

Abstract
5-exo, 5-exo Cyclizations of conformationally unbiased propargylic aminyl radicals proceed with excellent yield, chemoselectiv ity, and diastereoselectivity under tin-free reductive cyclization conditions, regardless of the electronic environments and intermediate radical stabilization resulting from various olefin substituents. These conditions avoid the need for slow addition of initiator and reductant. By contrast, analogous 6-exo, 5-exo cyclizations require substituents capable of intermediate radical stabilization to avoid premature reduction products. These experimental results are corroborated by computations that further establish the reactivity of these aminyl radicals upon exposure to tin-free cyclization conditions.
Graphical Abstract
Tertiary amines are privileged structures in pharmaceutical lead targets because they improve solubility and decrease lipophilicity, while maintaining potency.1 Polycyclic molecules bearing tertiary amines are of special interest because they possess the rigid structural framework, high density of sp3 centers, and low molecular weight profile associated with “drug-like” molecules.2 We were attracted to the potential utility of neutral aminyl radicals to construct these frameworks because radical cascades have well established reactivity with predictable bond construction outcomes. While amidyl,3 iminyl,4 and aminium5 radicals are more reactive and tend to cyclize more efficiently6,7 than the more stable neutral aminyl radicals,8,9 the latter are strategically appealing because their cyclization reactions lead directly to aliphatic amine-containing products.10 In this work, we demonstrate the utility of our recently reported11 tin-free conditions for the cyclization of neutral aminyl radicals. We obtained good yields of 5-exo-initiated12 tandem cyclization products employing electronically differentiated olefins and demonstrated the electronic dependence of 6-exo cyclizations of conformationally unbiased13 substrates. Lastly, we computed the barrier heights and thermodynamics of these cyclizations in the gas phase, toluene, and tetrahydrofuran (THF).
We recently reported the development of tin-free conditions to access the ABC core of the calyciphylline A alkaloids via tandem cyclization of a neutral aminyl radical.11 In these cyclizations, an existing 6-membered ring (ring A) reduced the conformational degrees of freedom in the substrate (Scheme 1). The cyclization of N-chloroamine 1 is initiated via 6-exo cyclization, which is a rare mode of reactivity for aminyl radicals. An electron deficient olefin was required in these cyclizations as allylic alcohol derivatives led only to N–Cl reduction products.14 Finally, our experiences indicated that internal and terminal alkynes performed differently in otherwise analogous tandem cyclizations. Because of the unique features of this substrate relative to known cyclizations of aminyl radicals, we sought to establish the utility of these tin-free cyclization conditions, which also avoid slow addition15 of initiator and H-atom donor, in conformationally unbiased systems.
Scheme 1.

Structural features of substrate for calyciphylline A alkaloid synthesis.
We hypothesized that cascades beginning with cyclization of the relatively nucleophilic16 aminyl radicals would have lower barriers when cyclizing with electron deficient olefins than with non-activated olefins.17 Findings that enones analogous to substrate 1 cyclized efficiently, while the corresponding allylic alcohol14 led only to N–Cl reduction (R2N–Cl → R2N–H) were consistent with our hypothesis but were insufficient to provide insight into 5-exo and 6-exo cyclizations of conformationally unbiased substrates. Thus, we selected representative unactivated and activated olefin substituents to investigate under these conditions. For convenience, we retained the terminal alkyne in the second cyclization, thus minimizing potential complications arising from formation of multiple diastereomers. We found that unactivated substrates 3a–3b18 afforded good to excellent yields