

Abstract
The chromatin fiber is the control panel of eukaryotic cells. Chromatin is mostly composed of DNA, which contains the genetic instruction for cell phenotype, and histone proteins, which provide the scaffold for chromatin folding and part of the epigenetic inheritance. Histone writers/erasers “flag” chromatin regions by catalyzing/removing covalent histone post-translational modifications (PTMs). Histone PTMs chemically contribute to chromatin relaxation or compaction and recruit histone readers to modulate DNA readout. The precursors of protein PTMs are mostly small metabolites. For instance, acetyl-CoA is used for acetylation, ATP for phosphorylation, and S-adenosyl methionine for methylation. Interestingly, PTMs such as acetylation can occur at neutral pH also without their respective enzyme when the precursor is sufficiently concentrated. Therefore, it is essential to differentially quantify the contribution of histone writers/erasers vs the effect of local concentration of metabolites to understand the primary regulation of histone PTM abundance. Aberrant phenotypes such as cancer cells have misregulated metabolism and thus the composition and the modulation of chromatin is not only driven by enzymatic tuning. In this review, we discuss the latest advances in mass spectrometry (MS) to analyze histone PTMs and the most adopted quantification methods for related metabolites, both necessary to understand PTM relative changes.
Keywords: acetyl-CoA, epigenetics, histone, metabolism, post-translational modifications
Introduction
Epigenetics is the scientific field investigating inheritable changes in gene regulation not coded in the DNA sequence. It is currently one of the fastest growing fields in biology and medical science, specifically because epigenetic alterations play a causal role in many human disorders [1,2]. Uncovering the role of epigenetic changes in human diseases and normal human traits has thus become a priority. DNA is packaged into basic units known as nucleosomes, which form the basic structural elements of chromatin [3,4]. Each nucleosome is composed of 147 base pairs of DNA and two copies each of the four major nucleosomal histones (H2A, H2B, H3, and H4), and is linked by a linker histone called H1 [5]. These nucleosomes are then folded into more complex three-dimensional structures. Histones are small proteins containing a high number of positively charged amino acid residues (lysine and arginine) that facilitate binding to negatively charged DNA. Chromatin structure is tightly modulated by histones and their covalent post-translational modifications (PTMs) [6]. While some of these modifications have been identified at the histone fold domains, most of them occur on the flexible N-terminal tails, which extend outward from the nucleosome core [7]. Histone PTMs can alter chromatin structure, influencing several processes such as gene transcription, DNA replication, DNA damage repair and alternative splicing [8,9]. Every process that requires access to genomics information must overcome this tight packaging of chromatin.
Lysine acetylation (ac) and methylation (me) were the first histone PTMs to be discovered (1960s), and they are the most abundant and well-characterized [10]. Lysine acetylation neutralizes the positive charge on histones, reducing their interactions with negatively charged DNA and allowing transcription activators to access DNA and promote gene expression [11]. Lysine acetylation is therefore commonly associated with transcriptionally active chromatin [12,13]. Histone lysine methylation has been correlated with either activation or repression of chromatin, depending on the localization of the methylated residue [14,15]. Since the discovery of these two marks, an ever-growing number of histone modifications has been identified, including ubiquitination (ub) [16], biotinylation (bio) [17], ADP-ribosylation (Ar) [18], and phosphorylation (ph) [19]. More recently, several less-characterized lysine acylations were included in the already long list of histone PTMs, including malonylation (ma) [20], succinylation (suc) [21], crotonylation (cr) [22], propionylation (pr) [23], butyrylation (bu) [23], glutarylation (glu) [24], 2-hydroxyisobutyrylation (hib) [25], and β-hydroxybutyrylation (bhb) [26]. For an updated review of all histone PTMs identified to date, we refer readers to the “Comprehensive Catalog Of Currently Documented Histone Modifications” [27]. Altogether, more than 400 modifications are known to occur on histone proteins to date, many of which have still uncharacterized function.
