6. As expected, these compounds also inhibited c-Jun phosphorylation in treated cells derivatives (6a, 6c, 6e, 6f, 6i, 7a, 7b, 8, 10c, and tryptanthrin-6-oxime) all had IC50 values close to that of SP600125 for inhibition of LPS-induced NF-κB/AP-1 activity and IL-6 secretion in biological assays (Table 1). Consistent with the JNK binding assay, 6b, 6h, 9a-d, and 10a-b did not inhibit NF-κB/AP-1 activity or IL-6 production (Table 1; examples are shown in Fig. 6), supporting the specificity of our assays. In contrast to the active oximes, ketone derivatives (2a2j, 3a, 5) (data not shown), as well as tryptanthrin (Fig. 6), did not inhibit LPS-induced NFκB/AP-1 activity or IL-6 production, even at concentrations up to 50 µM.
Figure 6.
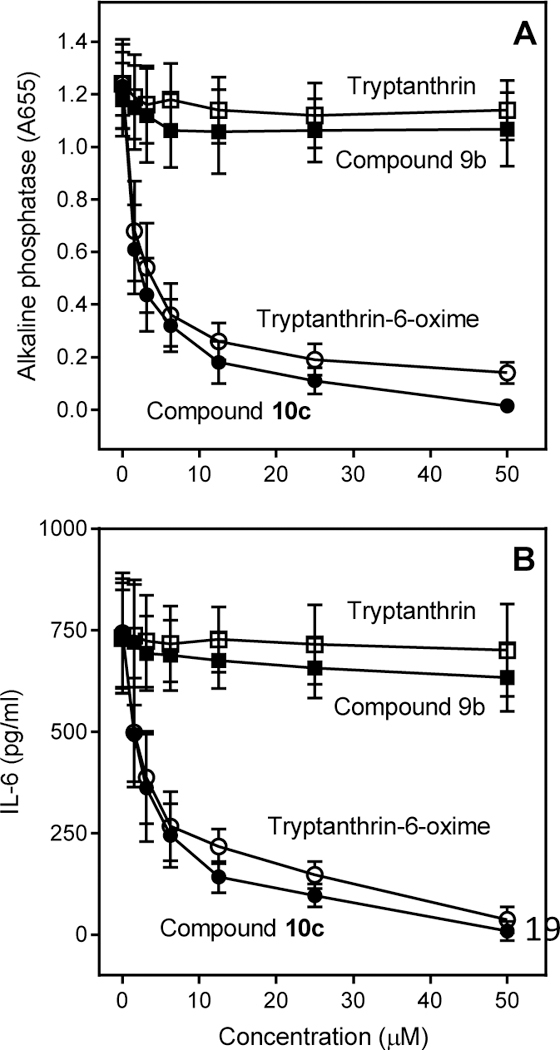
Effect of selected compounds on NF-κB/AP1 activation and IL-6 production. Panel A. THP-1Blue cells were pretreated with the indicated compounds or DMSO for 30 min, followed by addition of 250 ng/ml LPS or buffer for 24 h. NF-κB/AP-1 activation was monitored by measuring secreted alkaline phosphatase activity spectrophotometrically in the cell supernatants (absorbance at 655 nm). Panel B. MonoMac-6 cells were pretreated with the indicated compounds or DMSO for 30 min, followed by addition of 250 ng/ml LPS or buffer for 24 h. Production of IL-6 in the supernatants was evaluated by ELISA. The data are presented as the mean ± S.D. of triplicate samples from one experiment that is representative of three (examples shown in Fig. 7). Although compound 10c had higher JNK1/3 binding affinity than tryptanthrin-6-oxime, they both inhibited c-Jun phosphorylation, NF-κB/AP-1 activity, and IL-6 production over a similar concentration range (Figs. 6 and 7). This may be due to the higher cell permeability of tryptanthrin-6-oxime. It is also possible that JNK2 may play a greater role in these cellular responses and is targeted differently by the two inhibitors. Note that the oxime
To verify that the results were not influenced by possible toxicity, cytotoxicity of the compounds was evaluated at concentrations up to 50 µM in MonoMac-6 and THP-1Blue cells during a 24-h incubation with the compounds. None of the compounds affected cell viability, even at the highest tested concentrations, thereby verifying that these compounds were not cytotoxic during the 24-h incubation period of our assays (data not shown).
It should be noted that many aryl oxime derivatives, including IQ-1, release nitric oxide (NO) during their oxidoreductive bioconversion to ketones [31, 54, 55]. Thus, biological activities of NO and these ketone precursors, including trypthantrin, should also be considered in biological experiments. For example, although compound 1 (ketone corresponding to IQ-1) has not been shown to be a DNA intercalator [56], the ketone precursors of compounds 9a, 6i, and 7a have cytotoxicity against some cancer cell lines, probably because of their topoisomerase I inhibitory activity [36]. Tryptanthrin is a natural alkaloid found in Polygonum tinctorium and Isatis tinctoria [57, 58] and has been reported to have various pharmacological effects, such as anti-inflammatory [59–61], antimicrobial [62], and anti-tumor activity [63, 64]. Tryptanthrin has also been reported to suppress NO and prostaglandin E synthesis in macrophages exposed to oxidative stress [65] and inhibit enzymatic activity of 5-lipoxygenase, cyclooxygenase-2, and indoleamine 2,3-dioxygenase [66–68]. Previously, several tryptanthrin derivatives with different substituents have been reported, including compounds with antiplasmodium and antitoxoplasma activities, inhibitors of indoleamine 2,3-dioxygenase, and DNA triplex stabilizing agents [68–73].
We synthesized novel 11H-indeno[1,2-b]quinoxalin-11-one oxime analogs and tryptanthrin-6-oxime (indolo(2,1-b)quinazoline-6,12-dion-6-oxime) and evaluated their effects on JNK activity. Several compounds exhibited sub-micromolar JNK binding affinity and were selective for JNK1/JNK3 versus JNK2. The most potent compounds were 10c (11H-indeno[1,2b]quinoxalin-11-one O-(O-ethylcarboxymethyl) oxime) and tryptanthrin-6-oxime, which had dissociation constants (Kd) for JNK1 and JNK3 of 22 and 76 nM and 150 and 275 nM, respectively. Molecular modeling suggested a mode of binding interaction at the JNK catalytic site and that the selected oxime derivatives were potentially competitive JNK inhibitors. JNK binding activity of the compounds correlated with their ability to inhibit lipopolysaccharide (LPS)-induced nuclear factor-κB/activating protein 1 (NF-κB/AP-1) activation in human monocytic THP-1Blue cells and interleukin-6 (IL-6) production by human MonoMac-6 cells. Thus, oximes with indenoquinoxaline and tryptanthrin nuclei can serve as specific smallmolecule modulators for mechanistic studies of JNK, as well as potential leads for the development of anti-inflammatory drugs.
3. Conclusion
Synthesis and analysis of novel 11H-indeno[1,2-b]quinoxalin-11-one oxime analogs and tryptanthrin-6-oxime demonstrated that several of these compounds had high affinity for JNK and were selective for JNK1/JNK3 versus JNK2. These analogs also inhibited LPS-induced nuclear NF-κB/AP-1 activation and IL-6 production in human monocytic cells. Our molecular modeling showed that oxygen atoms of the oxime or ester groups participated in the formation of strong H-bonds with residues in the JNK1/JNK3 binding sites. Thus, it is reasonable to suggest that further modification of the O-substituent in the oxime moiety could lead to more specific inhibitors with higher JNK selectivity. Our results also suggest that pan-JNK inhibition may be suitable for suppression of the production of proinflammatory cytokines by human monocytic cells. Finally, the identified oximes represent new chemical tools that may be useful in further development of JNK and/or TRK inhibitors and could find application in the treatment of inflammatory diseases, neurodegenerative pathologies, and cancer.