of cyclized products 4a–4b when treated with azobisisobutyronitrile (AIBN) and the weak H-atom donor (iPr)3 SiH at 100°C in THF.14,19 Bicyclic amine 4b was formed as a single diastereomer. The relative stereochemistry is consistent with previous stereochemical studies of 5-exo, 5-exo tandem cyclizations.20 Enone substrate 3c also cyclized efficiently, and following thermal isomerization of the exocyclic olefin, bicyclic amine 4c was isolated in 87% yield. Cyclization of trans-3d led to a 69% yield of 4d with 95:5 dr. The cis-isomer of the styrenyl substrate (cis-3d) performed similarly to enone 3c with an isolated yield of 86% and 91:9 dr. Isomerization of the exocyclic olefin was not observed in either case, and the major product diastereomer was trans for both cyclizations.21
Previously, we observed that cyclization of N-chloroenone 1 in THF with (iPr)3SiH as an H-atom donor led to fewer reduction products than did the same cyclization in toluene with Bu3SnH.11 We presumed that the change in H-atom donor was the primary contributor to this reactivity; however, both solvents can also donate hydrogen atoms. The calculated energy barrier for intermolecular H-atom transfer from the solvent to radical 8a was 13.4 kcal/mol in implicit THF and 15.4 kcal/mol in implicit toluene (calculations were performed using the Gaussian 09 software22 and the ωB97xD level of theory and 6–31+G(d,p) basis set at 100°C).23 The barrier to hydrogen atom transfer from each reducing agent to radical 8a was also calculated at 100°C. For Et3 SnH;24 the barrier is 4.8 kcal/mol in implicit THF and 5.0 kcal/mol in implicit toluene. For (iPr)3SiH, the barrier is 13.7 kcal/mol in implicit THF and 14.4 kcal/mol in implicit toluene. Thus, the reduction products observed in toluene/tin hydride conditions likely arise due to H-atom transfer from the stannane, not toluene. These labeling experiments and computational data support our hypothesis that a weaker H-atom donor should favor tandem cyclization products in preference to N–H bond formation.11
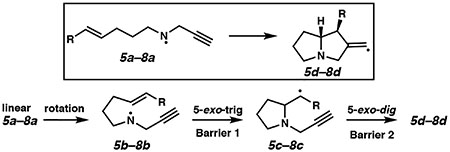
We next calculated the energetics of the cyclization pathway from the aminyl radicals 5a–8a to vinylic radicals 5d–8d via the Beckwith-Houk transition states of each substrate (Table 1).25,26 For 5-exo-initiated cyclizations, the initial cyclization barriers (i.e., Barrier 1 = ΔG‡ for 5b–8b → 5c–8c) range from 9.3 to 14.2 kcal/mol in implicit solvent, with the barrier for 7b in THF as an apparent outlier. These data are consistent with published computational results for aminyl radical monocyclization.7a The reverse reaction barriers range from 15.4–26.9 kcal/mol depending on the substrate and solvent. These data suggest that the first 5-exo cyclization event is likely irreversible for most of these substrates. These results are consistent with some7,27,28 previous experimental rate studies.29 For all evaluated substrates, the transformation of the N-centered radical (5a–8a) to the vinylic radical (5d–8d) is exothermic owing to the formation of two C–C bonds and a fused bicyclic system.
Table 1.
Activation barriers and relative energies of radical intermediates in the 5-exo, 5-exo pathway (ωB97xD functional, Gibbs free energies were calculated at 100°C, all values in kcal/mol).
 | ||||||
|---|---|---|---|---|---|---|
| R | solvent | 5–8b | barrier 1a | Int 5–8cb | barrier 2 | Int 5–8d |
| H(5) | (gas) | 0.0 | 13.2 | –6.2 | 12.4 | –26.6 |
| H | PhMe | 0.5 | 13.9 | –4.2 | 11.6 | –24.5 |
| H | THF | 0.9 | 14.2 | –3.1 | 10.8 | –23.0 |
| CH2OMe (6) | (gas) | 2.3 | 11.8 | –6.3 | 6.5 | –26.4 |
| CH2OMe | PhMe | 2.4 | 12.0 | –5.8 | 6.9 | –24.8 |
| CH2OMe | THF | 3.1 | 12.7 | –6.8 | 7.2 | –20.9 |
| COEt (7) | (gas) | –1.0 | 11.4 | –12.6 | 12.4 | –25.6 |
| COEt | PhMe | –0.3 | 11.4 | –11.6 | 13.9 | –23.3 |
| COEt | THF | 0.8 | 7.3 | –10.4 | 13.3 | –21.5 |
| Ph (8) | (gas) | –1.2 | 10.1 | –15.0 | 15.6 | –24.4 |
| Ph | PhMe | 0.7 | 10.1 | –17.5 | 16.2 | –21.7 |
| Ph | THF | 0.6 | 9.3 | –12.7 | 16.3 | –20.4 |
Barrier 1 is calculated from the prearranged conformation (5–8b).
