

SUMMARY
Extracellular microRNAs (miRNAs) and other small RNAs are implicated in cellular communication and may be useful as disease biomarkers. We systematically compared small RNAs in 12 human biofluid types using RNA sequencing (RNA-seq). miRNAs and tRNA-derived RNAs (tDRs) accounted for the majority of mapped reads in all biofluids, but the ratio of miRNA to tDR reads varied from 72 in plasma to 0.004 in bile. miRNA levels were highly correlated across all biofluids, but levels of some miRNAs differed markedly between biofluids. tDR populations differed extensively between biofluids. Y RNA fragments were seen in all biofluids and accounted for >10% of reads in blood plasma, serum, and cerebrospinal fluid (CSF). Reads mapping exclusively to Piwi-interacting RNAs (piRNAs) were very rare, except in seminal plasma. These results demonstrate extensive differences in small RNAs between human biofluids and provide a useful resource for investigating extracellular RNA biology and developing biomarkers.
Graphical Abstract
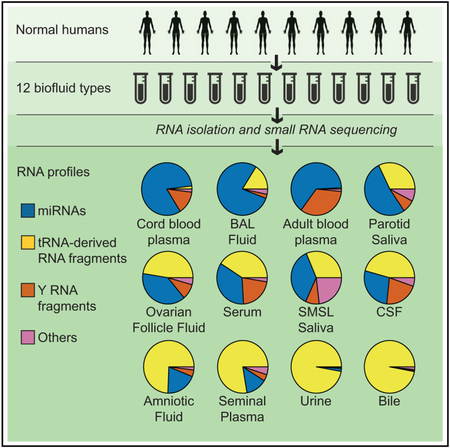
In Brief
Using a standardized sequencing-based approach, Godoy et al. characterize small RNAs in 12 normal human biofluids. They find that each biofluid contains an extensive collection of small RNAs that belong to multiple biotypes. The relative abundance of these RNAs varies widely between biofluids.
INTRODUCTION
RNAs released from cells have been detected in many biofluids (Patton et al., 2015). Although some reports suggest that large RNAs, including functional mRNAs, can be present in biofluids (Ni et al., 2002; Skog et al., 2008), most extracellular RNAs (exRNAs) are small RNAs (Hoy and Buck, 2012). Many reports have focused on microRNAs (miRNAs), which are small (typically 21–22 nt) RNAs produced by intracellular processing of larger precursor RNAs (Argyropoulos et al., 2013; Lee et al., 2010; Mitchell et al., 2008; Weber et al., 2010; Williams et al., 2013). Several pathways have been proposed to lead to release of miRNAs from cells, and extracellular miRNAs can be found within exosomes and in complexes containing argonaute proteins (Arroyo et al., 2011) or lipoproteins (Patton et al., 2015). Extracellular miRNAs can enter cells and may target mRNAs in those cells (Patton et al., 2015). The potential of miRNAs as disease biomarkers is being explored by many investigators (Argyropoulos et al., 2013; Barger et al., 2016; Gray et al., 2017; Mitchell etal., 2008).
Small RNA biotypes other than miRNAs have also been detected in biofluids. Piwi-interacting RNAs (piRNAs) are ~23- to 30-nt RNAs involved in transcriptional and post-transcriptional silencing of transposons and other targets in germ cells (Czech and Hannon, 2016). Sequences mapping to piRNAs have been reported in human seminal fluid plasma (Hong et al., 2016) as well as in blood plasma (Freedman et al., 2016), saliva (Bahn et al., 2015), and urine (Yeri et al., 2017). Many small RNAs found in biofluids are thought to be derived from larger RNAs by specific processing events or by nonspecific degradation. tRNA-derived RNAs (tDRs) are generated by cleavage of tRNAs at specific sites (Gebetsberger and Polacek, 2013). The variety of tDRs is extensive since there are many (>600) human tRNA genes, and multiple fragment types, including 5’ halves, 3’ halves, 5’ tRNA fragments (tRFs), 3’ tRFs, and internal tRFs (i-tRFs), have been identified (Loher et al., 2017). Small RNAs derived from mRNAs, long non-coding RNAs, rRNAs (Semenov et al., 2004), and Y RNAs (Dhahbi et al., 2013) have also been detected in some biofluids. Their functions are not yet well understood.
Extracellular RNAs have been detected in at least 15 biofluids to date (Sohel, 2016). The total concentration of RNA varies widely between biofluids, with certain biofluids such as breast milk and seminal fluid being more concentrated than more dilute biofluids like cerebrospinal fluid (CSF) and urine (Weber et al., 2010). Given the difficulties inherent in absolute quantification of exRNAs, most analyses have focused on relative quantification of specific RNAs. This approach has identified some differences in small RNA content between biofluid types. For example, a PCR-based study of 714 miRNAs identified certain miRNAs that were abundant in most of the 12 biofluids studied along with other miRNAs that were enriched in specific biofluids (Weber et al., 2010). However, a more complete understanding of small RNA differences between biofluids is problematic, since published studies rely on a diverse set of methods with different biases that preclude direct comparisons. Furthermore, most studies have focused primarily or exclusively on miRNAs, and much less information is available about other RNA biotypes.
One objective of the NIH Extracellular RNA Communication Consortium (ERCC) is to identify the range of RNAs present in human biofluids. To address this goal, we compared the small RNA populations of a large and diverse collection of human biofluids using one standard RNA sequencing (RNA-seq) approach that was validated as part of a multicenter ERCC study (Giraldez et al., 2017). This approach was designed for miRNAs but can also detect other small RNAs with a 5’ phosphate and a 3’ hydroxyl group. Our analysis of a total of 129 samples of 12 biofluid types from human donors reveals the presence of complex RNA repertoires in all biofluids and major differences in RNA composition between biofluid types. These results are publicly available through the exRNA Atlas (https://exrna-atlas.org).
RESULTS
Characteristics of the Study Population
We obtained samples of 12 different biofluid types (Table 1). With the exception of bile samples, which were obtained from patients who had previously undergone liver transplantation and had intact liver function, all samples were obtained from healthy subjects. For each biofluid, we obtained 5–15 samples. In general, each sample of a given biofluid was obtained from a different participant, except that the 10 ovarian follicle fluid samples were obtained from five participants who each provided two samples. Plasma, serum, and urine samples were obtained from a separate cohort of 12 individuals who each provided samples of each of these three biofluids. For saliva, another cohort of 15 participants provided samples of both parotid saliva and submandibular and sublingual (SMSL) saliva. For amniotic fluid, bronchoalveolar lavage (BAL) fluid, bile, cord blood plasma, CSF, and seminal fluid, 10–12 participants from separate cohorts each provided a single sample of one biofluid.
Table 1.