4. Experimental section
4.1. Chemistry
4.1.1. Reagents and general procedures
Indenoquinoxaline ketones 2b, 2d, 2f, and 2g were purchased from Vitas-M Laboratory (Moscow, Russia); 2e and 2h were from Maybridge (Cornwall, United Kingdom); and 2i was from Specs (Delft, The Netherlands). Tryptanthrin was purchased from Combi-Blocks (San Diego, CA). All other starting reagents were purchased from Sigma Aldrich. The chemicals were analytical grade and used without further purification. Compounds 1 (11H-indeno[1,2b]quinoxalin-11-one) and IQ-1 (11H-indeno[1,2-b]quinoxalin-11-one oxime) were synthesized, as described previously [32]. Ketone 2a was synthesized according to [34, 35], and compound 2c was synthesized according to [36]. Reaction progress was monitored by thin-layer chromatography (TLC) with UV detection using pre-coated silica gel F254 plates (Merck) or a Silufol UV-254. The synthesized structures were confirmed on the basis of analytical and spectral data. The melting points (m.p.) were determined using an electrothermal Mel-Temp capillary melting point apparatus. Elemental analysis was performed with a Carlo Erba instrument. GC-MS analysis was performed on an Agilent 7890A GC combined with an Agilent 5975C mass detector (Agilent Technologies, USA); carrier gas was helium. LC-MS analysis was performed on an Agilent 1260 Infinity combined with an Agilent 6530 Accurate Mass Q-TOF detector. Compounds dissolved in 3-nitrobenzyl alcohol were subjected to fast atom bombardment (FAB) ionization using a 10 kV argon beam, and the mass spectra were recorded with a VG 70–70 EQ spectrometer. IR spectra were recorded on a FT-IR spectrometer Nicolet 5700 with KBr pellets. 1H NMR spectra were recorded on Bruker 400 or 600 MHz spectrometers. For atom numbering detaails, see Supplementary Fig. 3S. Representative NMR spectra for compounds 8 and 10c are provided in supplementary material.
4.1.2. 6-Methyl-11H-indeno[2,3-b]quinoxalin-11-one oxime (6a) and general procedure for the synthesis of 6b-i.
A mixture of 2a (2.17 g, 9.45 mmol), hydroxylamine hydrosulfate (3.06 g, 23.6 mmol), and NaOH (1.0 g, 25 mmol) in EtOH (100 mL) was heated for 8 h at 60 oC. After cooling, the mixture was poured into water (600 mL), the precipitate was filtered out, dried, and recrystallized from EtOH. Yield 2.26 g (92%). M.p. 303–304°. 1H NMR (600 MHz, DMSO-d6), δ, ppm: 2.85 (s, 3H, CH3), 7.38–7.64 (m, 4H, H-2, H-3, H-7, H-8), 7.95 (d, 1H, 3J = 8 Hz, H-9), 8.20 (d, 1H, 3J = 7.6 Hz, H-4), 8.85 (d, 1H, 3J = 7.6 Hz, H-1), 13.31 (s, 1H, OH). M.w. 261.29. C16H11N3O. LC-MS – m/z (I, %): 262.04377 (100) [MH]+; 244.03701 (83) [MH – H2O]+. FABMS – m/z (I, %): 262 (100) [MH]+. A similar procedure was used for synthesis of the following 11H-indeno[2,3-b]quinoxaline-11-one oximes. To isolate sodium oximates of 6b and 6h, a 2fold excess of NaOH was added (with respect to hydroxylamine salt) after completion of the reaction.
4.1.3. 6,8-Dichloro-11H-indeno[2,3-b]quinoxalin-11-one oxime (6b).
Yield of sodium salt 93%.M.p. 344о. 1H NMR (600 MHz, DMSO-d6), δ, ppm: 7.55–7.70 (m, 2H, H-2, H-3), 7.77 (s, 1H, H7), 7.92 (s, 1H, H-9), 8.2 (d, 1H, 3J = 8 Hz, H-4), 8.55 (d, 1H, 3J = 7.5 Hz, H-1). M.w. 338.13. C15H6Cl2N3NaO. LC-MS – m/z (I, %): 316.91 (100) [MH]+; 298.84 (30) [MH – H2O]+.
4.1.4. 7,8-Dimethyl-11H-indeno[2,3-b]quinoxalin-11-one oxime (6c).
Yield 89%. M.p. 323–324°. 1H NMR (600 MHz, DMSO-d6), δ, ppm: 2.46 (s, 3H, CH3), 2.50 (s, 3H, CH3), 7.66–7.73 (m, 2H, H-2, H-3), 7.90 (s, 1H, H-7 or H-9), 7.91 (s, 1H, H-7 or H-9), 8.14 (d, 3J = 8 Hz, H-4), 8.53 (d, 1H, 3J = 8 Hz, H-1), 13.26 (s, 1H, OH). M.w. 275.31. C17H13N2O. LC-MS – m/z (I, %):276.0828 (100) [MH]+. FAB-MS – m/z (I, %): 276 (100) [MH]+; 258 (40) [MH – H2O]+.
4.1.5. 8-Carboxy-11H-indeno[2,3-b]quinoxalin-11-one oxime (6d).
Yield 90%. M.p. 323–324°.1H NMR (600 MHz, CDCl ), δ, ppm: 7.68–7.76 (m, 2H, H-2, H-3), 8.03 (d, 1H, 3J = 7 Hz, H-6),8.20 (d, 1H, 3J = 8 Hz, H-4), 8.32 (d, 1H, 3J = 7 Hz, H-7), 8.58 (s, 1H, H-9), 8.60 (d, 1H, 3J = 7 Hz, H-1), 13.65 (s, 1H, =N-OH). M.w. 291.27. C16H9N3O3. LC-MS – m/z (I, %): 292.03225 (100) [MH]+; 274.02414 (50) [MH – H2O]+.
4.1.6. 7-Methyl-11H-indeno[2,3-b]quinoxaline-11-one oxime (6e).
Yield 80%. M.p. 297–298°.1H NMR (600 MHz, DMSO-d6), δ, ppm: 2.58 (s, CH3), 7.65–7.74 (m, 3H, H-2, H-3, H-8), 7.94 (s, 1H, H-6), 8.03 (d, 1H, 3J = 7.6 Hz, H-9), 8.17 (d, 1H, 3J = 7 Hz, H-4), 8.55 (1H, 3J = 7 Hz, H1), 13.30 (s, OH). M.w. 261.29. C16H11N3O. LC-MS – m/z (I, %): 262.27436 (100) [MH]+. FABMS – m/z (I, %): 262 (100) [MH]+.
4.1.7. 7-Ethoxy-11H-indeno[2,3-b]quinoxalin-11-one oxime (6f).
Yield 87%. M.p. 303°−304°. 1H NMR (600 MHz, DMSO-d6), δ, ppm: 1.44 (t, 3H, 3J = 5 Hz, CH3), 4.26 (q, 2H, 3J = 5 Hz, CH2), 7.30–7.59 (m, 3H, H-2, H-3, H-8), 8.00 (d, 1H, 3J = 7.5 Hz, H-9), 8.06 (s, 1H, H-6), 8.15 (d, 1H, 3J = 7 Hz, H-4), 8.78 (d, 1H, J = 7 Hz, H-1), 13,17 (s, 1H, OH). M.w. 291.31. C17H13N3O2. LCMS – m/z (I, %): 292.07027 (100) [MH]+; 274.06221 (41) [MH – H2O]+. FAB-MS – m/z (I, %):292 (100) [MH]+.
4.1.8. 8-Nitro-11H-indeno[2,3-b]quinoxalin-11-one oxime (6g).
Yield 84%. M.p. > 360°. 1H NMR (600 MHz, DMSO-d6), δ, ppm: 7.42–7.67 (m, 2H, H-2, H-3), 8.18 (d, 1H, 3J = 7 Hz, H-4),8.31 (d, 1H, 3J = 7 Hz, H-6), 8.87 (d, 3J = 8 Hz, H-1), 8.90 (d, 3J = 7 Hz, H-7), 8.99 (s, 1H, H-9), 13.47 (s, 1H, OH). M.w. 292.26. C15H8N4O3. LC-MS – m/z (I, %): 293.54 (100) [MH]+; 275.50 (41) [MH – H2O]+.
4.1.9. 8-Trifluoromethyl-11H-indeno[2,3-b]quinoxalin-11-one oxime (6h).
Yield of sodium salt 81%. M.p. 303 . H NMR (600 MHz, DMSO-d6), δ, ppm: 7.60–7.75 (m, 2H, H-2, H-3), 8.02 (d, 1H, 3J = 7 Hz, H-6), 8.26 (d, 1H, 3J = 8 Hz, H-4), 8.34 (d, 1H, 3J = 7 Hz, H-7), 8.48 (s, 1H, H-9), 8.74 (d, 1H, 3J = 8 Hz, H-1). M.w. 337.24. C16H7F3N3NaO. LC-MS – m/z (I, %): 316.04372 (100) [MH]+.
4.1.10. 7-Chloro-11H-indeno[2,3-b]quinoxalin-11-one oxime (6i).
