

Abstract
Ribosome profiling has emerged as a powerful technique to study mRNA translation. Ribosome profiling has the potential to determine the relative quantities and locations of ribosomes on mRNA genome wide. Taking full advantage of this approach requires accurate measurement of ribosome locations. However, experimental inconsistencies often obscure the positional information encoded in ribosome profiling data. Here, we describe the Ribodeblur pipeline, a computational analysis tool that uses a maximum likelihood framework to infer ribosome positions from heterogeneous datasets. Ribodeblur is simple to install, and can be run on an average modern Mac or Linux-based laptop. We detail the process of applying the pipeline to high-coverage ribosome profiling data in yeast, and discuss important considerations for potential extension to other organisms.
1. Introduction
As the last step between transcription and protein production, mRNA translation is a crucial process in gene expression. Translation is highly regulated, and translation rates can have important effects on protein folding. Furthermore, translation rates are coupled to mRNA decay through quality control processes that respond to stalled ribosomes [1]. Ribosome profiling involves nuclease footprinting of ribosome positions on mRNA [2]. The millions of ribosome footprint locations that result from this powerful technique facilitate genome-wide analyses of translation processivity.
One of the most useful features of ribosome profiling is the 3-nucleotide periodicity often observed in footprint sequence reads. 3-nt periodicity results from consistent protection of specific lengths of mRNA corresponding to the molecular footprint of the ribosome. For example, most yeast ribosomes protect 28 nucleotides of mRNA from digestion with the nuclease RNase I [2]. Such “perfect” footprints show a high degree of 3-nt periodicity that can be used to infer the locations of ribosome A-sites by simply adding 15 to the location of read 5′ ends. This has been used to predict frame shifts, open reading frames (ORFs) and other gene features [3]. However, technical and experimental inconsistencies reduce the utility of the constant-nucleotide offset heuristic used in most studies.
The first challenge is that the read length from ribosome profiling experiments can span a wide range, from shorter than 20 nt to longer than 36 [3], with less than 15% being 28-nt reads [3]. The A-site position for 28-nt reads might not be suitable for other read lengths. Secondly, the locations of the 5′ end of reads with different read lengths are spread differently across the three reading-frames, indicating that the A-site location might not be constant for a given read length [6].
Many factors contribute to experimental inconsistencies in ribosome profiling datasets. The proportion of reads that have a 28-nt length can vary greatly due to differences in experimental design. Treatment with different translation inhibiting drugs can alter the conformation of ribosomes, and hence the length of protected RNA [4]. Finally, differences in fragment size selection can change the length distribution of protected RNA fragments [5]. Variation in RNase I digestion time or activity can also affect the distribution of footprint sizes. These differences in read-length and frame distributions complicate the analysis of ribosome profiling data.
We previously published a computational method to recover A-site locations from ribosome profiling reads [6]. Our method does not assume a constant A-site offset for a given read length, nor do we assume any prior knowledge of the RNase I digestion pattern, but learn such pattern directly from the data. Our method produces ribosome profiles with sub-codon resolution, with consistent profiles among different read lengths, and with a high concentration of read pileups towards a single reading frame, therefore successfully reduces the noise in ribosome profiling data caused by the complicated experiment procedure.
Here, we re-engineered our method into a command-line-based package, the Ribodeblur pipeline, to recover more accurate A-site locations from ribosome profiling reads of multiple lengths (Fig. 1). Ribodeblur assumes that the observed location of ribosome profiling reads reflects a blurring of actual ideal locations resulting from experimental variation in nuclease digestion [6]. Ribodeblur uses a maximum expectation framework to estimate functions that best fit the blurring process for each relevant read length, and then uses these functions to deblur the data and infer the most probable A-sites from ribosome profiling reads.
Fig. 1.