Study Participant Demographic Summary
| Race |
Ethnicity |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Biofluid | Sex | Age (y)a | African American |
Asian | White | Other (mixed) |
Unknown | Pacific Islander |
Hispanic | Non-Hispanic | Declined to State |
Unknown |
| Amniotic fluid | 10F | Unknown | – | – | – | – | 10 | – | – | – | – | 10 |
| BAL fluid | 4F, 6M | 29.5 (28.3–33.0) | 3 | 1 | 6 | – | – | – | 1 | 9 | – | – |
| Bile | 6F, 6M | 57.5 (53.3–60.3) | – | – | 12 | – | – | – | – | 11 | 1 | – |
| Cord blood plasma (maternal demographics) | 10F | 31 (29.5–33.5) | 1 | – | 4 | 2 | 2 | – | 3 | 5 | – | 2 |
| Cord blood plasma (fetal demographics) | 6F, 4M | 0 (0–0) | 1 | 2 | 2 | 3 | 2 | – | 3 | 4 | – | 3 |
| CSF | 3F, 8M | 40 (37.5–43.0) | 6 | 1 | 4 | – | – | – | – | 11 | – | – |
| Ovarian follicle fluid | 5F | 28 (26–28) | – | 2 | 3 | – | – | – | 5 | – | – | |
| Plasma | 9F, 3M | 26 (24.8–35.3) | – | 7 | 4 | – | – | 1 | 2 | 10 | – | – |
| Serum | 8F, 3M | 26 (24.5–30.0) | – | 7 | 3 | – | – | 1 | 1 | 10 | – | – |
| Urine | 8F, 2M | 27.5 (25.3–43.8) | – | 7 | 2 | – | – | – | 1 | 9 | – | – |
| Parotid saliva | 8F, 5M | 29 (24–30) | – | 3 | 8 | – | 2 | – | 2 | – | – | 11 |
| Submandibular and sublingual saliva | 8F, 7M | 29 (27–33) | – | 3 | 10 | – | 2 | – | 2 | – | – | 13 |
| Seminal fluid | 10M | 40.5 (33.8–40.5) | 1 | 3 | 6 | – | – | – | 1 | 9 | – | – |
F, female; M, male. Dashes indicate no data.
Median (interquartile range).
Sequencing and Quality Control
RNA-seq reads were aligned to the human transcriptome using the Genboree/exceRpt pipeline (http://www.genboree.org/index.html). This approach resulted in a substantial proportion of read counts assigned to piRNAs in adult blood plasma (median 13% assigned to piRNA), cord blood plasma (5%), and serum (6%) samples. Large proportions of reads that mapped to piRNA in these three biofluids mapped to a single piRNA sequence (piRNAbase hsa_piR_016658, GenBank: DQ592931; 79% of piRNA reads in adult blood plasma, 48% in cord blood plasma, and 43% in serum). However, these reads also map to a Y RNA (RNY4), and each of these biofluids also contains a large number of other reads that map to other regions of RNY4. In the aggregate, >99.7% of plasma or serum reads mapping to piRNAs also mapped to RNA(s) from a different biotype. Based upon these observations, we changed from the default order of read assignment to align to Gencode RNAs (which include Y RNAs) before aligning to piRNAs. Using this approach, 0.3% of seminal plasma reads mapped to piRNAs, whereas <0.1% of reads in other biofluids mapped to piRNAs.
A total of 145 samples were analyzed. 16 samples failed quality control despite repeat analysis (see Experimental Procedures for description of quality control criteria). The remaining 129 samples were used for the analyses presented here. For these samples, the mean number of reads that passed quality control was 12.8 × 106, and mean reads exceeded 7 × 106 per sample for each biofluid type (Table S1). The proportions of reads that were shorter than 18 nt (no mapping attempted) and reads mapped to rRNA differed between biofluid types (Table S1). In three biofluid types (parotid and submandibular and sublingual saliva and CSF), the proportion of reads that mapped to the human transcriptome was relatively low. Saliva contains a relatively high proportion of reads that map to bacterial genomes (Yeri et al., 2017). The basis of the low CSF alignment rate is unclear but our miRNA mapping rate for CSF (2.5% of reads passing quality control) was similar to the 1.5% rate reported in a previous study of CSF from healthy controls and individuals with amyotrophic lateral sclerosis (Waller et al., 2018). 54.7% of CSF reads aligned to genomes of other species. These included bacterial (11.3%), fungal (7.6%), and archaeal (0.05%) sequences as well as many sequences that could not be unambiguously assigned even at the kingdom level. These non-human sequences likely represent contaminants that were relatively frequent in normal CSF libraries due to a low concentration of exRNAs in this biofluid.
Relative Abundances of RNA Biotypes Differ Widely between Biofluids
The fraction of transcriptome-aligned reads mapping to different biotypes of human RNAs varied markedly between biofluid types (Figure 1; Table S1). The median rate of mapping to miRNAs was >50% for adult and cord blood plasma and BAL fluid, whereas tDRs represented >50% of reads in bile, urine, seminal plasma, and amniotic fluid. miRNAs and tDRs each accounted for >25% of reads in the remaining biofluids (parotid saliva and submandibular and sublingual saliva, ovarian follicle fluid, serum, and CSF). The relative abundance of miRNA to tRNA reads varied by >103-fold (from 72 in plasma to 0.004 in bile). Y RNA fragments represented >10% of mapped reads in adult and cord blood plasma, serum, and CSF but <0.8% of reads in urine and bile, with intermediate levels in other biofluids. miRNAs, tRNAs, and Y RNAs accounted for >90% of all mapped reads except in submandibular and sublingual saliva, which had the largest proportion of reads mapping to portions of protein coding genes (mRNAs, 7.6%), small nuclear RNAs (snRNAs) (2.3%), retained introns (1.5%), and Gencode “processed transcripts” (1.2%). This may reflect the presence of RNA degrading enzymes, cellular debris, or microbial-derived small RNAs that map to the human genome in saliva samples. Reads that mapped to piRNAs, but not to miRNAs, tRNAs, Y RNAs, or other Gencode transcripts, represented 0.30% of reads in seminal plasma and <0.10% of reads in other biofluids. These results demonstrate that multiple classes of small RNAs are represented in each biofluid type studied, but the relative abundance of these classes varies widely between biofluids.
Figure 1. Distribution of RNA Biotypes Differs between Biofluids.

Reads mapping to miRNAs, tRNAs, Y-RNAs, piRNAs, mRNAs, or other RNA biotypes as a fraction of total reads mapping to the human transcriptome. Boxes represent median and interquartile ranges, whiskers represent 1.5 times the interquartile range, and dots represent outliers.
miRNA Profiles
In each biofluid, we detected hundreds of miRNAs, but a small number of miRNAs accounted for a large proportion of miRNA read counts (Figures 2A, 2B, and S1; Table S2). Between 395 (parotid saliva) and 541 (CSF) miRNAs had a median of ≥;10 reads per million total miRNA reads in each biofluid. The 10 most frequent miRNAs represented between 39% (CSF) and 62% (serum) of total miRNA reads (Table S3). Despite marked differences in small RNA classes between biofluids, pairwise correlation coefficients for miRNA read counts between biofluids were generally high (Figures 2C–2E). Correlations were highest between blood-derived biofluids (plasma, cord plasma, and serum; R = 0.94–0.98) and between saliva samples from the two different sites (R = 0.98). Seminal fluid and cord blood plasma were the least correlated (R = 0.79). Using all 2,153 miRNAs detected in any sample, transfer stochastic neighbor embedding (tSNE) analysis revealed that samples of most biofluid types formed distinct clusters (Figure 2F). Samples of saliva from two different sites (parotid and submandibular and sublingual glands) formed overlapping clusters and two serum samples could not be clearly distinguished from the cluster of adult plasma samples. Therefore, although correlations in miRNA levels between biofluids were high, each biofluid (with the exception of the saliva samples from two different sites) had a distinct pattern of miRNAs.