Yield 89%. M.p. 323–324°. 1H NMR (600 MHz, DMSO-d ), δ, ppm: 7.47–7.64 (m, 2H, H-2, H-3), 7.76 (d, 1H, 3J = 8 Hz, H-8), 8.13 (d, 1H, 3J = 8 Hz, H-9), 8.21 (d, 1H, 3J = 7 Hz, H-4), 8.85 (d, 1H, 3J = 7 Hz, H-1), 13.64 (s, 1H, OH). M.w. 281.70. C15H8ClN3O. LC-MS – m/z (I, %): 282.14929 (100) [MH]+.
4.1.11. 6H-Indeno[1,2-b]pyrido[3,2-e]pyrazin-6-one (3a).
A mixture of 2,2-dihydroxyindane1,3-dione (ninhydrin, 0.39 g, 2.2 mmol) and 2,3-diaminopyridine (0.22 g, 2.0 mmol) in EtOH (50 mL) was heated for 10 h (TLC monitoring) at 60 °C. The mixture was then cooled, and the resulting precipitate (mixture of isomers 3a and 4a, 90:10 %) was filtered and recrystallized from EtOH to give 3a (0.50 g, 81% yield) as a yellow solid. M.p. 266–268°С. 1H NMR (500 MHz, CDCl3), δ, ppm: δ 7.68 (td, 1H, 3J = 7.5 Hz, 4J = 1 Hz, H-8), 7.73 (dd, 1H, 3J = 8 Hz, 4J = 4.5 Hz, H-3), 7.83 (td, 1H, 3J =7.5 Hz, 4J = 1 Hz, H-9), 7.97 (d, 1H, 3J = 7.5 Hz, H-7), 8.25 (d, 1H, J = 7.5 Hz, H-10), 8.60 (dd, 1H, 3J = 8 Hz, 4J = 2 Hz, H-4), 9.17 (dd, 1H, 3J = 4.5 Hz, 4J = 1.5 Hz, H2). NMR 13C (125 MHz, CDCl3), δ, ppm: 123.8 (C-10), 125.1 (C-3), 125.6 (C-7), 133.6 (C-8), 137.4 (C-6a), 137.4 (C-9), 138.3 (C-4a), 140.4 (C-4), 141.2 (C-10a), 150.6 (C-5a), 152.0 (C11a), 155.6 (C-2), 160.0 (C-10b), 188.8 (C-6). IR bands, cm−1: 1720 (C=O), 1612, 1602, 1502, 1153, 787. Found, %: C 72.24, H 3.18, N 18.30. C14H7N3O. Calculated, %: C 72.10, H 3.03, N 18.02.
4.1.12. 3-Chloro-6H-indeno[1,2-b]pyrido[3,2-e]pyrazin-6-one (3b).
Compound 3b was synthesized, as described under 4.1.1.12 from ninhydrin and 3,4-diamino-5-chloropyridine. Yield 58 %, M.p. 293–295. 1H NMR (400 MHz, CDCl3), δ, ppm: δ 7.72 (t, 1H, 3J = 7.2 Hz, H-8),7.87 (t, 1H, 3J = 7.2 Hz, H-9), 8.00 (d, 1H, 3J = 7.6 Hz, H-7), 8.26 (d, 1H, 3J = 7.6 Hz, H-10),8.55 (s, 1H, 4J = 2.4 Hz, H-4), 9.08 (s, 1H, 4J = 2.4 Hz, H-2). IR bands, cm−1: 1725 (C=O), 1610, 1596, 1481, 1134, 803, 752. Found, %: C 62.93, H 2.04, N 15.85. C14H6ClN3O. Calculated, %: C 62.82, H 2.26, N 15.70.
4.1.13. 10H-Indeno[1,2-b]pyrido[3,4-e]pyrazin-10-one (5).
Compound 5 was synthesized, as described under 4.1.1.12 from ninhydrin and 3,4-diaminopyridine. Yield 72%, M.p. 270–272°С. 1H NMR (600 MHz, CDCl ), δ, ppm: δ 7.72 (t, 1H, 3J = 7.8 Hz, H-8), 7.86 (t, 1H, 3J = 7.8 Hz, H37), 7.99 (d, 1H, 3J = 5 Hz, H-4), 8.00 (d, 1H, 3J = 7.2 Hz, H-9), 8.19 (d, 1H, 3J = 7.8 Hz, H-6), 8.89 (d, 1H, 3J = 5 Hz, H-3), 9.62 (s, 1H, H-1). NMR 13C (150 MHz, CDCl3), δ, ppm: 122.4 (C-4), 123.6 (C-6), 125.3 (C-9), 134.0 (C-8), 137.4 (C-7), 137.7 (C-11a), 137.8 (C-9a), 140.7 (C5b), 146.5 (C-4a), 149.8 (C-3), 151.2 (C-10a), 155.8 (C-1), 160.4 (C-5a), 188.5 (C-10). IR bands, cm−1: 1728 (C=O), 1559, 1572, 1430, 1123, 744. Found, %: C 72.37, H 2.89, N 18.24. C14H7N3O. Calculated, %: C 72.10, H 3.03, N 18.02.
4.1.14. 6H-Indeno[1,2-b]pyrido[3,2-e]pyrazin-6-one oxime (7a).
A mixture of 3a (0.41 g, 1.74 mmol) and hydroxylamine hydrochloride (0.30 g, 4.33 mmol) in EtOH (50 mL) was heated for 10 h (TLC monitoring) at 60 °C. The mixture was then cooled and poured into H2O (500 mL).The resulting precipitate was filtered, washed with water, and recrystallized from EtOH to give 7a (0.35 g, 80% yield) as a colorless solid. M.p. 291–293°С. 1H NMR (400 MHz, pyridine-d5), δ, ppm: 7.65 (td, 1H, 3J = 7.6 Hz, 4J = 1.2 Hz, H-8), 7.71 (td, 1H, 3J = 7.6 Hz, 4J = 1.2 Hz, H-3),8.06 (d, 1H, J = 5.6 Hz, H-9), 8.35 (d, 1H, 3J = 7.2 Hz, H-7), 8.92 (d, 1H, 3J = 5.6 Hz, H-10), 8.96 (d, 1H, 3J = 7.6 Hz, H-4), 9.78 (1H, H-2). NMR 13C (100 MHz, pyridine-d5), δ, ppm: 122.6,123.5, 129.6, 132.1, 133.7, 135.4, 138.0, 145.9, 147.9, 148.2, 153.6, 155.9, 157.5. IR bands, cm-1:1631 (C=N), 1604, 1575, 1470, 1381 (O–H), 1096, 905 (N–O), 776. Found, %: C 67.47, H 3.02, N 22.69. C14H8N4O. Calculated, %: C 67.74, H 3.25, N 22.57.
4.1.15. 3-Chloro-6H-indeno[1,2-b]pyrido[3,2-e]pyrazin-6-one oxime (7b).
Compound 7b was obtained from 3b and hydroxylamine hydrochloride, as described under 4.1.1.15. Yield 56%,M.p. 296–298°C. 1H NMR (400 MHz, DMSO-d6), δ, ppm: 7.77 (m, 2H, H-9, H-8), 8.25 (dd, 1H,3J = 8 Hz, J = 4 Hz, H-10), 8.57 (dd, 1H, J = 8 Hz, J = 4 Hz, H-9), 8.77 (s, 1H, J = 2.8 Hz, H4), 9.13 (s, 1H, 4J = 2.4 Hz, H-2), 13.58 (s, 1H, OH). IR bands, cm−1: 1639 (C=N), 1559, 1572, 1475, 1371 (O–H), 1091, 951 (N–O), 784, 739 (C–Cl). Found, %: C 59.18, H 2.47, N 19.93. C14H7ClN4O. Calculated, %: C 62.82, H 2.26, N 15.70.
4.1.16. 10H-Indeno[1,2-b]pyrido[3,4-e]pyrazin-10-one oxime (8).
Compound 8 was obtained from 5 and hydroxylamine hydrochloride, as described under 4.1.1.15. Yield 78%, M.p. > 300°С. 1H NMR (400 MHz, pyridine-d5), δ, ppm: 7.66 (t, 1H, 3J = 5 Hz, H-8), 7.71 (t, 1H, 3J = 5 Hz, H-7), 8.05 (d, 1H, 3J = 5 Hz, H-4), 8.34 (d, 1H, 3J = 5 Hz, H-9), 8.88 (d, 1H, 3J = 5 Hz, H-6),8.91 (d, 1H, 3J = 5 Hz, H-3), 9.71 (s, 1H, H-1). NMR 13C (100 MHz, pyridine-d5), δ, ppm: 121.2,122.1, 128.2, 130.8, 132.4, 133.9, 136.5, 144.4, 146.4, 146.8, 152.2, 153.5, 156.0. IR bands, cm−1: 1631 (C=N), 1575, 1540, 1486 (C–N), 1362 (O–H), 1005, 956 (N–O), 814 (see Supplementary Figs. 6S and 7S for 1H NMR and 13C NMR spectra, respectively). Found, %: C 68.02, H 3.47, N 22.21. C14H8N4O. Calculated, %: C 67.74, H 3.25, N 22.57.