There are at least four mechanisms by which histone modifications are regulated on the chromatin (Figure 1); they divide in (i) regulation of histone writers/erasers by signaling cascade led by external events, (ii) abundance of metabolite precursors of PTMs, (iii) chromatin accessibility due to pre-existing modifications, and (iv) protein-nucleic acid interactions to facilitate enzyme recruitment. Interestingly, these aspects can all be investigated using mass spectrometry (MS)-based proteomics, including protein-nucleic acid interaction [28]. In this review, we mainly focus on (ii) and (iii). The wide chemical variety of histone PTMs rely on central key metabolites that act as substrates for their catalysis in common. Acetyl-CoA, the main donor for histone acetylation, arises mostly from glucose-derived citrate by the intervention of ATP citrate lyase (ACL) or by the enzymatic activation of acetate by acetyl-CoA synthase 1 (AceCS1) (Figure 4, end of the manuscript) [29]. Likewise, there are other acyl-CoAs that act as co-substrates for newly-identified histone acyl-PTMs. This includes propionyl-, butyryl-, crotonyl-, glutaryl-, succinyl- and malonyl-CoA, all derived from either β-oxidation of fatty acids or from the catabolism of amino acids [30]. ATP, the main source of energy in the cell, is also the donor for protein phosphorylation. Moreover, both histone and DNA methylation require the methionine-derived cofactor S-adenosylmethionine (SAM) as a source of methyl groups. It is therefore intuitive that metabolic changes impacting the availability of these cofactors affect histone PTMs, and thereby influence cell function.
Figure 1: Pathways leading to histone modification catalysis.
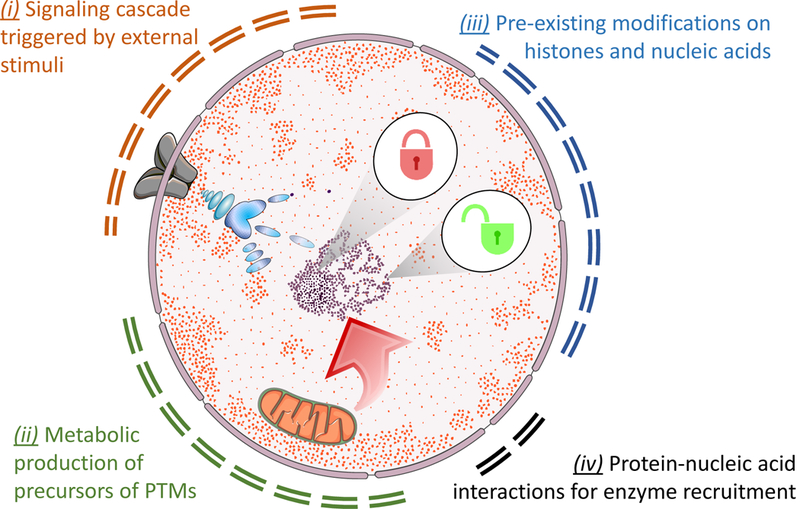
(A) Cartoon representation on the mechanisms leading to chromatin regulation. (i) External stimuli to the cell generate signaling cascade through pathways that eventually modify and modulate the activity of histone writers and erasers. Phosphorylation is the most common PTM used as messenger for signaling cascade via kinase/phosphatase catalysis and removal. (ii) The production of metabolites influences the abundance of histone and other protein PTMs. This phenomenon is frequently not considered in studies investigating global changes of protein PTMs. (iii) Chromatin can be modified depending on its accessibility, and by modifications already present that recruit transcription factors and enzymes. (iv) Other mechanisms for chromatin modification are possible, including regulation through protein-RNA binding and chromatin localization within the nucleus. This last has, as typical example, sequestration of chromatin on the nuclear periphery, leading to local inhibition of gene expression.
Figure 4: List of common histone PTMs, their immediate metabolic precursor and their estimated turnover rate.