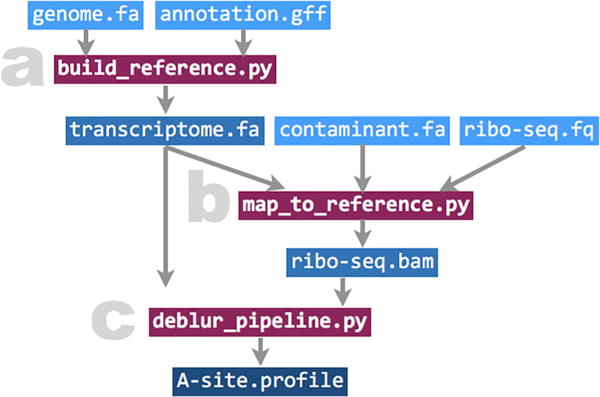
Diagram of the Ribodeblur pipeline. Input and output files are marked in blue, and scripts for each step are marked in red. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
2. Materials and methods
The essential step in the Ribodeblur pipeline is the deblur step ( deblur_pipeline.py). This step takes a transcriptome reference fasta file and a ribo-seq alignment bam file as inputs and outputs transcript-specific A-site profiles. Ribodeblur requires the transcriptome reference to have both coding regions (CDS) and untranslated regions (UTR) to be included, with the CDS range described in the fasta headers. A helper script ( build_reference.py) is included in the package to generate such a transcriptome fasta file. Ribodeblur also includes another helper script ( map_to_reference.py) to align ribo-seq reads to the transcriptome (Fig. 1). Here we provide detailed instructions to install and run the Ribodeblur pipeline.
- Collect the Ribodeblur source files and data.
- Supported Operating Systems: A computer running either Apple Mac OS X or Linux is required for Ribodeblur. On a laptop with a 4-core Intel i7 processor and 4 Gb of RAM, the pipeline takes less than one hour to process aligned ribosome profiling reads and create deblurred ribosome profiles.
- Download Ribodeblur source files: The programs required for the pipeline can be downloaded from the Kingsford lab Github repository (https://github.com/Kingsford-Group/ribodeblur). Download the pipeline as a ZIP file and unzip it to create a directory containing all the programs needed for Ribodeblur.
-
Download references: Ribodeblur requires ribosome profiling data in Illumina fastq format, a yeast fasta formatted transcriptome file, and a yeast fasta formatted contaminants (rRNA and other ncRNA) file as input. Ribodeblur comes with a shell script ( download_Refs.sh) that automatically grabs the reference genome, annotation (gff), and ncRNA files from the Ensembl ftp server. Simply run:
./download_Refs.sh
The resulting files will be placed in the user’s $HOME directory under data/Ribodeblur/Refs./
-
Set up the deblur pipeline prerequisites.
Ribodeblur requires Python 3.6 and Python packages numpy (1.13.0), scipy (0.19.1), bcbiogff(0.6.4), biopython (1.68) and pysam (0.11.2.2), and the STAR aligner (2.5.3a) [7]. While users can install these individually, we recommend using conda (https://conda.io/docs/) to setup a local environment with all necessary packages. Please follow the instructions below to setup the prerequisites.- Install miniconda:
- First, miniconda needs to be installed. If miniconda is not yet installed, follow these steps to set it up: To get the miniconda installation executable for Linux, run: curl https://repo.continuum.io/miniconda/Miniconda3-latest-Linux-x86_64.sh.
- Or for Mac, run: curl https://repo.continuum.io/miniconda/Miniconda3-latest-MacOSX-x86_64.sh. Alternatively, the user can google miniconda and get the most appropriate file.
- To install miniconda, run:
bash Miniconda3-latest-Linux-x86_64.sh –b –p $HOME/miniconda
- This step will install miniconda by running the shell script being downloaded. $HOME is the user’s home directory. Miniconda will be installed in a subdirectory called “miniconda”.
- Finally, to export path, run:
export PATH=“$HOME/miniconda/bin:$PATH
- This step will place the miniconda files in the user’s path. Unless this command is placed in bash.profile, the user will need to export the path each time Ribodeblur is used.
