

Abstract
Rhodopsins hosting synthetic retinal protonated Schiff base analogues are important for developing tools for optogenetics and high-resolution imaging. The ideal spectroscopic properties of such analogues include long-wavelength absorption/emission and fast/hindered photoisomerization. While the former may be achieved, for instance, by elongating the chromophore π-system, the latter requires a detailed understanding of the substituent effects (i.e. steric or electronic) on the chromophore light-induced dynamics. Below, we compare the results of QM/MM excited state trajectories of native and analogue-hosting microbial rhodopsins from the eubacterium Anabaena. The results uncover a relationship between nature of the substituent on the analogue (i.e. electron donating (a Me group) or withdrawing (a CF3 group)) and rhodopsin excited state lifetime. Most importantly, we show that electron donating or With drawing substituents cause a decrease or an increase in the electronic mixing of the first two excited states which, in turn, controls the photoisomerization speed.
Graphical Abstract
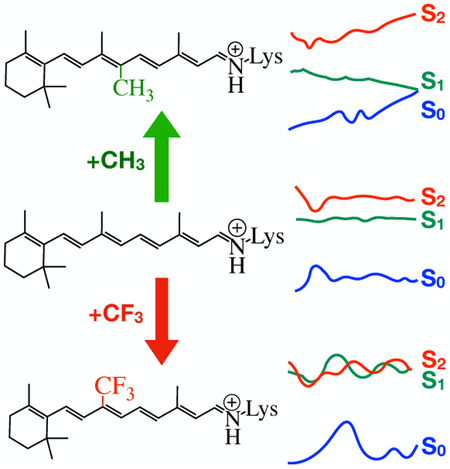
The use of retinal proteins (simply, rhodopsins) hosting synthetic retinal protonated Schiff base (rPSB) analogues in optogenetics and fluorescence microscopy is receiving increasing attention.1,2 Such analogues not only allow achieving long wavelength sensitivity which is essential for the biological applications3; but also provide a way of engineering fast or slow photoreactivity into the protein. In principle, the latter is highly important, especially in designing rhodopsin based fluorescent probes where one should hinder the isomerization of rPSB in order to achieve a higher excited state lifetime and therefore a higher fluorescence.4,5 Thus, understanding the impact of different rPSB substituents on absorption wavelength and photoisomerization timescale is of great importance. While the wavelength tuning is often achieved by increasing the π-system of the rPSB or introducing electron donating/withdrawing substituents, controlling the photoreactivity using such strategies requires a better understanding of their effects on the rPSB photodynamics. More specifically, one must understand which effects of the substitution (i.e. electronic, steric and etc.) cause a change in the photodynamics. Among the recent studies aiming to achieve such knowledge, the works by Kukura et.al and Garavelli et.al are significant.6–8 Kukura et.al investigated the photoisomerization of several artificial rPSB pigments in methanol solution using time-resolved spectroscopy.6,7 An interesting result from their experiments is that the introduction of a Me group at the C10 position (10MerPSB) of the native rPSB backbone (see Scheme 1A) redshifted the absorption wavelength by only few nm but decreases the original excited state lifetime from 4 to 0.7 ps. Later, Garavelli et. al reported a computational study indicating that the fast decay of 10Me-rPSB in solution is the result of an electron releasing effect of the Me substituent that increases the energy gap between first (S1) and second (S2) electronic singlet excited states.8 While the above investigations have provided basic information on rPSB photoreactivity tuning in solution, no information is presently available on the chromophore analogue control of the photoisomerization inside the protein environment (from now on, opsin cavity). In the past, bacteriorhodopsin (bR) hosting different artificial rPSB pigments have been employed in various experimental studies.9–13 One of such studies have provided evidence that electron withdrawing groups such as CF3 inhibit the photoisomerization.10 However, a computational/mechanistic study looking at which specific effects (i.e. steric or electronic) speed up or slow down the rPSB photoisomerization in the opsin cavity remains unexplored.
Scheme 1.