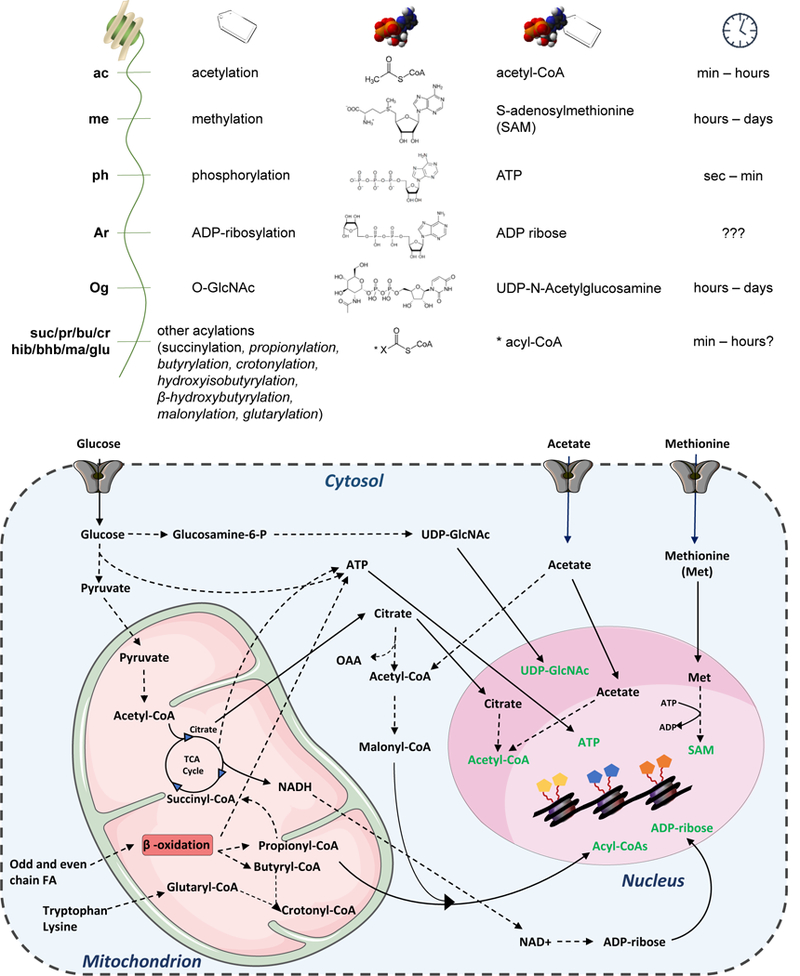
The most common histone modifications have as precursors small molecules derived from metabolism, with the exception for ubiquitinylation. In the figure, histone PTMs are listed as common symbol, full name, structure of immediate precursor metabolite, name of precursor metabolite, and estimated turnover rate of catalysis. Question marks indicate that no study was yet performed to estimate the turnover rate of the given PTMs. Below, major pathways leading to the synthesis of the described biomolecules.
Over the last decade, the implication of metabolism in various aspects of epigenetic control has been revealed in multiple systems. For example, it has been shown that siRNA-mediated silencing of ACL directly affects the transcription of genes regulating the metabolism of glucose [31]. In fact, silencing of ACL results in a decrease of nuclear acetyl-CoA pools and consequently a decrease in the levels of histone acetylation. Although less profound than acetylation, examples of metabolic control have also been seen for histone methylation. Decreasing the levels of SAM through threonine-depleted media in mouse embryonic stem cell (mESCs) was found to significantly decrease H3K4me3 levels, slowing the growth and increasing the differentiation of mESCs [32]. However, the catabolism of threonine is exclusive to mESCs; it is thus unclear whether the influence of metabolism in methylation can affect human stem cells as well.
PTMs are frequently co-localized on histones, which has led to the hypothesis of the “histone code”. This hypothesis proposes that histone PTMs can serve either alone or in combination as a signaling platform to recruit a network of histone-modifying enzymes and nucleosome remodeling complexes to fine-tune gene regulation [33,34]. In fact, dysregulation of combinations of histone PTMs has been implicated in the pathology of many human diseases including cancer [35–39]. For instance, low levels of H3K9ac, H3K9me3 and H4K16ac are positively correlated with tumor recurrence in patients with lung cancer, whereas high levels of H3K18ac and H3K4me2 are associated with an increased risk of prostate cancer progression and metastasis [40]. Nevertheless, the lack of a full understanding of PTM-mediated chromatin processes remains a challenge, as new analytical tools for the comprehensive study of the combinatorial nature of the code are just emerging. This review will give a brief summary of the MS-based approaches that are commonly applied for the analysis of histone PTMs and the possibility of employing MS-based metabolomics to help unravel the role of metabolism in epigenetic control. A detailed discussion of the basic principles of MS for the analysis of proteins is outside the scope of this paper, as such, we refer the interested reader to specialized reviews [41–43].