- Set up prerequisites: This is done by creating a conda virtual environment. Run:
-
conda env create –f ribodeblur.yml
The ribodeblur.yml file is included in the Github download unzipped in step 1. This is the conda environment yaml file and this command will setup the conda environment with all of the required packages listed above in step 2.
-
- Activate the conda environment: In the command line, run:
-
source activate Ribodeblur
This will activate the Ribodeblur virtual environment. The advantage of doing this is that it separates all the Ribodeblur required packages from the rest of the computer system. In other words, if the user has Python programs that require different versions of (e.g.) numpy, the above process will not overwrite the system numpy version with the version used by Ribodeblur.
-
- Run the deblur pipeline.
- Generate a transcriptome fasta file. Ribodeblur requires ribosome profiles from both coding regions (CDS) and a small portion of untranslated regions (UTR). To generate a transcriptome fasta file with UTR regions included, run:
-
python build_reference.py -f GENOME.FA -a ANNOTATION.GFF -o TRANSCRIPTOME.FA
where GENOME.FA is the genome fasta file and ANNOTATION.GFF is the annotation file, both should be downloaded from Ensembl. TRANSCRIPTOME.FA is the transcriptome reference the script generated. The optional parameter -p specifies the UTR padding regions added to each transcript. The default padding is 100 nucleotides.
-
- Align ribo-seq fastq sequence reads to the transcriptome. As a test case, Ribodeblur includes a shell script ( download_riboseq.sh) that can be used to download published example data [8] from National Center for Biotechnology Information Sequence Read Archive (NCBI SRA).
- Ribodeblur aligns the raw ribo-seq reads to the transcriptome with the STAR aligner [7]. It automatically builds a STAR index of the reference transcriptome, filters potential non-mRNA contaminants, and aligns the remaining reads to the transcriptome. To align ribo-seq reads to the transcriptome, run:
python map_to_reference.py -c CONTAMINANT.FA -t TRANCRIPTOME.FA -r ‘ RIBOSEQ.FQ -id STAR_IDX_DIR -ad STAR_ALIGN_DIR -p NTHREAD
- where CONTAMINANT.FA is the fasta formatted non coding RNA (ncRNA) file from Ensembl (see step 1c), TRANSCRIPTOME.FA is the fasta formatted transcriptome file created in step 3a, and RIBOSEQ.FQ is the fastq formatted ribosome profiling data. The –id option defines the name of the STAR index directory, and the –ad option defines the directory to output STAR alignments. The –p option allows the user to choose the number of threads used for alignment (the number of CPU cores on the computer that can be used).
- Deblur the ribosome profiles and output A-site profiles. The final step of Ribodeblur is the deblur step, where the read alignments are processed and grouped by read length for each transcript. Then for each read length, a blur vector is learned from a meta-profile combining all transcripts, and each transcript profile is deblurred independently. Lastly, the deblurred profiles from different read length are merged together to obtain a final ribosome A-site profile.
- During the deblur process, only profiles with enough coverage are used. This is because the core of Ribodeblur is reconcentrating off-frame ribo-seq reads back to their in-frame locations, and only profiles with high coverage have enough reads to be moved around. If the profile is too sparse, there will not be enough reads to be redistributed. Here, only profiles with more than half of the loci with non-zero read counts are kept. For example, a transcript with length of 120 bp (40 codons) need to have >60 nucleotide positions with non-zero reads to be included in the deblur pipeline. To generate deblurred A-site profiles, run:
python deblur_pipeline.py -r TRANSCRIPTOME.FA -b RIBOSEQ.BAM -o OUTPUT_PREFIX
- where TRANSCRITPOME.FA is the transcriptome reference generated from step 3a, RIBOSEQ.BAM is the alignment file generated from step 3b (located in the STAR_ALIGN_DIR), and OUTPUT_PREFIX is the user-defined prefix of the output for the A-site profile (what the user wants to call the output file). The final output of this step is an A-site profile starts with OUTPUT_PREFIX and ends with .profile. The resulting output file has the following format:
YCR024C-A: 47 116 57 1341 6 1 12 6 16 1986 10 28 3568 29 18 802 53 38 . . .