All intermediate values are relative to the energy of the fully elongated, staggered aminyl radical (5–8a).
As expected, Barrier 1 tracks the stability of the intermediate radicals 5c–8c. The barrier for the terminal olefin (5b) was 1.4–1.9 kcal/mol higher in energy than for methoxymethyl-substituted 6b. The barrier for enone 7b was 1.8–2.5 kcal/mol lower than for 5b in the gas phase or implicit toluene, while in THF, the gap is larger than expected at 6.9 kcal/mol. The barrier for styrenyl substrate 8b was 2.5–3.5 kcal/mol lower than for 5b. While these energy differences follow the expected trend, they are relatively small, and the high temperature for cyclization is sufficient to enable cyclization even in the highest barrier case (R = H). Interestingly, Barrier 1 is higher in THF than in toluene for 5b and 6b and lower for 7b and 8b. For substrates capable of generating a stabilized C-centered radical intermediate (i.e., 7c and 8c), the barrier height for the second cyclization (Barrier 2) is significantly larger than for radicals 5c and 6c. α-Keto radical 7c is 7.3–7.4 kcal/mol more stabilized than primary radical 5c in implicit THF and toluene solvent, respectively. For styrenyl substrate 8c, these values increase to 9.6–13.3 kcal/mol, respectively.
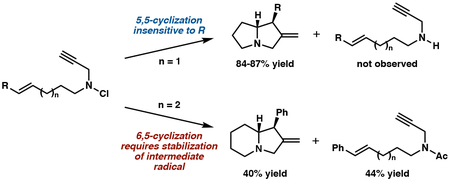
Based on the computational results outlined above, we hypothesized that 6-exo-initiated cyclizations would be more sensitive to substitution changes because of the significantly reduced reactivity of these substrates. We selected H and Ph groups as representative electron-normal and electron-withdrawing substituents. Attempted cyclization of N-hexenyl-N-propargyl chloroamine 9 with THF/(iPr)3SiH conditions led to recovery of 77% reduced amine 11 (Scheme 3A). Bicycle 10 was not observed in the crude NMR. For the styrenyl analog of this substrate (12), cyclization product 13 was isolated in 40% yield after an acetic anhydride workup, which was required to facilitate isolation of 14 (44% yield, Scheme 3B). Presumably, formation of the reduced products (pre-11, pre-14) in these cases occurs via both intramolecular and intermolecular H-atom transfer. This is particularly true in the styrenyl system where the allylic radical is highly stabilized.20a
Scheme 3.

Sensitivity of 6-exo, 5-exo initiated cyclizations to olefin electronics.
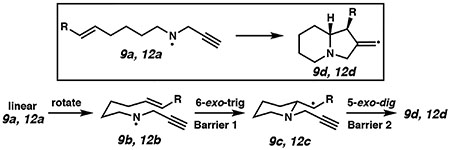
The computational analysis for these two cyclizations is consistent with both the drop in overall reactivity for 6-exo cyclizations and in the relative success of the two substrates (Table 2). Interestingly, once the substrate (9a and 12a) adopts the appropriate conformation for cyclization (9b and 12b), the Barrier 1 values for 6-exo cyclization are not significantly different from the 5-exo substrates. However, the energy required to adopt these precyclization conformations is significant. In THF, combining the uphill conformational change with the activation barrier gives an overall transition state energy (ΔG‡) of 15.3 kcal/mol. Thus, it is unsurprising that the reduction pathway, which had a barrier of 13.4 kcal/mol for H-atom transfer from THF to 8a, outcompetes the cyclization. However, for the styrenyl substrate, the ΔG‡ is 12.3 kcal/mol. It is thus expected to be competitive with reduction, and a mixture of products (13 and 14) is observed. As expected, the Barrier 2 heights and overall thermodynamics are similar to the 5,5-cyclization pathway. Overall, these computational data suggest that the rate differences between these 5-exo and 6-exo radical cyclizations are primarily derived from entropic factors associated with substrate pre-arrangement rather than an inherent decrease in reactivity for 6-exo cyclization resulting from an issue with alignment of the participating molecular orbitals.