PESs and the associated electronic characters of ASRAT models at the FC point. A. Structure of the native all-trans rPSB chromophore and representation of its photoisomerization reaction to the 13-cis rPSB chromophore. B. Resonance structures showing electronic characters dominating the S0 (bottom), S1 (middle) and S2 (top) states of ASRAT in its vertical excitation region C. Schematic representation of the effect of electron releasing (10-methyl, Me-ASRAT) or withdrawing (9-trifluoromethyl, CF3-ASRAT) substituents on the relationship between the PESs of ASRAT at the FC region.
Below we demonstrate how electronic effects introduced by Me or CF3 in specific position of the all-trans rPSB chromophore backbone control the photoreactivity of rPSB inside the opsin cavity. In order to do so, we construct hybrid quantum mechanics/molecular mechanics (QM/MM) models of a selected microbial opsin with native and artificial rPSBs featuring electron releasing or withdrawing substituents. The selected rhodopsin, Anabaena sensory rhodopsin (ASR), is found in fresh water cyanobacterium Anabaena (Nostoc) PCC7120 and known to be naturally hosting both all-trans and 13-cis rPSBs (hereafter ASRat and ASR13C).14,15 As convenient rPSB analogues, we have chosen 10-methylated and 9-trifluoromethylated all-trans and 13-cis rPSBs. From now on, ASR hosting these rPSB analogues will be referred to as Me-ASRX and CF3-ASRX respectively; where X stands for AT or 13C. The corresponding QM/MM models are then used to compute the wavelength of the absorption maximum (λmax) of each rhodopsin. More specifically we compute the corresponding vertical excitation energies at the Franck-Condon (FC) points. The same models are then used to investigate their photoisomerization dynamics. More specifically, QM/MM models are used to simulate the photodynamics of rPSB along short (<150 fs long) trajectories initiated on S1 or S2 potential energy surface (PES) with zero kinetic energies (see SI for details) called FC trajectory. We assume that in a sub-fs timescale, FC trajectories represent the center of a vibrational wave packet and thus provide information of the population dynamics. This assumption is consistent with our previous photodynamics studies.16,17
In our QM/MM photodynamics simulations, geometrical progression is computed using 3-root-state-averaged CASSCF/AMBER gradients.18 The effect of dynamic electron correlation is then introduced by re-computing the S0, S1 and S2 energy profiles at CASPT219 level of theory.
Substituent effects on λmax originate from competing steric and electronic effects.
Below we employ the ASRAT, Me-ASRAT and CF3-ASRAT models to look at the absorption spectroscopy of these corresponding rhodopsin. However, before discussing the computational results, it is necessary to revise our general understanding on the excited states of the rPSB chromophore. As depicted in Scheme 1B the electronic structures (from now on called “characters”) contributing to the 3-root-state-average wavefunctions (S0, S1 and S2) of each model are termed 1Ag (covalent), 1Bu (charge transfer) and 2Ag (diradical) respectively.16,17,20 In ASRAT, the S0 PES at the FC point displays dominating 1Ag character where the positive charge of the chromophore is mostly distributed in the Schiff base (-CH=NH-) region. At the same location, the S1 and S2 PESs display a dominating1Bu and 2Ag characters respectively. Therefore, S1 is characterized by the majority of the positive charge located in the β-ionone side of the rPSB and it is reactive as the S0 double bond have become, effectively, single bonds. On the other hand, S2 displays a similar charge distribution to the covalent S0 state with the charge mainly residing on the Schiff base moiety and it is not reactive as the double bonds still have the bonding character.
As illustrated in Scheme 1C (see the computed vertical excitation energies in Table S1), the introduction of electron releasing or withdrawing substituents leads to a relative stabilization or destabilization of the three different PESs. In fact, 9-trifluoromethylation destabilizes the S1 PES with respect to the S0 PES due to its 1Bu character and, for the same reason, decreases the S2-S1 gap. Consistently, the introduction of an electron donating substituent (Me-ASRAT) results in an increased S1-S2 energy gap. However, contrary to what is expected on the basis of electronic effects, we observe an increase in the S0-S1 excitation energy with respect to the rhodopsin with the native chromophore. This is due to steric effects. In fact, we find that the 10-methyl substituted rPSB becomes highly distorted around C6–C7 and C8–C9 single bonds (see Figure S2A) leading to partial deconjugation and, therefore, a λmax blue-shift.