4.1.17. 6-(Hydroxyimino)indolo[2,1-b]quinazolin-12(6H)-one (tryptanthrin-6-oxime).
A mixture of tryptanthrin (2.48 g, 10 mmol) and hydroxylamine hydrochloride (2.09 g, 30 mmol) in 30 mL of pyridine was stirred at 60 °C for 2 h (TLC monitoring). The reaction mixture was poured into 300 mL of water and the resulting precipitate was filtered, washed with water, and dried to give 2.50 g (95 %) of a slightly yellow solid, m.p. 280–282 °C. NMR 1H (500 MHz, DMSO-d6), δ, ppm: 7.44 (td, 1H, 3J = 7.5 Hz, 4J = 1 Hz, H-8), 7.44 (td, 1H, 3J = 7.5 Hz, 4J = 1 Hz, H-9), 7.64 (td, 1H, 3J = 7.5 Hz, 4J = 1 Hz, H-2), 7.80 (d, 1H, 3J = 7.5 Hz, H-4), 7.87 (td, 1H, 3J = 7 Hz, 4J =1.5 Hz, H-3), 8.27 (dd, 1H, 3J = 8 Hz, 4J = 1.5 Hz, H-7), 8.35 (dd, 1H, 3J = 7.5 Hz, 4J = 0.5 Hz, H-1), 8.35 (d, 1H, 3J = 8 Hz, H-10), 13.63 (s, 1H, C=N–OH). NMR 13C (125 MHz, DMSO-d6) δ, ppm: 116.2 (C-10), 118.8 (C-6a), 121.5 (C-12a), 126.5 (C-8), 126.6 (C-7), 127.4 (C-9), 127.5 (C-1), 128.1 (C-4), 132.0 (C-2), 134.7 (C-3), 139.3 (C-10a), 144.2 (C-5a), 147.0 (C-4a), 148.3 (C-6), 158.5 (C-12). IR bands, cm−1: 3114, 1690 (C=N), 1592, 1448, 1354, 1326, 1268, 1227, 1196, 1127, 1084, 1037, 924, 774, 688, 662. Found, %: C 68.70, H 3.31, N 15.65. C15H9N3O2. Calculated, %: C 68.44, H 3.45, N 15.96.
4.1.18. 11H-indeno[1,2-b]quinoxalin-11-one O-methyl oxime (9a) and general procedure for synthesis of the 9b-d.
A mixture of 1 (0.120 g, 0.52 mmol) and O-methylhydroxylamine hydrochloride (0.216 g, 2.6 mmol) in EtOH (10 ml) was heated for 9 h (TLC monitoring) at 78 °C. The mixture was then cooled and poured into water (100 ml). The resulting precipitate was filtered, washed with water, and recrystallized from EtOH to give 9a (0.120 g, yield 88%) as a colorless solid. M.p. 172–174°. 1H NMR (400 MHz, CDCl3), δ, ppm: 4.32 (s, 3H, CH3), 7.457.57 (m, 2H, H-2, H-3), 7.70–7.61 (m, 2H, H-7, H-8), 8.03 (d, 1H, 3J = 7.6 Hz, H-9), 8.11 (d, 1H, 3J = 7.2 Hz, H-6), 8.17 (d, 1H, J = 7.6 Hz, H-4), 8.34 (d, 1H, J = 7.2 Hz, H-1). Found, %: C 73.82, H 4.14, N 15.86. C16H11N3O. Calculated, %: C 73.55, H 4.24, N 16.08. The same procedure was used for the synthesis of the following oximes from corresponding O-substituted hydroxylamine hydrochlorides (see Scheme 1) and 1.
4.1.19. 11H-Indeno[1,2-b]quinoxalin-11-one O-ethyl oxime (9b).
Yield 90%, a colorless solid. M.p. 169–170°. 1H NMR (400 MHz, CDCl3), δ, ppm: 1.46 (t, 3H, 3J = 7.2 Hz, CH3), 4.60 (q, 2H, 3J = 7.2 Hz, CH2), 7.47–7.57 (m, 2H, H-2, H-3), 7.61–7.70 (m, 2H, H-7, H-8), 8.04 (d, 1H, J = 7.6 Hz, H-9), 8.12 (d, 1H, 3J = 7.2 Hz, H-6), 8.19 (d, 1H, 3J = 8.2 Hz, H-4), 8.38 (d, 1H, 3J = 6.8 Hz, H-1). Found, %: C 74.29, H 4.65, N 15.01. C17H13N3O. Calculated, %: C 74.17, H 4.76, N 15.26.
4.1.20. 11H-indeno[1,2-b]quinoxalin-11-one O-benzyl oxime (9с).
Yield 92%, a colorless solid. M.p. 191–193o. 1H NMR (400 MHz, CDCl3), δ, ppm: 5.59 (s, 2H, CH2), 7.28–7.57 (m, 7H, H-2, H-3, C6H5), 7.62–7.71 (m, 2H, H-7, H-8), 8.05 (d, 1H, 3J = 8.4 Hz, H-9), 8.12 (d, 1H, 3J = 7.6 Hz, H-6), 8.19 (d, 1H, 3J = 7.6 Hz, H-4), 8.33 (d, 1H, 3J = 7.6 Hz, H-1). Found, %: C 78.63, H 4.26, N 12.71. C22H15N3O. Calculated, %: C 78.32, H 4.48, N 12.46.
4.1.21. 11H-Indeno[1,2-b]quinoxalin-11-one O-allyl oxime (9d).
Yield 91%, a colorless solid in the form of needles. M.p. 123–124o. 1H NMR (400 MHz, CDCl3), δ, ppm: 5.02 (d, 2H, 3J = 5.6 Hz, OCH2), 5.27 (d, 1H, 3J = 10.4 Hz, =CH2, Htrans), 5.40 (d, 1H, 3J = 17.2 Hz, =CH2, Hcis), 6.066.18 (m, 1H, CH=CH2), 7.40–7.53 (m, 2H, H-2, H-3), 7.58–7.67 (m, 2H, H-7, H-8), 8.00 (d, 1H, 3J = 8.4 Hz, H-9), 8.06 (d, 1H, J = 6.8 Hz, H-6), 8.15 (d, 1H, J = 8 Hz, H-4), 8.33 (d, 1H, 3J = 7.2 Hz, H-1). Found, %: C 75.32, H 4.39, N 14.51. C18H13N3O. Calculated, %: C 75.25, H 4.56, N 14.63.
4.1.22. 11H-Indeno[1,2-b]quinoxalin-11-one O-(O-ethylcarboxymethyl) oxime (10c) and general procedure for synthesis of 10a,b,d. Method A:
To a suspension of IQ-1 (0.247 g, 1.0 mmol) and KOH (0.112 g, 2.0 mmol) in 5 ml DMSO, a solution of ethyl chloroacetate (0.183 g, 1.50 mmol, in 5 ml DMSO) was added dropwise. The mixture was stirred for 1 h at room temperature and poured into 150 ml of water. The precipitate was filtered out and recrystallized from EtOH to give 10c (0.28 g, 84% yield) as colorless crystals. Method B: Similar to Method A, but Na2CO3 (1.2:1.0 molar ratio to IQ-1) was used instead of KOH, and stirring was continued for 10 h. Yield 56%. M.p. 193–195o. 1H NMR (400 MHz, CDCl3), δ, ppm: 1.34 (t, 3H, 3J = 7.2 Hz, CH3),4.31 (q, 2H, 3J = 7.2 Hz, CH2CH3), 5.18 (s, 2H, =N-O-CH2), 7.59–7.69 (m, 2H, H-2, H-3), 7.70–7.80 (m, 2H, H-7, H-8), 8.14 (d, 1H, 3J = 8 Hz, H-9), 8.22 (d, 1H, 3J = 7.6 Hz, H-6), 8.27 (d, 1H,3J = 8 Hz, H-4), 8.58 (d, 1H, J = 7.6 Hz, H-1) (see Supplementary Figs. 4S and 5S for NMR H and 13C NMR spectra, respectively of E-isomer obtained by Method B). A mixture of Z- and Eisomers obtained by Method A gave additional signals in the 1H NMR spectrum: 1.27 (t, 3J = 7.2 Hz, CH3), 3.74 (q, 3J = 7.2 Hz, CH2CH3), 5.31 (s, =N-O-CH2). 13C NMR (100 MHz, CDCl3), δ, ppm: 14.24, 61.34, 72.71, 122.30, 129.41, 129.65, 129.94, 130.35, 130.54, 132.16, 132.59, 133.07, 137.40, 141.88, 142.72, 149.32, 150.37, 153.60, 168.93. Found, %: C 68.78, H 4.32, N 12.34. C19H15N3O3. Calculated, %: C 68.46, H 4.54, N 12.61.