Table 2.
Activation barriers and relative energies of radical intermediates in the 6-exo, 5-exo pathway (ωB97xD functional, Gibbs free energies were calculated at 100°C, all values in kcal/mol).
 | ||||||
|---|---|---|---|---|---|---|
| R | solvent | 9b, 12b | barrier 1a | Int 9c, 12cb | barrier 2 | Int 9d, 12db |
| H (9) | (gas) | 3.1 | 10.5 | −8.3 | 8.9 | −29.2 |
| H | PhMe | 2.1 | 12.3 | −7.1 | 8.7 | −28.3 |
| H | THF | 2.6 | 12.7 | −5.8 | 8.6 | −26.8 |
| Ph (12) | (gas) | 1.0 | 8.4 | −17.4 | 12.9 | −28.3 |
| Ph | PhMe | 2.9 | 8.1 | −16.2 | 13.8 | −25.2 |
| Ph | THF | 4.0 | 8.3 | −14.0 | 13.3 | −23.2 |
Barrier 1 is calculated from the prearranged conformation (9b, 12b).
All intermediate values are relative to the energy of the fully elongated, staggered aminyl radical (9a, 12a).
We also investigated the applicability of these conditions to the 5-exo, 6-exo substrate chloroamine 15 (Scheme 4). The desired product was formed in 39% yield and 77:23 dr. The lower dr here presumably arises from competitive rotation of the Ph group to the pseudo-equatorial position in the transition state. Interestingly, despite the initial formation of a 5-membered ring, significant amounts of reduction product (isolated as the acetate, 17) were observed (35% yield). The dependence of the formation of reduced amine on the size of the second ring suggests reversibility in the first cyclization. Meanwhile, our computational results from Table 1 support irreversible cyclization of the N-centered radical with a reverse barrier of ~23 kcal/mol for 8c → 8b. The retention of stereochemistry in reduction product 17 suggests that intermolecular H-atom transfer to the aminyl radical is faster than 5-exo cyclization in this substrate. By contrast, no reduction is observed in any of our 5-exo, 5-exo tandem cyclizations. Combining previous rate studies with these new computational and experimental data, one can conclude that the reversibility of neutral aminyl cyclizations is dependent on the intermediate radical stability, the reaction conditions, and the effects of impurities27 in the reaction.
Scheme 4.

Sensitivity of 6-exo, 5-exo initiated cyclizations to olefin electronics.
In summary, kinetically favorable 5-exo, 5-exo cyclizations of propargyl-appended aminyl radicals proceed efficiently in tin-free conditions employing a weak H-atom donor in THF, regardless of the electronic nature of the substitution on the olefin. Importantly, these conditions avoid kinetic tricks such as slow addition of the radical initiator and reducing agent. By contrast, a radical stabilizing group is essential to facilitate cyclization in 6-exo, 5-exo cyclizations of propargylic aminyl radicals. These results are supported by computational data. Finally, application to a 5-exo, 6-exo cyclization reveals a dependence on the size of the second ring in determining the ratio of cyclization to reduction. Overall, the weak H-atom donating ability of (iPr)3SiH and THF result in an unusual, if apparently synergistic, solvent/H-atom donor combination that is effective for a range of tandem cyclizations. We anticipate that these experimental and computational data will be useful for future synthetic design using neutral aminyl radicals. Further investigation into the factors influencing reversibility are ongoing in our laboratories.
Supplementary Material
Scheme 2.

Tin-free cyclization of unactivated and activated N-chloro propargylamines.
ACKNOWLEDGMENTS
The authors thank the National Institutes of Health (R00-GM097095 for 5,5-cyclizations with R = H, R = Ph), the National Science Foundation (CAREER Award to JLS: CHE-1554752 for all remaining experiments, Grant to BHS: CHE-1464450), and Wayne State University for generous financial support (startup funds to JLS, Rumble-Schaap Fellowship to HSS and use of the computational grid). We thank Mate-ria, Inc. for the kind donation of all metathesis catalysts used in the substrate syntheses. We are grateful to the staff of the WSU Lumigen Instrument Center, especially Nicholas Peraino, for spectroscopic support, and we thank Dr. Ahmad A. Ibrahim (WSU) for helpful discussions.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures and spectroscopic data (PDF) Computational methodology and Cartesian coordinates (PDF)
The authors declare no competing financial interests.