Figure 2. miRNA Profiles in 12 Biofluid Types.

(A) Number of miRNAs detected as a function of read depth.
(B) Cumulative distribution of miRNA reads. See also Figure S1.
(C and D) Examples of pairwise correlations between biofluids for cord blood plasma versus adult blood plasma (C) and cord blood plasma versus seminal plasma (D) Each point represents the median normalized read count for a single miRNA for the indicated biofluids. One normalized read count was added to each measurement to allow representation of log read counts for miRNAs with no reads.
(E) Correlations for all pairs of biofluids.
(F) tSNE plot produced using miRNA read counts. Each point represents a single biofluid sample.
To validate the ability of our RNA-seq method to detect differences in miRNA levels between biofluids, we used quantitative PCR (qPCR) to measure 103 miRNAs in independent samples of two biofluid types: adult blood plasma and BAL fluid (Table S4). qPCR and RNA-seq results were correlated (R = 0.72, p < 2.2 × 10−16; Figure S2; Table S4). Of these 103 miRNAs, 38 were at least 2-fold more abundant in one of these two biofluids compared with the other by RNA-seq (false discovery rate [FDR] < 0.05). For 28 of these 38 miRNAs, concordant and statistically significant (FDR <0.05) differences were also found by qPCR.
A total of 15 miRNAs had much higher relative abundance in one biofluid than any other biofluid (≥ 103 reads/106 total miRNA reads, >10-fold higher in one biofluid than all other biofluids, adjusted p < 0.05 for pairwise comparisons with all other biofluids by negative binomial Wald test; Table S5). In some cases, these levels are likely explained by high expression of the miRNAs by cell types that are in direct contact with the biofluid. Three of the four miRNAs with higher levels in amniotic fluid (miR-483-5p, miR-1247-5p, and miR-433-3p) are highly enriched in extraembryonic cells (amniotic epithelial cells, placental epithelial cells, or chorionic membranes) (de Rie et al., 2017). The three miRNAs with much higher levels in CSF are miR-9-3p, which is highly enriched in the vertebrate nervous system; miR-1911-5p, a brain-specific miRNA that was detected in CSF exosomes, but not blood plasma exosomes (Yagi et al., 2017); and miR-1298-5p, which is among the most abundant miRNAs in CSF exosomes (Yagi et al., 2017). miR-891a, which had much higher expression in seminal plasma, has been reported to be among the most abundant miRNAs in epididymis (Li et al., 2012). Combining results all blood-derived biofluids (adult and cord blood plasma and serum) into a single group and comparing with results each of the other biofluids did not identify any miRNAs that met the above criteria for much higher expression in blood-derived biofluids versus all other biofluids. Similarly, combining results from parotid and submandibular and sublingual saliva samples did not identify any miRNAs with much higher expression in saliva versus all other biofluids.
A comparison of umbilical cord blood plasma and adult blood plasma revealed that 18 miRNAs differed in relative abundance (Table S6). Three miRNAs (miR-487b-3p, miR-376c-3p, and miR-127-3p) were >5-fold higher in cord blood plasma. A previous report detected similar differences between cord and adult blood plasma and showed relatively high levels of each of these miRNAs in placenta (Williams et al., 2013). One miRNA, let-7b-5p, was >10-fold lower in cord blood plasma; this large difference was also seen previously (Williams et al., 2013).
We identified six groups of five or more miRNAs with similar abundance patterns across biofluids using Bayesian relevance network analysis (Ramachandran et al., 2017) (Figure 3). The largest group (group 1) contained 45 miRNAs. A subgroup (1A) containing 31 of these 45 miRNAs had levels that were highest in amniotic fluid and cord blood plasma. All 31 subgroup 1A miRNAs are derived from the 14q32 cluster, which is the largest miRNA cluster in the human genome (54 miRNAs) (Hill et al., 2017). Subgroup 1B contained 14 miRNAs that were highest in cord blood plasma and adult blood plasma and serum but low in amniotic fluid. These include two pairs of miRNAs produced from the same pre-miRNA (miR-486-5p and miR-486-3p and miR-126-5p and miR-126-3p) and three miRNAs produced from a cluster on chromosome 17 (miR-451a, miR-144-3p, and miR-4732-3p). Group 2 (8 miRNAs) includes 5 miRNAs from the X chromosome miR-506-514 cluster; these miRNAs were highest in ovarian follicle fluid and were also relatively abundant in seminal plasma. Group 3 (23 miRNAs) was enriched for miRNAs derived from the two miR-200 family clusters, miRs-200b/a/429 and miRs-200c/141. The miR-200 family has important roles in epithelial cells (Korpal and Kang, 2008) and miRNAs in this group were most abundant in seminal plasma and saliva and least abundant in blood-derived biofluids. Group 4 comprises 7 miRNAs that were relatively abundant in bile, and group 5 comprises 5 miRNAs that were relatively abundant in CSF. Group 6 (23 miRNAs) included a subgroup of 17 miRNAs with highest relative abundance in urine and a second subgroup with 6 members of the miR-34/449 family that were most abundant in CSF, BAL, and amniotic fluid.
Figure 3. miRNAs with Highly Correlated Read Counts across 12 Biofluids.

Hierarchical clustering heatmap depicting scaled miRNA read counts for six groups (1–6) of five or more miRNAs with similar abundance patterns across biofluids using Bayesian relevance network analysis. Z scores indicate levels of miRNA relative to levels of the same miRNA in other biofluids.