As we have discussed so far, addition of electron donating or withdrawing groups change the S1-S2 energy gap of PESs in the FC region. Below we show that such substitutions also lead to character variation along the S1 trajectory driving the photoisomerization of ASRAT to ASR13C (see Figure 1) decreasing or increasing reactive 1Bu or unreactive 2Ag characters. At this stage, it is essential to revise three important pieces of information that we fully detailed in ref. 17. These are: 1) the shape of the rPSB CASPT2 and CASSCF energy profiles are similar, 2) during the rPSB S1 relaxation starting at the FC point, the system may reach two conical intersections, namely CI1 and CI3, where first is a crossing between the S1 and S0 PESs associated with the 1Bu and 1Ag characters, while the second is a crossing between the S2 and S1 PESs associated with the 2Ag and 1Ag characters (a third conical intersection, CI2, between PESs dominated by 1Ag and 2Ag is higher in energy and it is not involved in rPSB photodynamics) and 3) the change in the total charge of a suitable rPSB moiety can be used to learn about the magnitude of the coupling between different PESs.
Figure 1.

Substituent effect on the relaxation of ASRAT. A. CASSCF and CASPT2 energy profiles (top panel) and evolution of CASSCF charge on the Schiff base side (bottom panel) of the Me-ASRAT. B. Same data for ASRAT. C. Same data for CF3-ASRAT. S0, S1 and S2 PESs are shown in blue, green and red colors respectively. The profiles shown in the bottom panels are the positive charge on the displayed molecular fragment.
In Figure 1, we present the results of our FC trajectory computations for ASRAT with and without retinal analogues. These include the evolution of the CASSCF and CASPT2 energies and of the total CASSCF Mulliken charges on a selected moiety of the chromophore (the Schiff base moiety -C14H-C15H=NH-). Note that such charges were computed at the CASSCF level to be consistent with geometrical progression.
Electron releasing substitution reduce the 1Bu/2Ag mixing and leads to a faster rhodopsin photoisomerization.
The results presented in Figure 1 are interpreted using the information 1–3 recalled above. Upon inspection of the energy profiles (Figure 1B top) one notes that in the case of ASRAT (central panels) the molecule remains on S1 for the whole simulation time. This observation is consistent with the fact that ASRAT decay takes experimentally more than 700 fs as reported by previous investigations.21,22 The charge variation displayed in Figure IB bottom shows that the Schiff base moiety of the rPSB carries an oscillating positive charge centered around ~0.6 on both S2 and S0 states and ~0.4 charge on S1 state. Recall that a large positive charge indicates 1Ag or 2Ag characters and a small charge indicates 1Bu character. Thus, in conclusion, the S1 PES displays an oscillating 1Bu character. These oscillations exhibit an out of-phase relationship with oscillations of S2 PES that is associated with the 2Ag character (i.e. 1Bu/2Ag coupling, see the framed region in Figure 1B bottom panel). Such a relationship indicates that S1 and S2 PESs are interacting with each other (i.e. the two states are mixed) and the S1 PES is only “partially” reactive.
As apparent from Figure 1A, the situation changes upon addition of a 10-methyl group in Me-ASRAT. The oscillatory relationship between 1Bu and 2Ag characters lasts for only about 60 fs and thereafter, 1Bu displays an out-of-phase oscillation relationship with 1Ag character. This means that S1 PES is interacting with S0 after 60 fs and continues until the molecule reach CI1 and decay to S0 at 110 fs eventually producing the 13-cis isomer. The slow decay of ASRAT and the fast decay of Me-ASRAT is consistent with the changes documented between all-trans-rPSB and 10Me-rPSB in methanol solution.6,8 In conclusion, in the protein cavity the 10-Me substituent induces the same effect seen in solution.
Electron withdrawing substituents increase 1Bu/2Ag mixing and leads to a slower (or blocked) photoisomerization.