REFERENCES
- 1.Ghose AK; Viswanadhan VN; Wendoloski JJ A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem 1999, 1, 55–68. [DOI] [PubMed] [Google Scholar]
- 2.Newman DJ; Cragg GM; Snadar KM Natural Products as Sources of New Drugs over the Period 1981–2002. J. Nat. Prod 2003, 66, 1022–1037. [DOI] [PubMed] [Google Scholar]
- 3.a) Sharp L; Zard SZ A Short Total Synthesis of (±) Aspidospermi-dine. Org. Lett 2006, 8, 831–834. [DOI] [PubMed] [Google Scholar]; b) Callier-Dublanchet A-C; Cassayre J; Gagosz F; Quiclet-Sire B; Sharp LA; Zard SZ Amidyls in Radical Cascades. The Total Synthesis of (±)-Aspidospermidine and (±)-13-Deoxyserratine. Tetrahedron 2008, 64, 4803–4816. [Google Scholar]; c) Biechy A; Hachisu S; Quiclet-Sire B; Ricard L; Zard SZ The Total Synthesis of (±)-Fortucine and a Revision of the Structure of Kirkine. Angew. Chem. Int. Ed 2008, 47, 1436–1438. [DOI] [PubMed] [Google Scholar]
- 4.Bowman WR; Cloonan MO; Fletcher AJ; Stein T Synthesis of Heteroarenes Using Cascade Radical Cyclization via Iminyl Radicals. Org. Biomol. Chem, 2005, 3, 1460–1467. [DOI] [PubMed] [Google Scholar]
- 5.a) Newcomb M; Marquardt DJ; Deep TM N-Hydroxypyridine-2-thione Carbamates. V. Syntheses of Alkaloid Skeletons by Aminium Cation Radical Cyclizations. Tetrahedron, 1990, 46, 2329–2344. [Google Scholar]; b) Cosgrove SC; Plane JMC; Mardsen SP Radical-mediated direct C–H amination of arenes with secondary amines. Chem. Sci 2018, 9, 6647–6652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Tadic-Bladatti M-HL; Caller-Dublanchet A-C; Horner JH, Quiclet-Sire B; Zard SZ; Newcomb M Absolute Rate Constants for Iminyl Radical Reactions. J. Org. Chem 1997, 62, 559–563. [DOI] [PubMed] [Google Scholar]; b) Horner JH; Musa OM; Bouvier A; Newcomb M Absolute Kinetics of Amidyl Radical Reactions. J. Am. Chem. Soc 1998, 120, 7738–7748. [Google Scholar]; c) Horner JH; Martinez FN; Musa OM; Newcomb M; Shahin HE Kinetics of Dialkylaminium Cation Radical Reactions: Radical Clocks, Solvent Effects, Acidity Constants, and Rate Constants for Reactions with Hydrogen Atom Donors. J. Am. Chem. Soc 1995, 117, 11124–11133. [Google Scholar]; d) Musa OM; Horner JH; Shahin H; Newcomb M A Kinetic Scale for Dialkylaminyl Radical Reactions. J. Am. Chem. Soc 1996, 118, 3862–3868. [Google Scholar]
- 7.a) Tsanaktsidis J; Maxwell BJ Cyclization of N-Butyl-4-pentenylaminyl: Implications for the Cyclization of Alkenylaminyl Radicals. J. Am. Chem. Soc 1996, 118, 4276–4283. [Google Scholar]; b) Maxwell BJ; Smith BJ; Tsanaktsidis J The Cyclization of N-Butylpent-4-enylaminyl Revisited: A Combined Theoretical and Experimental Study. J. Chem. Soc.; Perkin Trans. 2 2000, 425–431. [Google Scholar]