We were also interested in exploring the relationship between miRNA levels in biofluids and miRNA levels in organs and tissues. Analysis of data from a previous miRNA microarray study (Ludwig et al., 2016) showed that pairwise correlations for miRNA levels in nine different tissues or organs that are in contact with the biofluids we studied, like correlations between biofluids, were also high (R = 0.73–0.92; Figure S3). Since technical differences between small RNA-seq and microarrays precluded direct comparisons between these two datasets, we used our small RNA-seq method to analyze RNA from one human organ: the brain. Brain miRNA levels were correlated with our biofluids (R ranging from 0.63 in seminal fluid to 0.74 in CSF; Figure S3; Table S2). These data are consistent with the hypothesis that intracellular miRNA levels are one major determinant of extracellular miRNA levels. Further ERCC-supported studies of relationships between exRNA levels and levels of RNAs in different organs, tissues, and primary cell types are ongoing.
tDR Profiles
Sequences aligning to tRNAs were detected in all biofluids, although the frequencies of tDR reads varied considerably between biofluids (Figure 1). Assigning tDRs to the human genome and transcriptome is challenging, since many tDRs map to multiple tRNAs corresponding to the same anticodon, a different anticodon for the same amino acid, or even different amino acids. The Genboree pipeline aggregates tDR reads by amino acid. To analyze tDR reads in more detail, we used MINTmap (Loher et al., 2017), a tDR analysis tool that counts each unique sequence, including those that differ in length by as little as 1 nt, separately. Using this approach, between 954 (bile) and 4,997 (parotid saliva) tDRs had a median of ≥ 10 reads per million total tDR reads in each biofluid (Figures 4A and S4; Table S2). The 10 most frequent tDR reads represent between 18% (adult blood plasma) and 77% (BAL) of total tDR reads (Figure 4B). Pairwise correlation coefficients for tDR read counts between biofluids ranged from 0.08 to 0.89 (Figures 4C–4E) and were typically far lower than those found for pairwise correlations using miRNA reads. Correlations were highest between adult and cord blood plasma (R = 0.89) and between saliva samples from the two different sites (R = 0.84). The proportion of mapped reads that represented tDRs was much higher for serum (37%) than for adult plasma (0.8%), and the pairwise correlation for tDR read counts between serum and adult plasma was only moderate (R = 0.65). Using all 8,672 tDRs detected in any sample, tSNE analysis revealed that samples of most biofluid types formed largely distinct clusters (Figure 4E). As with miRNAs, parotid and submandibular and sublingual glands saliva samples formed overlapping clusters. Urine and CSF samples were also overlapping. Although tSNE analysis based on miRNAs produced overlapping clusters of serum and plasma samples, these biofluid types were clearly distinct when the tSNE analysis was based on tDRs.
Figure 4. tDR Profiles across 12 Biofluid Types.

(A) Number of tDRs detected as a function of read depth.
(B) Cumulative distribution of tDR reads. See also Figure S4.
(C and D) Examples of pairwise correlations between biofluids for cord blood plasma versus adult blood plasma (C) and cord blood plasma versus bile (D). Each point represents the median normalized read count for a single tDR for the indicated biofluids. One normalized read count was added to each measurement to allow representation of read counts for tDRs with no reads on a log scale.
(E) Correlations for all pairs of biofluids.
(F) tSNE plot produced using tDR read counts. Each point represents a single biofluid sample.
To further analyze tDRs, we grouped together tDRs based on the amino acid that corresponds to the tRNA of origin (Figures 5A and S5). tDRs that could not be unambiguously assigned to an amino acid were excluded from this analysis. Glycine tDRs were predominant in urine, serum, and CSF. Leucine tDRs were predominant in BAL (62%) but were much less frequently found (<15%) in other biofluids. Glutamic acid tDRs were predominant in amniotic fluid, bile, plasma from cord blood and adult blood, and both types of saliva, whereas methionine tDRs were predominant in seminal plasma. Glycine, glutamate, and methionine each represented >20% of tDR reads in ovarian follicle fluid. Median tDR reads for tyrosine, asparagine, phenylalanine, and isoleucine tDRs were <1% each for all biofluid types. Normalized read counts for many tDR-associated amino acids differed markedly between biofluids. For example, tRNA reads aligning to glycine made up 68.9% of tDR reads in urine but ≤43.4% of tDR reads in every other biofluid. A short (16 nt) read mapping to the 5’ end of leucine tRNAs accounted for the majority (59%) of all tDR reads in BAL fluid samples but was rare in all other biofluids (0.003%−1.6%).
Figure 5. tDR Abundance by Amino Acid, Anticodon, and Fragment Type.

(A) tDR abundance by amino acid. Boxes represent median and interquartile ranges, whiskers represent 1.5 × the interquartile range. Dots represent outliers.
(B) tDR abundance by anticodon.
(C) tDR abundance by fragment type.
Data are shown for tDRs from the five most highly represented tRNAs. Data for other tDRs are shown in Figures S5–S7.
For amino acids encoded by more than one codon, we examined the distribution of reads for each possible anticodon (Figures 5B and S6). For some amino acids (e.g., glycine), anticodon frequency varied substantially between biofluids, but for others (e.g., leucine), anticodon frequency was less variable. We also classified tDRs by fragment type (Loher et al., 2017). Fragment types differed substantially according to the tRNA of origin (Figures 5C and S7). For example, in most biofluids, glycine tDRs mapped primarily to the 5’ region (5’ tRFsand 3’ halves), whereas methionine and histidine tDRs mapped primarily to internal regions of tRNAs (i-tRFs, fragments beginning after the first position and ending before the non-templated CCA addition). To examine differences in tDR fragments between biofluids in more detail, we constructed read coverage maps for four tDRs with the largest numbers of mapped reads (Figure S7). Inspection of these coverage maps reveals large differences in fragment types across biofluids (e.g., increased 3’ coverage in amniotic fluid for three of these four tRNAs) and illustrates major differences in fragment sizes (e.g., shorter 5’ fragments for tRNA7 LeuAAG compared with the other three tRNAs).
Y RNA Profiles
We found reads mapping to each of the four human Y RNA genes in every biofluid (Figure 6A; Tables S2 and S7). In each biofluid except for BAL fluid, most reads that mapped to Y RNA mapped to RNY4. In BAL fluid, the mean mapping rate for RNY4 was 43%, and RNY5 accounted for 49%. Most reads mapped to either the 5’ region or the 3’ region of full-length Y RNAs, with relatively few reads mapping to the central portion of Y RNAs (Figure 6B). The proportion of 5’ reads to 3’ reads varied between biofluids and between different Y RNA genes. For example, RNY4 fragments more frequently mapped to the 5’ end in seminal plasma and CSF but more frequently mapped to the 3’ end in adult blood plasma, BAL fluid, and saliva.
Figure 6. Y RNA Fragments in 12 Biofluid Types.

(A) Distribution of Y RNA reads by Y RNA gene. Boxes represent median and interquartile ranges, whiskers represent 1.5 × the interquartile range. Dots represent outliers.
(B) Y RNA read-mapping positions. We determined the number of reads covering each nucleotide of each full-length Y RNA. Values are normalized to the position of each Y RNA with the largest number of reads in each biofluid.
piRNA Profiles
As discussed previously, some biofluids contained substantial proportions of reads that mapped to both piRNAs and other Gencode RNAs (especially RNY4). The majority of reads that mapped to piRNAs also mapped to other RNA biotypes, except in one biofluid: seminal plasma. 57% of seminal plasma reads that mapped to piRNAs did not map to another biotype. After exclusion of ambiguously mapped reads (see Experimental Procedures), the number of piRNAs detected was higher in seminal fluid (5,588 distinct piRNAs in one or more samples) than in other fluids (94–1,068 piRNAs). The most frequent 57 piRNAs accounted for 50% of seminal plasma reads that mapped exclusively to piRNAs (Table S2).