Comparison of Figure 1B and 1C shows that replacing the methyl group at C9 position with a CF3 leads to strong S2/S1 state mixing. At the FC point of CF3-ASRAT, the optically allowed 1Bu-like state is higher in energy than the 2Ag-like state. This is observed in both CASSCF and CASPT2 energy profiles. Therefore, while CF3-ASRAT relaxation along the S1 PES remains dominated by a 1Bu character leads, within 62 fs, to a CI3-like structure with largely mixed 1Bu/2Ag characters and hops to the 2Ag dominated S2 PES. From S2, the system will then return to S1 through another CI3-like conical intersection. Another interpretation would be that the system remains on the S1 PES but remains partially “trapped” on a highly bumpy surface which frequently change electronic character between reactive (1Bu) and unreactive (2Ag) thus slowing down the isomerization motion. More specifically, the strong state mixing results in a bumpy adiabatic S1 potential energy surface formed by several avoided crossings between diabatic states with covalent (unreactive character, 2Ag) and charge transfer (reactive character, 1Bu). Even if the molecule retains on S1, the observed molecular motion on such a bumpy S1 PES would be attempts to overcome a barrier (caused by an avoided crossing) resulting in a slower photoreactivity. It is important to note that S1 and S2 CASPT2 energy profiles show a similar shape to CASSCF until the S1 to S2 hop point; but exhibit different shapes thereafter. Further inspection of the CASPT2 energy profiles show sudden increases in energy around 65 fs and 85 fs of the trajectory. This indicates that S1 PES interacts with a higher excited state PES as supported by the inspection of the charge profiles. In fact, during first ~90 fs of the CF3-ASRAT relaxation, we observe a dominating out-of-phase relationship between S1 and S2 charge profiles (1Bu/2Ag state mixing) similar to ASRAT. However, such relationship occasionally disappears and this is attributed to a coupling with another higher state. In order to track the interacting higher state, we recomputed energy and charge profiles at the 4-root-state-average level along the same geometries. The results indicate that the S3 PES is interacting with S1 (see Figure S3). On the basis of the above results we predict that the electron withdrawing group that we have introduced has increased state mixing of rPSB resulting in a longer decay time with respect to ASRAT. In order to better demonstrate the increase in decay time, we propagated a ASR13C trajectory. In fact, ASR13C that incorporates a native 13-cis chromophore, has an observed excited state lifetime of ca. 150 fs.21 As apparent from Figure 2A, our ASR13C model decays to ground state within 150 fs consistently with the previous theoretical and experimental studies.16,21 Inspection of charge profiles suggest that 1Bu/2Ag mixing persists until about 75 fs and then 1Bu/1Ag mixing takes over until the hop point (see framed regions in bottom panel). Such situation is completely different from that found after introduction of CF3.
Figure 2.

Effect of CF3 substitution on the relaxation of ASR13C. A. CASSCF and CASPT2 energy profiles (top panel) and evolution of charge on the Schiff base side (bottom panel) of ASR13C. B. Same data for CF3-ASR13C. S0, S1 and S2 PESs are shown in blue, green and red colors. The profiles shown in bottom panels are the positive charge on the displayed molecular fragment.
In fact, the FC region of CF3-ASR13C display similar features to the CF3-ASRAT and the relaxation initiates from a S2 PES which is dominated by the lB u character. The first ~15 fs of relaxation is dominated by 1Bu/2Ag mixing but the molecule reaches a CB-like point and hops to the S1 PES which is associated with unreactive 2Ag character. During this time, the S1 PES appears to interact with another state. This is evident from the absence of a 1Bu-like state that displays an out-of-phase relationship with 2Ag-like state (see Figure 2B, bottom panel). Recomputing the energies at 4-root-state-average level suggest that the interacting PES is S3 (see Fig. S3). The unreactive 2Ag character associated with S1 PES explains why CF3-ASR13C does not undergo isomerization similar to ASR13C More specifically, 9-trifluoromethylation has contributed to an increase in the electronic state mixing of ASR13C·
In the past, the effect of 9-trifluoromethylation on bR (CF3-bR) photoisomerization has been experimentally documented by Sheves and coworkers.10 In such work, the authors carried out steady state absorption experiments and second harmonic generation experiments of CF3-bR and native bR. The formation of photoproduct intermediates was also studied. The results showed that CF3-bR absorption maximum is blue shifted by 48 nm with respect to bR. Furthermore, the second harmonic generation signal of the former (0.375) was significantly low with respect to the latter (1.0). However, the authors observed photoproduct intermediates in both cases. The fact that calculated absorption maxima of CF3-ASRAT and CF3-ASR13C above are blue shifted with respect to their native counterparts is consistent with the results of bR absorption experiments. We also constructed QM/MM models for CF3-bR and bR and studied the absorption spectroscopic properties. Consistently with the experiments, the computed maximum absorption wavelength of the former is blue-shifted by 66 nm in comparison to the latter (see Table S1). At the FC region, the S1-S2 energy gap of both models remains close to each other, however, the increased S1/S2 mixing of CF3-bR is clearly evident as the molecule is allowed to relax on S1 PES.