4.1.23. 11H-Indeno[1,2-b]quinoxalin-11-one O-isobutyl oxime (10a).
Compound 10a was synthesized similarly to 10c (Method A) by reaction of IQ-1 with isobutyl bromide (54% yield, M.p. 143–146°). 1H NMR (400 MHz, CDCl3), δ, ppm: 1.09 (d, 6H, 3J = 7 Hz, CH3), 2.29 (m, 1H, CH(CH3)2), 4.41 (d, 2H, 3J = 5.5 Hz, CH2), 7.5–8.5 (m, 8H, Har). The product contained Z-isomer, which gave additional signals in 1H NMR spectrum: 1.13 (d, 3J = 7 Hz, CH3), 2.59 (m, CH(CH3)2), 5.09 (d, 3J = 5.5 Hz, CH2). Found, %: C 74.95, H 5.38, N 13.54. C19H17N3O. Calculated, %: C 75.23, H 5.65, N 13.85. Compounds 10b, d were synthesized similarly to 10c (Method B) by reaction of IQ-1 with 2-chloroethanol or chloroacetic acid, and the following derivatives were obtained, respectively.
4.1.24. 11H-Indeno[1,2-b]quinoxalin-11-one O-carboxymethyl oxime (10b).
Yield 52%, M.p. 230–232°. 1H NMR (400 MHz, CDCl3), δ, ppm: 4.72 (s, 2H, CH2), 7.66–7.76 (m, 2H, H-2, H-3), 7.79–7.87 (m, 2H, H-7, H-8), 8.13 (d, 1H, 3J = 6.8 Hz, H-9), 8.19 (d, 1H, 3J = 8 Hz, H-6), 8.53 (d, 1H, 3J = 7.8 Hz, H-4), 8.61 (d, 1H, 3J = 7.6 Hz, H-1). Found, %: C 67.04, H 3.41, N 13.48. C17H11N3O3. Calculated, %: C 66.88, H 3.63, N 13.76.
4.1.25. 11H-Indeno[1,2-b]quinoxalin-11-one O-(2-hydroxyethyl) oxime (10d).
Yield 83%, M.p.194°, decomp. 1H NMR (400 MHz, CDCl3), δ, ppm: 4.24 (t, 2H, 3J = 4.4 Hz, CH2OH), 5.43 (t, 2H, 3J = 4.4 Hz, =N-O-CH2), 7.54–7.59 (m, 2H, H-2, H-3), 7.64–7.73 (m, 2H, H-7, H-8), 8.01 (d, 1H, 3J = 8.4 Hz, H-9), 8.07 (d, 1H, 3J = 8 Hz, H-6), 8.12–8.18 (m, 2H, H-1, H-4). Found, %: C 70.38, H 4.27, N 14.13. C17H13N3O2. Calculated, %: C 70.09, H 4.50, N 14.42.
4.2. Kinase profiling and Kd determination
Kinase profiling was performed by KINOMEscan (Eurofins Pharma Discovery, San Diego, CA, USA) using a panel of 97 protein kinases, as described previously [50, 51]. In brief, the kinases were produced and displayed on T7 phage or expressed in HEK-293 cells. Binding reactions were performed at room temperature for 1 h, and the fraction of kinase not bound to a test compound was determined by capture with an immobilized affinity ligand and quantified by quantitative polymerase chain reaction. Primary screening at fixed concentrations of compounds was performed in duplicate. Selected compounds were submitted for dissociation constant (Kd) determination using the same platform. For dissociation constant Kd determination, a 12-point half-log dilution series (a maximum concentration of 33 µM) was used. Assays were performed in duplicate, and their average mean value is displayed.
4.3. Cell culture
All cells were cultured at 37°C in a humidified atmosphere containing 5% CO2. THP1Blue cells obtained from InvivoGen (San Diego, CA, USA) were cultured in RPMI 1640 medium (Mediatech Inc., Herndon, VA, USA) supplemented with 10% (v/v) fetal bovine serum (FBS), 100 µg/ml streptomycin, 100 U/ml penicillin, 100 µg/ml phleomycin (Zeocin), and 10 µg/ml blasticidin S. Human monocyte-macrophage MonoMac-6 cells (Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH, Braunschweig, Germany) were grown in RPMI 1640 medium supplemented with 10% (v/v) FBS, 10 µg/ml bovine insulin, 100 µg/ml streptomycin, and 100 U/ml penicillin.
4.4. Analysis of AP-1/NF-κB activation
Activation of AP-1/NF-κB was measured using an alkaline phosphatase reporter gene assay in human monocytic THP1-Blue cells, which are stably transfected with a secreted embryonic alkaline phosphatase gene that is under the control of a promoter inducible by NFκB/AP-1. THP-1Blue cells (2 × 105 cells/well) were pretreated with test compound or DMSO for 30 min, followed by addition of 250 ng/ml LPS for 24 h, and alkaline phosphatase activity was measured in cell supernatants using QUANTI-Blue mix (InvivoGen) as absorbance at 655 nm and compared with positive control samples (LPS). For selected compounds, the concentrations of inhibitor that caused 50% inhibition of the NF-κB reporter activity (IC50) were calculated.
4.5. Cytokine analysis
A human IL-6 ELISA kit (BD Biosciences, San Jose, CA, USA) was used to assess the effect of selected compounds on IL-6 production. MonoMac-6 cells were plated in 96-well plates at a density of 2 × 105 cells/well in culture medium supplemented with 3% (v/v) endotoxin-free FBS. Cells were pretreated with test compound or DMSO for 30 min, followed by addition of 250 ng/ml LPS for 24 h. IC50 for IL-6 production was calculated by plotting percentage inhibition against the logarithm of inhibitor concentration (at least five points).
4.6. Cytotoxicity assay
Cytotoxicity was analyzed with a CellTiter-Glo Luminescent Cell Viability Assay Kit from Promega (Madison, WI, USA), according to the manufacturer’s protocol. Cells were treated with compound under investigation and cultivated for 24 h. After treatment, the cells were allowed to equilibrate to room temperature for 30 min, substrate was added, and the samples were analyzed with a Fluoroscan Ascent FL (Thermo Fisher Scientific, Waltham, MA, USA). The cell IC50 was calculated by plotting percentage inhibition against the logarithm of inhibitor concentration (at least five points).
4.7. Western blotting
MonoMac-6 monocytic cells were pretreated with different concentrations of the compounds under investigation for 30 min and treated with LPS (250 ng/ml) or vehicle for another 30 min. Cells were washed twice with Hanks’ balanced salt solution, and cell lysates were prepared using lysis buffer from the JNK kinase assay kit (Cell Signaling Technology, Danvers, MA). Cell lysates (from 5×106 cells) were separated on ExpressPlus 4–20% PAGE Gels (GenScript, Piscataway, NJ, USA) using TRIS-MOPS running buffer (GenScript) and transferred to nitrocellulose membranes. The blots were probed with antibodies against c-Jun, phospho-c-Jun (Ser73), and total c-Jun (Cell Signaling Technology, Danvers, MA, USA), followed by horseradish peroxidase-conjugated secondary antibody (Cell Signaling Technology). The blots were developed using SuperSignal West Femto chemiluminescent substrate (Thermo Fisher Scientific) and visualized with a FluorChem FC2 imaging system (Alpha Innotech Corporation, San Leandro, CA, USA). Quantitation of the chemiluminescent signal was performed using AlphaView software (ver. 3.0; Alpha Innotech).