DISCUSSION
We applied a standardized RNA-seq approach to identify small RNAs in 12 human biofluid types. This work extends previous analyses of biofluid exRNAs by applying a method that allows for relative quantification of small RNAs from multiple biotypes to a diverse collection of normal biofluids. In each biofluid tested, the majority of mapped reads could be assigned to miRNAs, tDRs, or Y RNAs. Additional reads mapping to piRNAs, mRNAs, long non-coding RNAs, snRNAs, and other RNAs were also detected but were less common. Relative levels of different RNA biotypes differed dramatically between biofluids. The relative abundance of the two major biotypes, miRNAs and tDRs, varied by >103-fold across the set of 12 biofluids. Our results are consistent with those from another recent study that found relatively high levels of tDRs in urine and relatively high levels of Y RNA fragments in plasma (Yeri et al., 2017). Despite differences in overall small RNA composition, miRNA levels were generally highly correlated between biofluids (R = 0.79–0.98). tDR levels were less well correlated (R = 0.08–0.89). Biofluids that had highly similar profiles for small RNAs of one biotype sometimes had very different profiles for RNAs of other biotypes. For example, adult blood plasma and serum miRNA levels were highly correlated (R = 0.98), but tDR levels in these biofluids were much less well correlated (R = 0.68). Our findings demonstrate the extensive diversity of small RNA populations within each biofluid type and highlight both similarities and differences between biofluids.
The methods we used have advantages and limitations compared with those used in previous studies. We used a uniform approach to RNA isolation, RNA-seq library preparation, sequencing library size selection, sequencing, and data analysis for all samples. The RNA-seq method we used reduces bias by employing oligonucleotides with random nucleotide sequences for adaptor ligation (Giraldez et al., 2017). Use of a permissive size selection strategy rather than one targeting miRNAs allowed us to identify RNAs of many biotypes. We used a publicly available analysis pipeline, and our data are publicly available, which allows for reanalysis of the primary data as databases and analysis tools are updated. To obtain a broad view of small RNAs in biofluids, we extracted RNA from unfractionated biofluids. Other studies have shown differences in small RNA content of specific biofluid compartments such as exosomes and argonaute-2-containing ribonucleoprotein complexes (Arroyo et al., 2011), and further work will be required to understand the compartmentalization of RNAs in the large set of biofluids we studied. A major limitation of the RNA-seq method we used is that this method is designed for sequencing of RNAs with a 5’ phosphate and a 3’ hydroxyl group. RNAs with various 5’ end modifications (hydroxyl, triphosphate, or 5’ cap) or 3’ end modifications (2’-3’-cyclic phosphate, 2’-O-methyl) as well as RNAs with certain structures are more difficult or impossible to detect with this method (Raabe et al., 2014). RNAs that undergo many types of post-translational modifications, including those that are commonly found in tRNAs or after miRNA editing, may not be detected or may fail to map to the human genome. The proportion of reads that aligned to the human genome was variable, and for some biofluids, the absolute number of reads aligning to certain biotypes was low. We focused our comparative analyses on large differences in RNAs that were abundant in at least one biofluid, and our approach may not detect differences in RNAs that less abundant but nonetheless have biological importance. Differences in centrifugation speeds of biofluids could also be responsible for some of the observed differences between biofluid types. In addition, although we excluded samples with apparent blood contamination, the presence of the erythrocyte miRNA miR-451a (among the top 10 miRNAs in ovarian follicle fluid and bile as well as plasma and serum) might reflect low-level blood contamination of some samples.
Our approach was designed to compare levels of each RNA between samples. A qPCR analysis (Figure S2) validated our ability to measure relative differences in RNA levels by small RNA-seq. For both RNA-seq and qPCR, detection of RNAs depends upon both RNA abundance and technical biases. Differences in technical bias are likely the major explanation of why miRNAs with the highest RNA-seq read counts in our study differ markedly from miRNAs that were most highly detected (lowest cycle threshold [Ct]) in a prior qPCR-based study of many biofluids (Weber et al., 2010). Therefore, detection rates should not be used as direct measures of the abundance of different RNAs within any sample.
miRNA profiles of these 12 biofluids were remarkably similar overall. Comparison of biofluid miRNA levels with miRNA levels in a single human organ (brain) suggests that cellular miRNA levels are a major determinant of biofluid miRNA abundance (Supplemental Experimental Procedures; Figure S3), but there may be other factors related to miRNA export from cells (Villarroya-Beltri et al., 2013) or extracellular miRNA stability that also affect miRNA abundance in biofluids (Taylor and Gercel-Taylor, 2008; Gilad et al., 2008). We identified large differences in levels of some miRNAs between biofluids. These may arise from differences in local production of cellular miRNAs, since we found higher levels of extraembryonic cell miRNAs in amniotic fluid, nervous system cell miRNAs in CSF, and an epididymal miRNA in seminal plasma. In many cases, miRNAs produced from the same pre-miRNA or the same miRNA cluster had highly correlated levels across the full set of biofluids, which also suggests that miRNA production is a major determinant of differences in miRNA abundance. In addition to differences in local production, differences in extracellular miRNAs could arise from preferential secretion of miRNAs from cells (Valadi et al., 2007) or differences in processing or elimination of extracellular miRNAs. Since extracellular miRNAs have been shown to be capable of entering cells may modulate gene expression (Patton et al., 2015), our results suggest that differences in miRNA composition between biofluids may be functionally important.
We found large differences in tDRs across biofluids. tRNAs can be processed into tDRs by sequence-specific enzymatic cleavage events (Lee et al., 2009), and tDRs have been previously identified in human biofluids, including human serum and urine, and in exosomes from human semen (Dhahbi, 2015; Yeri et al., 2017). We found differences in tRNA of origin and fragment type across the set of biofluids. Many factors, including tRNA levels, tRNA cleavage, tDR secretion, and biodistribution and elimination of extracellular tDRs, may contribute to determining levels of tDRs. Intracellular tDRs have been reported to have many roles, including inhibiting protein translation during cellular stress (Ivanov et al., 2011) and mediating intergenerational inheritance (Chen et al., 2016; Sharma et al., 2016). Roles of extracellular tDRs remain enigmatic, but the presence of relatively high levels of extracellular tDRs in biofluids such as bile, urine, seminal plasma, and amniotic fluid (where tDRs represented the majority of all mapped small RNA reads) suggests that extracellular tDRs may be functionally important. At least some of the functional roles of intracellular tDRs are quite specific. For example, a tDR from the Gly-GCC tRNA, but not other tDRs, was shown to regulate retroelement-driven transcription of highly expressed genes in preimplantation embryos (Sharma et al., 2016). Differences in tDR populations between biofluids may therefore have important functional implications.
Y RNA fragments represented a substantial proportion of small RNAs detected in some biofluids, including adult and cord blood plasma, serum, and CSF. A previous study also reported many Y RNA fragment reads in human serum and plasma (Dhahbi et al., 2013). That study found that >95% of fragments mapped to the 5’ end of Y RNAs. Our results, obtained using an RNA-seq method developed to reduce bias, revealed a substantially higher proportion of 3’ Y RNA fragments. Intact cellular Y RNAs are required for the initiation of DNA replication, regulation of RNA stability, and cellular responses to stress (Kowalski and Krude, 2015).The role ofYRNAfragmentsislesswell understood. It has been reported that RNY5 fragments produced in cancer cell extracellular vesicles trigger rapid cell death in primary cells, but not in cancer cells (Chakrabortty et al., 2015), suggesting that further studies of the biogenesis, trafficking, and function of Y RNA fragments found in biofluids are warranted.