- Each line starts with the transcript name followed by the read count for each nucleotide location within the coding region. The read count vector is the sum of all read-length-specific profiles post deblur. After the deblur process, ribosome profiling data will typically have much more of the resulting data in frame-0 (increased frame skewness, Fig. 2). For transcripts marked “(no deblur)”, all of the read-length-specific ribosome profiles lacked sufficient depth to deblur the profile. In those cases, only offset shifts (e.g. +15 nt for 28-mer reads) were performed.
Fig. 2.

One example of the A-site profile (0-based) generated by Ribodeblur for transcript YCR024C-A before deblur (A) and after deblur (B). Deblur greatly increases the frame skewness. In-frame read portion is boosted from 57% to 94% after deblur.
3. Discussion
The deblur process identifies the reading frame of many reads that would otherwise simply be found out-of-frame. It produces ribosome profiles with a 3-nt periodicity with a high concentration of reads in one reading frame. This is important for de novo ORF prediction, the identification of frameshifts, and evaluation of codon-specific ribosome dwell times. Therefore, deblurred ribosome profiles can in theory improve all of these analyses. Because the deblurring process is data-driven, the experimental heterogeneity among datasets can potentially be reduced, improving attempts to compare the results of datasets produced in different laboratories. However, there exist several challenges and limitations for the current deblur pipeline. Here, we discuss some important considerations in using the Ribodeblur pipeline and potential areas for future improvement.
As with any software package, some users may encounter difficulty running Ribodeblur. We anticipate the most common problems would involve syntax errors, in which case users should take care to enter the commands as shown in the manuscript. Improper implementation of the miniconda management system may also cause problems for some users. We chose to use miniconda to facilitate installing and running Ribodeblur. However, users must remember to activate the Ribodeblur environment each time they wish to use the pipeline, and may want to deactivate the environment afterwards. Additional troubleshooting and support is available by contacting the authors.
3.1. Expanding Ribodeblur for use in other species with modified transcriptome reference files
Although the deblur process improves the periodicity of yeast data, it is currently not directly suitable for use on ribosome profiling data from other species. The main reason is the pipeline runs on single-isoform genes. Genes with alternative splicing (i.e. most metazoan genes) are thus not compatible with Ribodeblur. While one tool (Ribomap) is available to de-convolve the ribosome profiles of overlapping isoforms [9], estimating deblur vectors is likely to be challenging due to confounding factors such as multi-mapping among isoforms.
One way to enable deblur for multi-isoform organisms is to select one representative isoform for each transcript after constructing the transcriptome fasta file. This way multi-mapping can be greatly reduced for organisms with prevalent alternative splicing (such as human). Since Ribodeblur only uses uniquely mapped reads, providing such a constrained set of transcripts allows enough data to be kept for extracting the blur vectors. Thanks to the modular design of Ribodeblur, users can perform reference construction and mapping with alternative settings. The deblur step only requires a transcript fasta file (with annotated CDS regions) and an alignment bam file.
3.2. Adjusting length-specific A-site offsets for other experimental protocols
Ribodeblur currently uses fixed offsets that vary by fragment length (e.g. +15 for 28-mer reads) to initialize the deblur process. These offsets were determined by observation in yeast data. However different offsets have been reported for mammalian ribosome profiling data [10]. Further, the necessary use of MNase (as opposed to RNase I) in insect ribosome profiling experiments carries distinct nucleotide digestion patterns [11]. Thus, using Ribodeblur on data generated with protocols different from the one used in yeast would require changes in the hard-coded offset parameters, and further validation is needed to extend the usefulness of the Ribodeblur pipeline towards correction of metazoan ribosome profiles.
3.3. Adjusting deblur output profile range for de novo ORF discovery
Ribodeblur is designed to reduce noise in ribosome profiles for known transcripts. Only CDS regions are included in the A-site profiles produced by Ribodeblur. However, users can include additional UTR regions for the output profiles by changing the hard-coded profile range. This can further assist uORF finding near any known transcripts.