4.8. Molecular modeling
Geometries of JNK1–3 proteins were obtained by downloading crystal structures from the Protein Data Bank (PDB entry codes 1UKI, 3NPC, and 1PMV for JNK1, JNK2, and JNK3, respectively) into Molegro software (Molegro ApS, Aarhus, Denmark). All solvent molecules were removed. Additionally, tryptanthrin and tryptanthrin-6-oxime molecules were docked into TRK-A binding site (PDB code 4AOJ). A search space was chosen for each of the receptors as a sphere centered on co-crystallized ligand present in the corresponding PDB structure. Radii of the spheres were equal to 8, 11, 10, and 10 Å for JNK1, JNK2, JNK3, and TRK-A binding sites, respectively. Each sphere completely encompassed the co-crystallized ligand and the binding site. Side chains of all amino acid residues of a receptor within the corresponding sphere were regarded as flexible during docking. The number of such residues was equal to 21, 31, 39, and 17 for 1UKI, 3NPC, 1PMV, and 4AOJ structures, respectively. The flexible residues were treated with default settings of “Setup Sidechain Flexibility” tool in Molegro, and a softening parameter of 0.7 was applied during flexible docking, according to the standard protocol using the Molegro Virtual Docker (MVD) program (MVD 2010.4.2).
Before docking, structures of compounds were pre-optimized using HyperChem software (HyperCube, Gainesville, FL) with the MM+ force field and saved in Tripos MOL2 format (Tripos, St. Louis, MO). The ligand structures were imported into MVD. The options “Create explicit hydrogens,” “Assign charges (calculated by MVD),” and “Detect flexible torsions in ligands” were enabled during importing. Appropriate protonation states of the ligands were also automatically generated at this step. Each ligand was subjected to 30 docking runs with respect to a given receptor structure using MVD software. The docking poses obtained were saved together with the corresponding optimal geometries for identified flexible residues. DFT calculations were performed with the use of Gaussian 09W (Revision D.01) software.
Supplementary Material
Figure 7.

Pharmacological inhibition of c-Jun (Ser73) phosphorylation by selected compounds. Human MonoMac-6 monocytic cells were pretreated with indicated concentrations of 10c and tryptanthrin-6-oxime for 30 min, followed by treatment with LPS (250 ng/ml) or vehicle for another 30 min. The cells were lysed, and the lysates were analyzed by Western blotting. Total JNK (non-phosphoryalted) was used as loading control for the lysates. A representative blot from two independent experiments is shown (Panel A). The blots were analyzed by densitometry, and the ratio of phospho-c-Jun/total cJun is shown in Panel B.
Highlights.
New c-Jun N-terminal kinase (JNK) inhibitors were synthesized.
Compounds 10c and tryptanthrin-6-oxime were the most potent JNK inhibitors.
Compounds 6i, 10c, and tryptanthrin-6-oxime exhibited high selectivity for JNK1/JNK3 versus JNK2.
The active JNK inhibitors inhibited NF-κB/AP-1 activation and IL-6 production by human monocytic cells.
Acknowledgments
This research was supported in part by Pfizer Investigator Initiated Research Agreement WI214720, National Institutes of Health IDeA Program COBRE Grant GM110732; USDA National Institute of Food and Agriculture Hatch project 1009546; the Montana State University Agricultural Experiment Station; and Tomsk Polytechnic University Competitiveness Enhancement Program Grant CEP - N. Kizhner Center - 213/2018. Organic synthesis and molecular modelling were supported by the Russian Science Foundation grant No. 17–15-01111. Synthesis and physical characterization of the tryptanthrin derivative were supported by the Ministry of Education and Science of the Russian Federation project No. 4.8192.2017/8.9.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest
The authors declare no conflict of interest.
References
- [1].Armstrong SC, Protein kinase activation and myocardial ischemia/reperfusion injury, Cardiovasc. Res, 61 (2004) 427–436. [DOI] [PubMed] [Google Scholar]
- [2].Bode AM, Dong ZG, The functional contrariety of JNK, Mol. Carcinog, 46 (2007) 591598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bogoyevitch MA, Ngoei KR, Zhao TT, Yeap YY, Ng DC, c-Jun N-terminal kinase (JNK) signaling: recent advances and challenges, Biochim. Biophys. Acta, 1804 (2010) 463–475. [DOI] [PubMed] [Google Scholar]
- [4].Duplain H, Salvage of ischemic myocardium: A focus on JNK, Hypoxia and Exercise, 588 (2006) 157–164. [DOI] [PubMed] [Google Scholar]
- [5].Gupta S, Barrett T, Whitmarsh AJ, Cavanagh J, Sluss HK, Derijard B, Davis RJ, Selective interaction of JNK protein kinase isoforms with transcription factors, The EMBO journal, 15 (1996) 2760–2770. [PMC free article] [PubMed] [Google Scholar]
- [6].Waetzig V, Herdegen T, Context-specific inhibition of JNKs: overcoming the dilemma of protection and damage, Trends Pharmacol. Sci, 26 (2005) 455–461. [DOI] [PubMed] [Google Scholar]
- [7].Ip YT, Davis RJ, Signal transduction by the c-Jun N-terminal kinase (JNK) - from inflammation to development, Curr. Opin. Cell Biol, 10 (1998) 205–219. [DOI] [PubMed] [Google Scholar]
- [8].Javadov S, Jang S, Agostini B, Crosstalk between mitogen-activated protein kinases and mitochondria in cardiac diseases: Therapeutic perspectives, Pharmacol. Ther, 144 (2014) 202225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Nijboer CH, van der Kooij MA, van BF, Ohl F, Heijnen CJ, Kavelaars A, Inhibition of the JNK/AP-1 pathway reduces neuronal death and improves behavioral outcome after neonatal hypoxic-ischemic brain injury, Brain Behav.Immun, 24 (2010) 812–821. [DOI] [PubMed] [Google Scholar]
- [10].Shvedova M, Anfinogenova Y, Atochina-Vasserman EN, Schepetkin IA, Atochin DN, c-Jun N-Terminal Kinases (JNKs) in Myocardial and Cerebral Ischemia/Reperfusion Injury, Front. Pharmacol, 9 (2018) 715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Johnson GL, Nakamura K, The c-jun kinase/stress-activated pathway: regulation, function and role in human disease, Biochim. Biophys. Acta, 1773 (2007) 1341–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Guma M, Ronacher LM, Firestein GS, Karin M, Corr M, JNK-1 deficiency limits macrophage-mediated antigen-induced arthritis, Arthritis Rheum, 63 (2011) 1603–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bogoyevitch MA, Boehm I, Oakley A, Ketterman AJ, Barr RK, Targeting the JNK MAPK cascade for inhibition: basic science and therapeutic potential, Biochim. Biophys. Acta, 1697 (2004) 89–101. [DOI] [PubMed] [Google Scholar]
- [14].Zhang GY, Zhang QG, Agents targeting c-Jun N-terminal kinase pathway as potential neuroprotectants, Expert Opin.Investig.Drugs, 14 (2005) 1373–1383. [DOI] [PubMed] [Google Scholar]
- [15].Ge HX, Zou FM, Li Y, Liu AM, Tu M, JNK pathway in osteoarthritis: pathological and therapeutic aspects, J. Recept. Signal Transduct, 37 (2017) 431–436. [DOI] [PubMed] [Google Scholar]