Blood plasma and serum contained a substantial number of small RNAs with sequences that map to piRNAs, but we urge caution about inferring that these are derived from piRNAs. The large majority of piRNA-mapped reads we obtained in our plasma and serum samples also mapped to other RNA biotypes (especially Y RNAs) that are abundant in these biofluids. In both our study and a previous study (Freedman et al., 2016), the piRNA with the largest number of mappings shares sequence with a fragment of RNY4, a Y RNA. We therefore confined our analysis of piRNAs to sequences that did not map to RNAs of other biotypes. piRNAs have prominent roles in the testis (Girard et al., 2006), and even using this more conservative approach, we found that a substantial number and variety of piRNA sequences were detected in seminal plasma. A previous report also identified piRNAs in seminal plasma and identified a set of piRNAs that were associated with infertility (Hong et al., 2016). Although previously suggested to be germline specific, piRNAs also play roles in non-gonadal cells (Ishizu et al., 2012). Non-gonadal cells might therefore be a source of piRNAs in biofluids outside the reproductive tract. However, we found relatively few readsthat mapped only to piRNAs in biofluids otherthan seminal plasma. The functional significance of extracellular piRNAs is not yet known.
By using a uniform RNA-seq-based approach, we produced an extensive catalog of small RNAs that are present in a large set of human biofluids. The RNA composition of biofluids differed widely, with major differences in the distribution of biotypes and of specific RNAs within each biotype. This work provides a resource for investigators seeking to understand the production, distribution, and function of exRNAs. Biofluid small RNAs are being explored as disease biomarkers, and our work also helps to identify RNAs that are present in multiple biofluids and may have potential as novel biomarkers.
EXPERIMENTAL PROCEDURES
Human Subjects
With the exception of bile samples, which were obtained at the Mayo Clinic following liver transplantation, all samples were obtained from healthy subjects enrolled in a variety of studies conducted at the University of California, San Francisco. Protocols were approved by institutional review boards at the Mayo Clinic (bile) and the University of California, San Francisco (all other specimen types). Except for cord blood plasma, which was obtained at birth, all other samples were obtained from studies enrolling subjects aged 18 years or older. Details of sample collection and storage for each sample type are provided in Supplemental Experimental Procedures.
RNA Isolation, Library Preparation, and Sequencing
Frozen biofluid samples used for small RNA-seq experiments were thawed and centrifuged at 2,000 × g for 5 min at 4°C. RNA was isolated from 200 μL of biofluid using the QIAGEN miRNEasy Micro Kit according to the manufacturer’s protocol, except that 1 mL Qiazol and 180 μL chloroform were used. Small RNA-seq libraries were prepared using 4N protocol D as previously described (Giraldez et al., 2017; Supplemental Experimental Procedures).
Sequence Alignment
All FASTQ files were processed using the exceRpt small RNA-seq pipeline version 4.6.2 available on the Genboree Workbench (http://www.genboree.org/index.html). Sequence readswere clipped 4 nt from the invariant portions of both 5’ and 3’ adapters to remove the four randomized nucleotide in the adapters. Clipped reads were aligned with a one-mismatch allowance. To exclude piRNAs that mapped to other biotypes, we changed the order of mapping assignment to count Gencode alignments in preference to piRNA alignments (alignment order: miRNAs, tRNAs, all annotations from Gencode, piRNAs, and circular RNA). We also eliminated piRNA mappings if the large majority (>98%) of reads mapping to a given piRNA also mapped to an RNA of a different biotype. To determine whether the high proportion of CSF reads that did not map to the human genome mapped to other genomes, we used the Genboree exogenous alignment option to map CSF samples to exogenous miRNAs in miRbase and all sequenced genomes in Ensembl and NCBI. No mismatches were permitted for exogenous alignments.
RNA-Seq Data Inclusion Criteria
RNA-seq results were included if (1) >100,000 reads mapped to the transcriptome, (2) the number of reads mapping to annotated transcripts represented at least 50% of the number of reads mapping to the human genome, and (3) R ≥ 0.85 for correlation of miRNA reads with most other samples of the same biofluid. For samples that failed to meet inclusion criteria on initial analysis, duplicate biofluid samples were used for a second round of RNA isolation and library preparation. If the repeat analysis met inclusion criteria, then these results were used for all analyses. If the repeat analysis did not meet the inclusion criteria, then data from that sample were not used for subsequent analyses. There was no case in which replicate analysis of a sample gave consistent results (R ≥ 0.85 for miRNAs), but these results were poorly correlated (R < 0.85) with most other samples of the same biofluid type.
RNA Biotype Distribution Analysis
We used the biotype counts file generated by the small RNA-seq pipeline on the Genboree Workbench. Since Y RNA reads represented a large proportion of Gencode transcript reads in some biofluids, we used the Gencode read counts file to quantify Y RNA reads separately from other Gencode reads.
miRNA Analysis
miRNA diversity analyses were performed using the mean normalized miRNA read counts for all samples of a given biofluid type. Read counts for each miRNA in each sample were normalized by dividing by the total number of miRNA read counts in that sample. For analyses of the numbers of miRNAs detected as a function of read depth, we excluded all miRNAs with mean read counts <1/106 total miRNA reads (lower limit of detection). For biofluids where miRNAs represented a small proportion of total RNA reads, the total number of miRNA reads for all samples was <106. For those biofluids, the lower limit of detection was defined as 106 divided by the total number of miRNA reads for that biofluid. For analyses of cumulative distributions, we ranked miRNAs in descending order of normalized mean read counts for a given biofluid and calculated the cumulative sum until we included enough miRNAs to account for 99.9% of miRNA reads.
For pairwise correlations and tSNE analyses, we normalized read counts by DESeq2 (Love etal., 2014) and determined mean read counts for all samples of each biofluid. We calculated the Pearson correlation coefficient of each biofluid pair using R. We generate tSNE plots using the Rtsne function of the Rtsne R package with perplexity 20 and a maximum iteration of 5,000. We plotted the results using the xyplot function from the lattice R package. We used Bayesian relevance networks (Ramachandran et al., 2017) to generate a list of co-expressing miRNAs. We normalized miRNA read counts with DESeq2, transformed the counts data using the varianceStabilizingTransformation function, identified the top 25% most frequent miRNAs (by mean), and then selected the top 13% most variable as input to the Bayesian relevance networks algorithm. We selected all co-expressing miRNAs with a Bayesian correlation ≥0.80, which had an estimated FDR of 0.012. We used the pheatmap function from the pheatmap R package to generate a hierarchical clustering diagram for all miRNAs belonging to networks with 3 or more miRNAs.