Considerations for designing artificial rPSB analogues that are useful in high-resolution imaging.
Above we have provided computational evidence for a correlation between the type of rPSB substitutions (i.e. electron withdrawing or donating) and electronic state mixing. More specifically, a microbial rhodopsin hosting an electron releasing, native and electron withdrawing rPSBs display low, moderate and high amount of state mixing respectively. The corresponding decay times increase as we go from electron rich rPSB to electron poor rPSB. The relationship that we have reported here may be used as a design principle to develop rPSB chromophore analogues that, after insertion into a suitable opsin, would provide fluorescent probes useful in high resolution imaging. In fact, as reported in the literature, creating an excited state barrier and preventing the photoisomerization of rPSB is a way of engineering fluorescence into a rhodopsin.4 Since increasing electronic state mixing appears to hinder the photoisomerization, we propose to search for other electron withdrawing substituents and suitable substitution positions on the chromophore chain to increase the electronic state mixing of rPSB is another way of engineering fluorescence. According to the results presented above ASRAT is a potential candidate for such reengineering resulting in novel fluorescent probes.
Supplementary Material
ACKNOWLEDGMENT
This work was supported in part by the Italian MIUR for funding (PRIN 2015) and, in part, by the National Science Foundation under Grant No. CHE-1710191 and National Institute of Health under Grant No. R15GM126627. M.O. is grateful to USIAS (University of Strasbourg) for a 2015 fellowship and to the Ohio Supercomputer Center for granting computer time. We are also grateful to Dr. Alessio Valentini for helpful discussion.
Footnotes
ASSOCIATED CONTENT
Supporting Information contains computational details for: methodology, absorption energies and oscillator strengths, comparison of rPSB geometries and cavity residues of ASRAT models, energy profiles computed at 4-root-state-average level, energy and charge profiles of bR models. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).AzimiHashemi N; Erbguth K; Vogt A; Riemensperger T; Rauch E; Woodmansee D; Nagpal J; Brauner M; Sheves M; Fiala A; et al. Synthetic Retinal Analogues Modify the Spectral and Kinetic Characteristics of Microbial Rhodopsin Optogenetic Tools. Nat. Commun 2014, 5, 5810. [DOI] [PubMed] [Google Scholar]
- (2).Herwig L; Rice AJ; Bedbrook CN; Zhang RK; Lignell A; Cahn JKB; Renata H; Dodani SC; Cho I; Cai L; et al. Directed Evolution of a Bright Near-Infrared Fluorescent Rhodopsin Using a Synthetic Chromophore. Cell Chem. Biol 2017, 24, 415–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Sineshchekov OA; Govorunova EG; Wang J; Spudich JL Enhancement of the Long-Wavelength Sensitivity of Optogenetic Microbial Rhodopsins by 3,4-Dehydroretinal. Biochemistry 2012, 51, 4499–4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Laricheva EN; Gozem S; Rinaldi S; Melaccio F; Valentini A; Olivucci M Origin of Fluorescence in 11-Cis Locked Bovine Rhodopsin. J. Chem. Theory Comput. 2012, 8, 2559–2563. [DOI] [PubMed] [Google Scholar]
- (5).Kandori H; Katsuta Y; Ito M; Sasabe H Femtosecond Fluorescence Study of the Rhodopsin Chromophore in Solution. J. Am. Chem. Soc 1995, 117, 2669–2670. [Google Scholar]
- (6).Sovdat T; Bassolino G; Liebel M; Schnedermann C; Fletcher SP; Kukura P Backbone Modification of Retinal Induces Protein-like Excited State Dynamics in Solution. J. Am. Chem. Soc 2012, 134, 8318–8320. [DOI] [PubMed] [Google Scholar]