- [16].Solinas G, Becattini B, JNK at the crossroad of obesity, insulin resistance, and cell stress response, Molecular Metabolism, 6 (2017) 174–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kumar A, Singh UK, Kini SG, Garg V, Agrawal S, Tomar PK, Pathak P, Chaudhary A, Gupta P, Malik A, JNK pathway signaling: a novel and smarter therapeutic targets for various biological diseases, Future Med. Chem, 7 (2015) 2065–2086. [DOI] [PubMed] [Google Scholar]
- [18].Haeusgen W, Herdegen T, Waetzig V, The bottleneck of JNK signaling: Molecular and functional characteristics of MKK4 and MKK7, Eur. J. Cell Biol, 90 (2011) 536–544. [DOI] [PubMed] [Google Scholar]
- [19].Vlahopoulos S, Zoumpourlis VC, JNK: A key modulator of intracellular signaling, Biochemistry-Moscow+, 69 (2004) 844–854. [DOI] [PubMed] [Google Scholar]
- [20].Shao ZL, Bhattacharya K, Hsich E, Park L, Walters B, Germann U, Wang YM, Kyriakis J, Mohanlal R, Kuida K, Namchuk M, Salituro F, Yao YM, Hou WM, Chen X, Aronovitz M, Tsichlis PN, Bhattacharya S, Force T, Kilter H, c-jun N-terminal kinases mediate reactivation of Akt and cardiomyocyte survival after hypoxic injury in vitro and in vivo, Circ. Res, 98 (2006) 111–118. [DOI] [PubMed] [Google Scholar]
- [21].Sabio G, Davis RJ, cJun NH2-terminal kinase 1 (JNK1): roles in metabolic regulation of insulin resistance, Trends Biochem. Sci, 35 (2010) 490–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Su C, Gao XJ, Yang WD, Zhao YL, Fu X, Cui XT, Zhang CY, Xin LB, Ren YY, Li LX, Shui WQ, Yang X, Wei MX, Yang J, Phosphorylation of Tudor-SN, a novel substrate of JNK, is involved in the efficient recruitment of Tudor-SN into stress granules, BbaMol Cell Res, 1864 (2017) 562–571. [DOI] [PubMed] [Google Scholar]
- [23].LoGrasso P, Kamenecka T, Inhibitors of c-jun-N-Terminal Kinase (JNK), Mini-Rev. Med. Chem, 8 (2008) 755–766. [DOI] [PubMed] [Google Scholar]
- [24].Petrov D, Luque M, Pedros I, Ettcheto M, Abad S, Pallas M, Verdaguer E, Auladell C, Folch J, Camins A, Evaluation of the Role of JNK1 in the Hippocampus in an Experimental Model of Familial Alzheimer’s Disease, Mol. Neurobiol, 53 (2016) 6183–6193. [DOI] [PubMed] [Google Scholar]
- [25].Park H, Iqbal S, Hernandez P, Mora R, Zheng K, Feng YB, LoGrasso P, Structural Basis and Biological Consequences for JNK2/3 Isoform Selective Aminopyrazoles, Sci. Rep, 5 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Koch P, Gehringer M, Laufer SA, Inhibitors of c-Jun N-Terminal Kinases: An Update, J. Med. Chem, 58 (2015) 72–95. [DOI] [PubMed] [Google Scholar]
- [27].Gehringer M, Muth F, Koch P, Laufer SA, c-Jun N-terminal kinase inhibitors: a patent review (2010–2014), Expert Opin. Ther. Pat, 25 (2015) 849–872. [DOI] [PubMed] [Google Scholar]
- [28].Messoussi A, Feneyrolles C, Bros A, Deroide A, Dayde-Cazals B, Cheve G, Van Hijfte N, Fauvel B, Bougrin K, Yasri A, Recent Progress in the Design, Study, and Development of c-Jun N-Terminal Kinase Inhibitors as Anticancer Agents, Chem. Biol, 21 (2014) 1433–1443. [DOI] [PubMed] [Google Scholar]
- [29].Schepetkin IA, Kirpotina LN, Khlebnikov AI, Hanks TS, Kochetkova I, Pascual DW, Jutila MA, Quinn MT, Identification and characterization of a novel class of c-Jun N-terminal kinase inhibitors, Mol. Pharmacol, 81 (2012) 832–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Schepetkin IA, Kirpotina LN, Hammaker D, Kochetkova I, Khlebnikov AI, yakhov SA, Firestein GS, Quinn MT, Anti-Inflammatory Effects and Joint Protection in Collagen-induced Arthritis Following Treatment with IQ-1S, a Selective c-Jun N-terminal Kinase Inhibitor, J. Pharmacol. Exp. Ther, 353 (2015) 505–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Atochin DN, Schepetkin IA, Klebnikov IA, Seledtsov VI, Swanson H, Quinn MT, Huang PL, A novel dual NO-donating oxime and c-Jun N-terminal kinase inhibitor protects against cerebral ischemia-reperfusion injury in mice. Neurosci. Lett, 618 (2016) 45–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Pearson BD, Indenoquinolines. III. Derivatives of 11H-Indeno-[1,2-b]quinoxaline and related indenoquinolines, J. Org. Chem, 27 (1962) 1674–1678. [Google Scholar]
- [33].Obot IB, Obi-Egbedi NO, Indeno-1-one [2,3-b]quinoxaline as an effective inhibitor for the corrosion of mild steel in 0.5 M H2SO4 solution, Mater. Chem. Phys, 122 (2010) 325–328. [Google Scholar]
- [34].Kaupp G, Naimi-Jamal MR, Schmeyers J, Quantitative reaction cascades of ninhydrin in the solid state, Chem. Eur. J, 8 (2002) 594–600. [DOI] [PubMed] [Google Scholar]
- [35].Deady LW, Desneves J, Ross AC, Synthesis of Some 11h-Indeno[1,2-B]Quinoxalin-11Ones, Tetrahedron, 49 (1993) 9823–9828. [Google Scholar]
- [36].Zhang C, Li SS, Ji LY, Liu S, Li ZJ, Li SC, Meng XB, Design, synthesis and antitumor activity of non-camptothecin topoisomerase I inhibitors, Bioorg. Med. Chem. Lett, 25 (2015) 4693–4696. [DOI] [PubMed] [Google Scholar]
- [37].Saintruf G, Some Derivatives of Indeno[2,1-G]Pteridine, J. Heterocycl. Chem, 11 (1974) 13–15. [Google Scholar]
- [38].Trofimov BA, Schmidt EY, Reactions of acetylenes in superbasic media. Recent advances, Russ Chem Rev+, 83 (2014) 600–619. [Google Scholar]
- [39].Bain J, McLauchlan H, Elliott M, Cohen P, The specificities of protein kinase inhibitors: an update, Biochem. J, 371 (2003) 199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wong V, Pavlick D, Brennan T, Yelensky R, Crawford J, Ross JS, Miller VA, Malicki D, Stephens PJ, Ali SM, Ahn H, Evaluation of a Congenital Infantile Fibrosarcoma by Comprehensive Genomic Profiling Reveals an LMNA-NTRK1 Gene Fusion Responsive to Crizotinib, J. Natl. Cancer Inst, 108 (2016) djv307. [DOI] [PubMed] [Google Scholar]
- [41].Rolfo C, Raez L, New targets bring hope in squamous cell lung cancer: neurotrophic tyrosine kinase gene fusions, Lab. Invest, 97 (2017) 1268–1270. [DOI] [PubMed] [Google Scholar]
- [42].Nishiyama A, Yamada T, Kita K, Wang R, Arai S, Fukuda K, Tanimoto A, Takeuchi S, Tange S, Tajima A, Furuya N, Kinoshita T, Yano S, Foretinib overcomes entrectinib resistance associated with the NTRK1 G667C mutation in NTRK1 fusion-positive tumor cells in a brain metastasis model, Clin. Cancer Res, 24 (2018) 2357–2369. [DOI] [PubMed] [Google Scholar]
- [43].Khotskaya YB, Holla VR, Farago AF, Shaw KRM, Meric-Bernstam F, Hong DS, Targeting TRK family proteins in cancer, Pharmacol. Ther, 173 (2017) 58–66. [DOI] [PubMed] [Google Scholar]
- [44].Retamales-Ortega R, Orostica L, Vera C, Cuevas P, Hernandez A, Hurtado I, Vega M, Romero C, Role of Nerve Growth Factor (NGF) and miRNAs in Epithelial Ovarian Cancer, Int J Mol Sci, 18 (2017) 507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Tajbakhsh A, Mokhtari-Zaer A, Rezaee M, Afzaljavan F, Rivandi M, Hassanian SM, Ferns GA, Pasdar A, Avan A, Therapeutic Potentials of BDNF/TrkB in Breast Cancer; Current Status and Perspectives, J. Cell. Biochem, 118 (2017) 2502–2515. [DOI] [PubMed] [Google Scholar]
- [46].Smith KM, Fagan PC, Pomari E, Germano G, Frasson C, Walsh C, Silverman I, Bonvini P, Li G, Antitumor Activity of Entrectinib, a Pan-TRK, ROS1, and ALK Inhibitor, in ETV6-NTRK3-Positive Acute Myeloid Leukemia, Mol. Cancer Ther, 17 (2018) 455–463. [DOI] [PubMed] [Google Scholar]