Methods used for qPCR miRNA validation experiments are described in Supplemental Experimental Procedures.
tDR Analysis
We used MINTmap (Loheret al., 2017) to analyze tDRs. We removed adapters, trimmed 4 nt from each end of the read and removed low-quality reads (using standard Genboree parameters) from all fastq files with the fastx-toolkit. Fastq files were then processed individually by MINTmap. We only counted alignments that mapped exclusively to annotated tRNA regions to reduce ambiguity. For analyses of amino acid or anticodon read counts, reads that mapped ambiguously (to multiple amino acids or anticodons) were deemed “undetermined.” All reads were aggregated by sum and normalized by the total number of tDR reads within each sample or biofluid type. To analyze tRNA coverage, we took the normalized mean of each sample and aggregated read counts by biofluid and the start and end position.
Y-RNA Analysis
Using the bam files generated by the Genboree pipeline using the “Upload Full Results” option, we searched for any alignment tothe 4 human Y RNAs (RNY1-201, RNY3-201, RNY4-201, and RNY5-201) and considered each unique fragment a distinct read. To plot read coverage, we followed the same strategy as outlined above for tDRs.
DATA AND SOFTWARE AVAILABILITY
The accession number for the raw sequence data for bile reported in this paper is GEO: GSE112343. The accession numbers for raw data for seminal plasma, amniotic fluid and cord blood plasma, CSF, and ovarian follicle fluid reported in this paper are dbGAP: phs001692.v1.p1, phs001693.v1.p1, phs001694.v1.p1, and phs001695.v1.p1, respectively. The accession numbers for the read counts for all samples reported in this paper are exRNA Atlas (https://exrna-atlas.org/): EXR-DERLE1PHASE1PRIOT-AN (parotid and submandibular and sublingual saliva) and EXR-DERLE1PHASE1OPEN-AN (all other biofluids). Due to issues related to patient confidentiality, raw data are not currently available for adult blood plasma, serum, urine, BAL fluid, and parotid and submandibular and sublingual saliva samples. If consent and approval are granted in the future, the raw data will be made available at that time.
Supplementary Material
Highlights.
RNA-seq of 12 human biofluids shows all have diverse small RNA repertoires
Relative abundance of miRNA to tRNA-derived RNA differs markedly across biofluids
Biofluid miRNA profiles are generally similar, but some miRNAs vary widely
There are large differences in tRNA-derived RNAs between biofluids
ACKNOWLEDGMENTS
This publication is part of the NIH ERCC paper package and was supported by the NIH Common Fund’s exRNA Communication Program. We thank the individuals who participated in the study by providing samples; Christine P. Nguyen and Bobby Antalek for assistance with collection and annotation of the BAL fluid, adult blood plasma, serum, and urine samples; Serena Spudich for access to CSF samples collected in studies that she directed; Michael Li and Suresh Garudadri for performing qPCR experiments; and William Thistlethwaite (Baylor College of Medicine) for help with depositing data. This work was supported by the NIH Extracellular RNA Communication Program (grant U01HL126493 to P.G.W. and D.J.E. and grants UH2TR000884 and UH3TR000884 to T.P.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and seven tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.10.014.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Argyropoulos C, Wang K, McClarty S, Huang D, Bernardo J, Ellis D, Orchard T, Galas D, and Johnson J (2013). Urinary microRNA profiling in the nephropathy of type 1 diabetes. PLoS ONE 8, e54662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arroyo JD, Chevillet JR, Kroh EM, Ruf IK, Pritchard CC, Gibson DF, Mitchell PS, Bennett CF, Pogosova-Agadjanyan EL, Stirewalt DL, et al. (2011). Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc. Natl. Acad. Sci. USA 108, 5003–5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahn JH, Zhang Q, Li F, Chan TM, Lin X, Kim Y, Wong DT, and Xiao X (2015). The landscape of microRNA, Piwi-interacting RNA, and circular RNA in human saliva. Clin. Chem. 61, 221–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barger JF, Rahman MA, Jackson D, Acunzo M, and Nana-Sinkam SP (2016). Extracellular miRNAs as biomarkers in cancer. Food Chem. Toxicol. 98 (Pt A), 66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabortty SK, Prakash A, Nechooshtan G, Hearn S, and Gingeras TR (2015). Extracellular vesicle-mediated transfer of processed and functional RNY5 RNA. RNA 21, 1966–1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Yan M, Cao Z, Li X, Zhang Y, Shi J, Feng GH, Peng H, Zhang X, Zhang Y, et al. (2016). Sperm tsRNAs contribute to intergenerational inheritance of an acquired metabolic disorder. Science 351, 397–400. [DOI] [PubMed] [Google Scholar]
- Czech B, and Hannon GJ (2016). One loop to rule them all: the ping-pong cycle and piRNA-guided silencing. Trends Biochem. Sci. 41, 324–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rie D, Abugessaisa I, Alam T, Arner E, Arner P, Ashoor H, Åström G, Babina M, Bertin N, Burroughs AM, et al. ; FANTOM Consortium (2017). An integrated expression atlas of miRNAs and their promoters in human and mouse. Nat. Biotechnol. 35, 872–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhahbi JM (2015). 5’ tRNA halves: the next generation of immune signaling molecules. Front. Immunol. 6, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhahbi JM, Spindler SR, Atamna H, Boffelli D, Mote P, and Martin DI (2013). 5’-YRNA fragments derived by processing of transcripts from specific YRNA genes and pseudogenes are abundant in human serum and plasma. Physiol. Genomics 45, 990–998. [DOI] [PubMed] [Google Scholar]
- Freedman JE, Gerstein M, Mick E, Rozowsky J, Levy D, Kitchen R, Das S, Shah R, Danielson K, Beaulieu L, et al. (2016). Diverse human extracellular RNAs are widely detected in human plasma. Nat. Commun. 7, 11106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebetsberger J, and Polacek N (2013). Slicing tRNAs to boost functional ncRNA diversity. RNA Biol. 10, 1798–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilad S, Meiri E, Yogev Y, Benjamin S, Lebanony D, Yerushalmi N, Benjamin H, Kushnir M, Cholakh H, Melamed N, et al. (2008). Serum microRNAs are promising novel biomarkers. PLoS ONE 3, e3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraldez MD, Spengler RM, Etheridge A, Godoy PM, Barczak AJ, Srinivasan S, De Hoff PL, Tanriverdi K, Courtright A, Lu S, et al. (2017). Accuracy, reproducibility and bias of next generation sequencing for quantitative small rna profiling: a multiple protocol study across multiple laboratories. bioRxiv. https://doi.org/10.1101/113050. [Google Scholar]
- Girard A, Sachidanandam R, Hannon GJ, and Carmell MA (2006). A germline-specific classofsmall RNAs binds mammalian Piwi proteins. Nature 442, 199–202. [DOI] [PubMed] [Google Scholar]