3.4. Evaluating experimental reproducibility by comparing deblurred ribosome profiles
Ribosome profiling is a complex procedure. Different labs use slightly different protocols that can produce different ribo-seq results. The Ribodeblur pipeline can extract the nucleotide digestion pattern directly from the ribosome profiling data. These blur vectors can be used to compare different data sets. Drastically different blur vectors between two replicates might indicate inconsistencies between the two experiments. Thus, this might serve as a sanity check for reproducibility. Since the deblur pipeline can automatically learn the digestion patterns and output corrected ribosome profiles with less noise, it should increase the consistencies of experiments on the same species across different labs. However, further analyses are needed to evaluate such a functionality.
4. Conclusion
We present the Ribodeblur package for correction of yeast ribosome profiles. Ribodeblur can be used on relatively modest computer hardware (e.g. modern laptop computers). The resulting deblurred ribosome profiles have improved 3-nucleotide periodicity, which should improve de novo ORF finding and codon-level analyses in yeast. Future improvements to the pipeline would allow extension of this approach to metazoans.
Acknowledgments
This research is funded in part by the Gordon and Betty Moore Foundation’s Data-Driven Discovery Initiative through Grant GBMF4554 to C.K., and by the US National Institutes of Health (R01HG007104 and R01GM122935 to CK and R01GM121895 to C.J.M.).
References
- 1.Hanson G, Coller J. Codon Optimality, Bias and Usage in Translation and mRNA Decay. Nature Publishing Group; 2017. pp. 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ingolia NT, Ghaemmaghami S, Newman JRS, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. https://doi.org/10.1126/science.1168978 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Calviello L, Ohler U. Beyond read-counts: Ribo-seq data analysis to understand the functions of the transcriptome. Trends Genetics. 2017;33:728–744. doi: 10.1016/j.tig.2017.08.003. https://doi.org/10.1016/j.tig.2017.08.003 . [DOI] [PubMed] [Google Scholar]
- 4.Lareau LF, Hite DH, Hogan GJ, Brown PO. Distinct stages of the translation elongation cycle revealed by sequencing ribosome-protected mRNA fragments. eLife. 2014;3:e01257. doi: 10.7554/eLife.01257. https://doi.org/10.7554/eLife.01257 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guydosh NR, Green R. Dom34 rescues ribosomes in 3’ untranslated regions. Cell. 2014;156:950–962. doi: 10.1016/j.cell.2014.02.006. https://doi.org/10.1016/j.cell.2014.02.006 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang H, McManus J, Kingsford C. Accurate recovery of ribosome positions reveals slow translation of wobble-pairing codons in yeast. J Comput Biol. 2017;24:486–500. doi: 10.1089/cmb.2016.0147. https://doi.org/10.1089/cmb.2016.0147 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. https://doi.org/10.1093/bioinformatics/bts635 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Albert FW, Muzzey D, Weissman JS, Kruglyak L. Genetic influences on translation in yeast. PLoS Genet. 2014;10:e1004692. doi: 10.1371/journal.pgen.1004692. https://doi.org/10.1371/journal.pgen.1004692.s016 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang H, McManus J, Kingsford C. Isoform-level ribosome occupancy estimation guided by transcript abundance with Ribomap. Bioinformatics. 2016;32:1880–1882. doi: 10.1093/bioinformatics/btw085. https://doi.org/10.1093/bioinformatics/btw085 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. https://doi.org/10.1016/j.cell.2011.10.002 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dunn JG, Foo CK, Belletier NG, Gavis ER, Weissman JS. Ribosome profiling reveals pervasive and regulated stop codon read-through in Drosophila melanogaster. eLife. 2013;2:e01179. doi: 10.7554/eLife.01179. https://doi.org/10.7554/eLife.01179.026 . [DOI] [PMC free article] [PubMed] [Google Scholar]