- (7).Bassolino G; Sovdat T; Liebel M; Schnedermann C; Odell B; Claridge TDW; Kukura P; Fletcher SP Synthetic Control of Retinal Photochemistry and Photophysics in Solution. J. Am. Chem. Soc 2014, 136, 2650–2658. [DOI] [PubMed] [Google Scholar]
- (8).Demoulin B; Altavilla SF; Rivalta I; Garavelli M Fine Tuning of Retinal Photoinduced Decay in Solution. J. Phys. Chem.Lett 2017, 4407–4412. [DOI] [PubMed] [Google Scholar]
- (9).Bayley H; Radhakrishnan R; Huang KS; Khorana HG Light-Driven Proton Translocation by Bacteriorhodopsin Reconstituted with the Phenyl Analog of Retinal. J. Biol. Chem 1981, 256, 3797–3801. [PubMed] [Google Scholar]
- (10).Zadok U; Khatchatouriants A; Lewis A; Ottolenghi M; Sheves M Light-Induced Charge Redistribution in the Retinal Chromophore Is Required for Initiating the Bacteriorhodopsin Photocycle. J. Am. Chem. Soc 2002, 124, 11844–11845. [DOI] [PubMed] [Google Scholar]
- (11).Ottolenghi M; Sheves M Synthetic Retinals as Probes for the Binding Site and Photoreactions in Rhodopsins. J. Membr. Biol 1989, 112, 193–212. [DOI] [PubMed] [Google Scholar]
- (12).Sheves M; Albeck A; Friedman N; Ottolenghi M Controlling the PKa of the Bacteriorhodopsin Schiff Base by Use of Artificial Retinal Analogues. Proc. Natl. Acad. Sci. U. S. A 1986, 83, 3262–3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Albeck A; Friedman N; Sheves M; Ottolenghi M Factors Affecting the Absorption Maxima of Acidic Forms of Bacteriorhodopsin. A Study with Artificial Pigments. Biophys. J 1989, 56, 1259–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Jung K-H; Trivedi VD; Spudich JL Demonstration of a Sensory Rhodopsin in Eubacteria. Mol. Microbiol 2003, 47, 1513–1522. [DOI] [PubMed] [Google Scholar]
- (15).Schapiro I; Ruhman S Ultrafast Photochemistry of Anabaena Sensory Rhodopsin: Experiment and Theory. Biochim. Biophys. Acta 2014, 1837, 589–597. [DOI] [PubMed] [Google Scholar]
- (16).Luk HL; Melaccio F; Rinaldi S; Gozem S; Olivucci M Molecular Bases for the Selection of the Chromophore of Animal Rhodopsins. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 15297–15302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Manathunga M; Yang X; Orozco-Gonzalez Y; Olivucci M Impact of Electronic State Mixing on the Photoisomerization Timescale of the Retinal Chromophore. J. Phys. Chem. Lett 2017, 8, 5222–5227. [DOI] [PubMed] [Google Scholar]
- (18).Frutos LM; Andruniów T; Santoro F; Ferré N; Olivucci M Tracking the Excited-State Time Evolution of the Visual Pigment with Multiconfigurational Quantum Chemistry. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 7764–7769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Andersson K; Malmqvist P-Å; Roos BO Second-Order Perturbation Theory with a Complete Active Space Self-Consistent Field Reference Function. J. Chem. Phys 1992, 96, 1218–1226. [Google Scholar]
- (20).Gozem S; Luk HL; Schapiro I; Olivucci M Theory and Simulation of the Ultrafast Double-Bond Isomerization of Biological Chromophores. Chem. Rev 2017, 117, 1350213565. [DOI] [PubMed] [Google Scholar]
- (21).Cheminal A; Leonard J; Kim S-Y; Jung K-H; Kandori H; Haacke S 100 Fs Photo-Isomerization with Vibrational Coherences but Low Quantum Yield in Anabaena Sensory Rhodopsin. Phys. Chem. Chem. Phys 2015, 17, 25429–25439. [DOI] [PubMed] [Google Scholar]
- (22).Wand A; Rozin R; Eliash T; Jung K-HH; Sheves M; Ruhman S Asymmetric Toggling of a Natural Photoswitch: Ultrafast Spectroscopy of Anabaena Sensory Rhodopsin. J. Am. Chem. Soc 2011, 133, 20922–20932. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.