- Gray C, McCowan LM, Patel R, Taylor RS, and Vickers MH (2017). Maternal plasma miRNAs as biomarkers during mid-pregnancy to predict later spontaneous preterm birth: a pilot study. Sci. Rep. 7, 815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill KE, Kelly AD, Kuijjer ML, Barry W, Rattani A, Garbutt CC, Kissick H, Janeway K, Perez-Atayde A, Goldsmith J, et al. (2017). An imprinted non-coding genomic clusterat 14q32 definesclinically relevant molecularsub-types in osteosarcoma across multiple independent datasets. J. Hematol. Oncol. 10, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong Y, Wang C, Fu Z, Liang H, Zhang S, Lu M, Sun W, Ye C, Zhang C-Y, Zen K, et al. (2016). Systematic characterization of seminal plasma piRNAs as molecular biomarkers for male infertility. Sci. Rep. 6, 24229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoy AM, and Buck AH (2012). Extracellularsmall RNAs: what, where, why? Biochem. Soc. Trans. 40, 886–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizu H, Siomi H, and Siomi MC (2012). Biology of PIWI-interacting RNAs: new insights into biogenesis and function inside and outside of germlines. Genes Dev. 26, 2361–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov P, Emara MM, Villen J, Gygi SP, and Anderson P (2011). Angiogenin-induced tRNA fragments inhibit translation initiation. Mol. Cell 43, 613–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korpal M, and Kang Y (2008). The emerging role of miR-200 family of microRNAs in epithelial-mesenchymal transition and cancer metastasis. RNA Biol. 5, 115–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalski MP, and Krude T (2015). Functional roles of non-coding Y RNAs. Int. J. Biochem. Cell Biol. 66, 20–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Shibata Y, Malhotra A, and Dutta A (2009). A novel class of small RNAs: tRNA-derived RNA fragments (tRFs). Genes Dev. 23, 2639–2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee LW, Zhang S, Etheridge A, Ma L, Martin D, Galas D, and Wang K (2010). Complexity of the microRNA repertoire revealed by next-generation sequencing. RNA 16, 2170–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wang H-Y, Wan F-C, Liu F-J, Liu J, Zhang N, Jin S-H, and Li J-Y (2012). Deep sequencing analysis of small non-coding RNAs reveals the diversity of microRNAs and piRNAs in the human epididymis. Gene 497, 330–335. [DOI] [PubMed] [Google Scholar]
- Loher P, Telonis AG, and Rigoutsos I (2017). MINTmap: fast and exhaustive profiling of nuclear and mitochondrial tRNA fragments from short RNA-seq data. Sci. Rep. 7, 41184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig N, Leidinger P, Becker K, Backes C, Fehlmann T, Pallasch C, Rheinheimer S, Meder B, Stähler C, Meese E, and Keller A (2016). Distribution of miRNA expression across human tissues. Nucleic Acids Res. 44, 3865–3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell PS, Parkin RK, Kroh EM, Fritz BR, Wyman SK, Pogosova-Agadjanyan EL, Peterson A, Noteboom J, O’Briant KC, Allen A, et al. (2008). Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 105, 10513–10518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni H, Capodici J, Cannon G, Communi D, Boeynaems JM, Karikó K, and Weissman D (2002). Extracellular mRNA induces dendritic cell activation by stimulating tumor necrosis factor-alpha secretion and signaling through a nucleotide receptor. J. Biol. Chem. 277, 12689–12696. [DOI] [PubMed] [Google Scholar]
- Patton JG, Franklin JL, Weaver AM, Vickers K, Zhang B, Coffey RJ, Ansel KM, Blelloch R, Goga A, Huang B, et al. (2015). Biogenesis, delivery, and function of extracellular RNA. J. Extracell. Vesicles 4, 27494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raabe CA, Tang T-H, Brosius J, and Rozhdestvensky TS (2014). Biases in small RNA deep sequencing data. Nucleic Acids Res. 42, 1414–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran P, Sánchez-Taltavull D, and Perkins TJ (2017). Uncovering robust patterns of microRNA co-expression across cancers using Bayesian Relevance Networks. PLoS ONE 12, e0183103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenov DV, Kuligina EV, Shevyrina ON, Richter VA, and Vlassov VV (2004). Extracellular ribonucleic acids of human milk. Ann. NY Acad. Sci. 1022, 190–194. [DOI] [PubMed] [Google Scholar]
- Sharma U, Conine CC, Shea JM, Boskovic A, Derr AG, Bing XY, Belleannee C, Kucukural A, Serra RW, Sun F, et al. (2016). Biogenesis and function of tRNA fragments during sperm maturation and fertilization in mammals. Science 351, 391–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skog J, Würdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, Curry WT Jr., Carter BS, Krichevsky AM, and Breakefield XO (2008). Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 10, 1470–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohel MH (2016). Extracellular/circulating microRNAs: release mechanisms, functions and challenges. Achiev. Life Sci. 10, 175–186. [Google Scholar]
- Taylor DD, and Gercel-Taylor C (2008). MicroRNA signatures of tumor-derived exosomes as diagnostic biomarkers of ovarian cancer. Gynecol. Oncol. 110, 13–21. [DOI] [PubMed] [Google Scholar]
- Valadi H, Ekström K, Bossios A, Sjöstrand M, Lee JJ, and Lötvall JO (2007). Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 9, 654–659. [DOI] [PubMed] [Google Scholar]
- Villarroya-Beltri C, Gutiérrez-Vázquez C, Sánchez-Cabo F, Pérez-Hernández D, Vázquez J, Martin-Cofreces N, Martinez-Herrera DJ, Pascual-Montano A, Mittelbrunn M, and Sánchez-Madrid F (2013). Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat. Commun. 4, 2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waller R, Wyles M, Heath PR, Kazoka M, Wollff H, Shaw PJ, and Kirby J (2018). Small RNA sequencing of sporadic amyotrophic lateral sclerosis cerebrospinal fluid reveals differentially expressed miRNAs related to neural and glial activity. Front. Neurosci. 11, 731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber JA, Baxter DH, Zhang S, Huang DY, Huang KH, Lee MJ, Galas DJ, and Wang K (2010). The microRNA spectrum in 12 body fluids. Clin. Chem. 56, 1733–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams Z, Ben-Dov IZ, Elias R, Mihailovic A, Brown M, Rosenwaks Z, and Tuschl T (2013). Comprehensive profiling of circulating microRNA via small RNA sequencing of cDNA libraries reveals biomarker potential and limitations. Proc. Natl. Acad. Sci. USA 110, 4255–4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi Y, Ohkubo T, Kawaji H, Machida A, Miyata H, Goda S, Roy S, Hayashizaki Y, Suzuki H, and Yokota T (2017). Next-generation sequencing-based small RNA profiling of cerebrospinal fluid exosomes. Neurosci. Lett. 636, 48–57. [DOI] [PubMed] [Google Scholar]
- Yeri A, Courtright A, Reiman R, Carlson E, Beecroft T, Janss A, Siniard A, Richholt R, Balak C, Rozowsky J, et al. (2017). Total extracellular small RNA profiles from plasma, saliva, and urine of healthy subjects. Sci. Rep 7, 44061. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.