- 8.a) Hioe J; Šakic D; Vrček V; Zipse H The Stability of Nitrogen-Centered Radicals. Org. Biomol. Chem 2015, 13, 157–169. [DOI] [PubMed] [Google Scholar]; b) Stella L Homolytic Cyclizations of N‐Chloroalkenylamines. Angew. Chem. Int. Ed 1993, 22, 337–350. [Google Scholar]; c) Boate D; Fontaine C; Guittet E; Stella L Synthe se Du Systeme Cyclique De L’Aza-Triquinane Lineaire Par Trois Cyclisations Radicalaires En Cascade. Tetrahedron. 1993, 49, 8397–8406. [Google Scholar]
- 9.a) Quiclet-Sire B; Zard SZ Some aspects of radical cascade and relay reactions. Proc. R Soc. A 2017, 473: 20160859. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chen J-R; Hu X-Q; Lu L-Q Xiao W-J Visible light photoredox-controlled reactions of N-radicals and radical ions. Chem. Soc. Rev 2016, 45, 2044–2056. [DOI] [PubMed] [Google Scholar]; c) Zard SZ Recent Progress in the Generation and Use of Nitrogen-Centered Radicals. Chem. Soc. Rev 2008, 37, 1603–1618. [DOI] [PubMed] [Google Scholar]
- 10.White LV; Schwartz BD; Banwell MG; Willis ACA A Chemoenzymatic Total Synthesis of (+)-Clividine. J. Org. Chem 2011, 76, 6250–6257. [DOI] [PubMed] [Google Scholar]
- 11.Lopez AM; Ibrahim AI; Rosenhauer GJ; Sirinimal HS; Stock-dill JL Tin-Free Access to the ABC Core of the Calyciphylline A Alkaloids and Unexpected Formation of a D-Ring-Contracted Tetracyclic Core. Org. Lett 2018, 20, 2216–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baldwin JE Rules of Ring Closure. J. Chem. Soc., Chem. Commun 1976, 734–736. [Google Scholar]
- 13.Beesley RM; Ingold CK; Thorpe JF The Formation and Stability of spiro-Compounds. Part I. Spiro-Compounds from Cyclohexane. J. Chem. Soc., Trans 1915, 107, 1080–1106. [Google Scholar]
- 14.Ibrahim AA; Golonka AN; Lopez AM; Stockdill JL Rapid Access to the Heterocyclic Core of the Calyciphylline A and Daphnicyclidin A-Type Daphniphyllum Alkaloids via Tandem Cyclization of a Neutral Aminyl Radical. Org. Lett 2014, 16, 1072–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jasperse CP; Curran DP; Fevig TL Radical Reactions In Natural Product Synthesis. Chem. Rev, 1991, 91, 1237–1286. [Google Scholar]
- 16.Kim S; Yoon KS Kinetics of Intramolecular Additions of the Aminyl Radicals to Carbonyl Groups and Subsequent Ring Openings. Tetrahedron 1995, 51, 8437–8446. [Google Scholar]
- 17.Roberts BP Polarity-reversal catalysis of hydrogen-atom abstraction reactions: concepts and applications in organic chemistry. Chem. Soc. Rev 1999, 28, 25–35. [Google Scholar]
- 18.See the Supporting Information for experimental details and substrate synthesis.
- 19.The cyclization of 3a was performed on 1.1 mmol scale, resulting in 81% yield of 4a. See the Supporting Information for details.