- [47].Bailey JJ, Schirrmacher R, Farrell K, Bernard-Gauthier V, Tropomyosin receptor kinase inhibitors: an updated patent review for 2010–2016-Part II, Expert Opin. Ther. Pat, 27 (2017) 831–849. [DOI] [PubMed] [Google Scholar]
- [48].Bailey JJ, Schirrmacher R, Farrell K, Bernard-Gauthier V, Tropomyosin receptor kinase inhibitors: an updated patent review for 2010–2016-Part I, Expert Opin. Ther. Pat, 27 (2017) 733–751. [DOI] [PubMed] [Google Scholar]
- [49].Sharma VM, Prasanna P, Seshua KVA, Renuka B, Rao CVL, Kumar GS, Narasimhulu CP, Babu PA, Puranik RC, Subramanyam D, Venkateswarlu A, Rajagopal S, Kumar KBS, Rao CS, Mamidi NVSR, Deevi DS, Ajaykumar R, Rajagopalan R, Novel indolo[2,1-b]quinazoline analogues as cytostatic agents: Synthesis, biological evaluation and structure-activity relationship, Bioorg. Med. Chem. Lett, 12 (2002) 2303–2307. [DOI] [PubMed] [Google Scholar]
- [50].Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, Faraoni R, Floyd M, Hunt JP, Lockhart DJ, Milanov ZV, Morrison MJ, Pallares G, Patel HK, Pritchard S, Wodicka LM, Zarrinkar PP, A quantitative analysis of kinase inhibitor selectivity, Nat.Biotechnol, 26 (2008) 127–132. [DOI] [PubMed] [Google Scholar]
- [51].Fabian MA, Biggs WH III, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG, Carter TA, Ciceri P, Edeen PT, Floyd M, Ford JM, Galvin M, Gerlach JL, Grotzfeld RM, Herrgard S, Insko DE, Insko MA, Lai AG, Lelias JM, Mehta SA, Milanov ZV, Velasco AM, Wodicka LM, Patel HK, Zarrinkar PP, Lockhart DJ, A small molecule-kinase interaction map for clinical kinase inhibitors, Nat. Biotechnol, 23 (2005) 329336. [DOI] [PubMed] [Google Scholar]
- [52].Gong LY, Tan YC, Boice G, Abbot S, McCaleb K, Iyer P, Zuo FR, Dal Porto J, Wong B, Jin S, Chang A, Tran P, Hsieh G, Niu LH, Shao A, Reuter D, Lukacs CM, Kammlott RU, Kuglstatter A, Goldstein D, Discovery of a novel series of 4-quinolone JNK inhibitors, Bioorg. Med. Chem. Lett, 22 (2012) 7381–7387. [DOI] [PubMed] [Google Scholar]
- [53].Lee E, Jeong KW, Shin A, Jin B, Jnawali HN, Jun BH, Lee JY, Heo YS, Kim Y, Binding model for eriodictyol to Jun-N terminal kinase and its anti-inflammatory signaling pathway, Bmb Rep, 46 (2013) 594–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Andronik-Lion V, Boucher JL, Delaforge M, Henry Y, Mansuy D, Formation of nitric oxide by cytochrome P450-catalyzed oxidation of aromatic amidoximes, Biochem. Biophys. Res. Commun, 185 (1992) 452–458. [DOI] [PubMed] [Google Scholar]
- [55].Caro AA, Cederbaum AI, Stoyanovsky DA, Oxidation of the ketoxime acetoxime to nitric oxide by oxygen radical-generating systems, Nitric Oxide, 5 (2001) 413–424. [DOI] [PubMed] [Google Scholar]
- [56].Tzeng CH, Chen YR, Tzeng CC, Liu WT, Chou CK, Chiu CC, Chen YL, Discovery of indeno[1,2-b]quinoxaline derivatives as potential anticancer agents, Eur. J. Med. Chem, 108 (2016) 258–273. [DOI] [PubMed] [Google Scholar]
- [57].Honda G, Tosirisuk V, Tabata M, Isolation of an antidermatophytic, tryptanthrin, from indigo plants, Polygonum tinctorium and Isatis tinctoria, Planta Med, 38 (1980) 275–276. [DOI] [PubMed] [Google Scholar]
- [58].Scovill J, Blank E, Konnick M, Nenortas E, Shapiro T, Antitrypanosomal activities of tryptanthrins, Antimicrob. Agents Chemother, 46 (2002) 882–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Recio MC, Cerda-Nicolas M, Potterat O, Hamburger M, Rios JL, Anti-inflammatory and antiallergic activity in vivo of lipophilic Isatis tinctoria extracts and tryptanthrin, Planta Med, 72 (2006) 539–546. [DOI] [PubMed] [Google Scholar]
- [60].Iwaki K, Ohashi E, Arai N, Kohno K, Ushio S, Taniguchi M, Fukuda S, Tryptanthrin inhibits Th2 development, and IgE-mediated degranulation and IL-4 production by rat basophilic leukemia RBL-2H3 cells, J. Ethnopharmacol, 134 (2011) 450–459. [DOI] [PubMed] [Google Scholar]
- [61].Pathania AS, Kumar S, Guru SK, Bhushan S, Sharma PR, Aithagani SK, Singh PP, Vishwakarma RA, Kumar A, Malik F, The synthetic tryptanthrin analogue suppresses STAT3 signaling and induces caspase dependent apoptosis via ERK up regulation in human leukemia HL-60 cells, PLoS One, 9 (2014) e110411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Honda G, Tabata M, Tsuda M, The antimicrobial specificity of tryptanthrin, Planta Med, 37 (1979) 172–174. [DOI] [PubMed] [Google Scholar]
- [63].Kimoto T, Hino K, Koya-Miyata S, Yamamoto Y, Takeuchi M, Nishizaki Y, Micallef MJ, Ushio S, Iwaki K, Ikeda M, Kurimoto M, Cell differentiation and apoptosis of monocytic and promyelocytic leukemia cells (U-937 and HL-60) by tryptanthrin, an active ingredient of Polygonum tinctorium Lour, Pathol. Int, 51 (2001) 315–325. [DOI] [PubMed] [Google Scholar]
- [64].Liao X, Leung KN, Tryptanthrin induces growth inhibition and neuronal differentiation in the human neuroblastoma LA-N-1 cells, Chem. Biol. Interact, 203 (2013) 512–521. [DOI] [PubMed] [Google Scholar]
- [65].Ishihara T, Kohno K, Ushio S, Iwaki K, Ikeda M, Kurimoto M, Tryptanthrin inhibits nitric oxide and prostaglandin E(2) synthesis by murine macrophages, Eur. J. Pharmacol, 407 (2000) 197–204. [DOI] [PubMed] [Google Scholar]
- [66].Pergola C, Jazzar B, Rossi A, Northoff H, Hamburger M, Sautebin L, Werz O, On the inhibition of 5-lipoxygenase product formation by tryptanthrin: mechanistic studies and efficacy in vivo, Br. J. Pharmacol, 165 (2012) 765–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Danz H, Stoyanova S, Wippich P, Brattstrom A, Hamburger M, Identification and isolation of the cyclooxygenase-2 inhibitory principle in Isatis tinctoria, Planta Med, 67 (2001) 411–416. [DOI] [PubMed] [Google Scholar]
- [68].Yang SS, Li XS, Hu FF, Li YL, Yang YY, Yan JK, Kuang CX, Yang Q, Discovery of Tryptanthrin Derivatives as Potent Inhibitors of Indoleamine 2,3-Dioxygenase with Therapeutic Activity in Lewis Lung Cancer (LLC) Tumor-Bearing Mice, J. Med. Chem, 56 (2013) 8321–8331. [DOI] [PubMed] [Google Scholar]
- [69].Onambele LA, Riepl H, Fischer R, Pradel G, Prokop A, Aminake MN, Synthesis and evaluation of the antiplasmodial activity of tryptanthrin derivatives, Int J Parasitol Drugs Drug Resist, 5 (2015) 48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Krivogorsky B, Grundt P, Yolken R, Jones-Brando L, Inhibition of Toxoplasma gondii by Indirubin and Tryptanthrin Analogs, Antimicrob. Agents Chemother, 52 (2008) 4466–4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Chen GS, Bhagwat BV, Liao PY, Chen HT, Lin SB, Chern JW, Specific stabilization of DNA triple helices by indolo[2,1-b]quinazolin-6,12-dione derivatives, Bioorg. Med. Chem. Lett, 17 (2007) 1769–1772. [DOI] [PubMed] [Google Scholar]
- [72].Krivogorsky B, Nelson AC, Douglas KA, Grundt P, Tryptanthrin derivatives as Toxoplasma gondii inhibitors-structure-activity-relationship of the 6-position, Bioorg. Med. Chem. Lett, 23 (2013) 1032–1035. [DOI] [PubMed] [Google Scholar]
- [73].Bandekar PP, Roopnarine KA, Parekh VJ, Mitchell TR, Novak MJ, Sinden RR, Antimicrobial Activity of Tryptanthrins in Escherichia coli, J. Med. Chem, 53 (2010) 35583565. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.