- 20.a) Bowman WR, David NC, Robert JM Synthesis of pyrrolizidines using aminyl radicals generated from sulfenamide precursors. Tetrahedron 1994, 50, 1295–1310. [Google Scholar]; b) Hasegawa H; Hisanori S; Yoshinori K; Kazuhiko O; Masao T Stereoselective synthesis of 2-methylenepyrrolizidines by tandem cyclization of N-propargylaminyl radicals. Tetrahedron 2003, 59, 827–832. [Google Scholar]; c) Tsuchida S; Atsunori K; Tokutaro O; Hiromi B; Yasutomo Y; Kiyoshi T Consecutive Cyclization of Allylaminoalkene by Intramolecular Aminolithiation− Carbolithiation. Org. Lett 2008, 10, 3635–3638. [DOI] [PubMed] [Google Scholar]
- 21.Hasegawa H; Senboku H; Kajizuka Y; Orite K; Tokuda M Stereoselective synthesis of 2-methylenepyrrolizidines by tandem cyclization of N-propargylaminyl radicals. Tetrahedron 2003, 59, 827–832. [Google Scholar]
- 22.Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Petersson GA; Nakatsuji H; Caricato M; Li X; Hratchian HP; Izmaylov AF; Bloino J; Zheng G; Sonnenberg JL; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Montgomery JA; Peralta JE Jr.; Ogliaro F; Bearpark M; Heyd JJ; Brothers E; Kudin KN; Staroverov VN; Kobayashi R; Normand J; Raghavachari K; Rendell A; Burant JC; Iyengar SS; Tomasi J; Cossi M; Rega N; Millam JM; Klene M; Knox JE; Cross JB; Bakken V; Adamo C; Jaramillo J; Gomperts R; Stratmann RE; Yazyev O; Austin AJ; Cammi R; Pomelli C; Ochterski JW; Martin RL; Morokuma K; Zakrzewski VG; Voth GA; Salvador P; Dannenberg JJ; Dapprich S; Daniels AD; Farkas Ö; Foresman JB; Ortiz JV; Cioslowski J; Fox DJ Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford CT, 2016. [Google Scholar]
- 23.See the Supporting Information for computational details.
- 24.Ethyl is employed to expedite the computation.
- 25.a) Gilmore K; Igor VA Cyclizations of alkynes: revisiting Baldwin’s rules for ring closure. Chem. Rev 2011, 111, 6513–6556. [DOI] [PubMed] [Google Scholar]; b) Beckwith ALJ; Christopher J. Easton.; Algirdas KS Some guidelines for radical reactions. J. Chem. Soc., Chem. Commun 1980, 11, 482–483. [Google Scholar]; c) Julia M Free radical cyclizations. Pure. Appl. Chem 1967, 15, 167–184. [Google Scholar]; d) Baldwin JE Rules for ring closure. J. Chem. Soc., Chem. Commun 1976, 18, 734–736. [Google Scholar]; e) Beckwith ALJ; Tony L; Algirdas KS Stereoselectivity of ring closure of substituted hex-5-enyl radicals. J. Chem. Soc., Chem. Commun 1980, 11, 484–485. [Google Scholar]; f) Beckwith ALJ; Carl HS Regio-and stereo-selectivity of alkenyl radical ring closure: A theoretical study. Tetrahedron. 1985, 41, 3925–3941. [Google Scholar]; g) Spellmeyer DC, Houk KN Force-field model for intramolecular radical additions. J. Org. Chem 1987, 52, 959–974. [Google Scholar]
- 26.Clark AJ; Filik RP; Thomas GH; Sherringham J Anti-Beckwith stereoselectivity in amidyl radical cyclisations: Bu3SnH-mediated 5-exo-trig acyl mode cyclisation of 2-substituted pent-4-enamide radicals. Tetrahedron Lett. 2013, 54, 4094–4097. [Google Scholar]
- 27.Maxwell BJ; Tsanaktsidis J Influence of bis(tributyltin) Oxide on Aminyl Radical Cyclizations. J. Chem. Soc., Chem. Commun 1994, 533–534. [Google Scholar]
- 28.a) Musa OM; Horner JH; Shahin H; Newcomb M A Kinetic Scale for Dialkylaminyl Radical Reactions. J. Am. Chem. Soc 1996, 118, 3862–3868. [Google Scholar]; b) Newcomb M; Musa OM; Martinez FN; Horner JH Kinetics of 5-exo Cyclizations of N-Alkyl-4-pentenaminyl Radicals and b-Fragmentations of b-(Dialkylamino)alkyl Radicals. J. Am. Chem. Soc 1997, 119, 4569–4577. [Google Scholar]; c) Newcomb M; Horner JH; Shahin H Rate Constants for Aminyl Radical Reactions. Tetrahedron Lett. 1993, 34, 5523–5526. [Google Scholar]
- 29.Bowman WR; Clark DN; Marmon RJ Generation of Aminyl Radicals using Sulfenamides as Synthetic Precursors. Tetrahedron Lett. 1994, 50, 1275–1294. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.