Overview of all methods for the analysis of histone PTMs
Over the past decade, advances in high-resolution MS have revolutionized the analysis of histone PTMs. The ability to perform high-throughput analysis of single and/or combinatorial patterns of histone PTMs has made MS the most suitable technique for the comprehensive analysis of histone proteoforms, where “proteoform” is defined as all potential isoforms comprising different combinations of PTMs and sequence variations in which a protein can be found [44]. MS provides by far the most accurate quantification and highest throughput in terms of how many PTMs can be identified and quantified in a single analysis. However, histone PTMs can also be investigated by means of site-specific antibody-based methods such as Western Blot, Immunofluorescence and Chromatin Immunoprecipitation (ChIP). The latter can be coupled to DNA microarrays (ChIP-chip) or highly parallel DNA sequencing (ChIP-seq) for genome-wide analysis [45,46]. Nonetheless, it is important to specify that antibody-based techniques have well-known biases in epitope recognition when co-existing PTMs are present (Figure 2) [47]. For instance, site-specific antibodies for H3S10ph fail to recognize the phosphorylated epitope when the nearby H3K9 or H3K14 residues are acetylated, giving rise to false negative results [48]. Moreover, antibody cross-reactivity is often observed with different modification states (e.g., mono-, di-, or trimethylation), especially when they are placed within the same amino acid context, as is the case for H3K9 and H3K27, which have a common A-R-K-S sequence motif [49]. Cross-reactivity has also been reported for pan-anticrotonyl-lysine and pan anti-butyryl-lysine antibodies with structurally related acyl marks including lysine acetylation, propionylation, crotonylation and butyrylation [50].
Figure 2: Potential biases of antibody-based strategies for the analysis of histone PTMs.
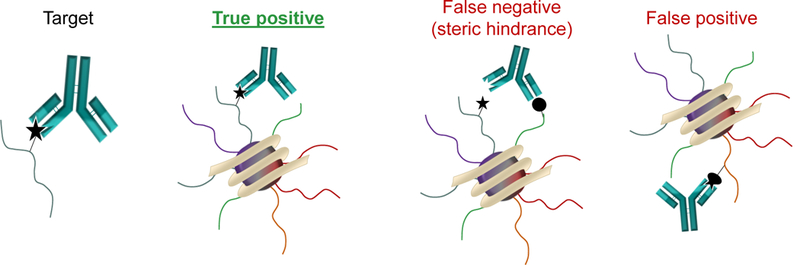
Recognition of a specific histone modification by an antibody can be hindered by the presence of nearby PTMs, generating a false negative. Generating highly specific antibodies for small modifications is also a challenging task, potentially leading to false positive identifications. For instance, it is difficult to generate antibodies that specifically recognize one type of acyl-PTM and not bind another (e.g. acetyl, propionyl, crotonyl). Moreover, antibodies for these “exotic” modifications are rarely site specific.
As a result, several efforts have been made to provide researchers with resources to assess the quality of histone modification-specific antibodies [51–55]. One such resource is the recently published “Histone Antibody Specificity Database,” where authors have evaluated the reactivity of more than 100 commercial PTM-specific antibodies against 250 modified histone peptides and have made the results available on an open-access website [55]. In this report, the evaluated antibodies displayed various degrees of cross-reactivity and influence by neighboring PTMs, highlighting the importance of carefully validating antibodies to avoid misleading conclusions. A recent work employing antibody-based strategies has succesfully identified p300, a histone acetyltransferase (HAT), as able to perform histone crotonylation [56]. The study also showed that p300-catalyzed histone crotonylation is able to stimulate transcription to a greater degree than histone acetylation and that this mechanism is dependent on the metabolic concentrations of the cofactor crotonyl-CoA. The study did not find remarkable differences in the genome-wide localization between acetyl and crotonyl, raising doubts on whether the two marks have different biological function.
In contrast to PTM-specific antibodies, MS represents a more unbiased technique that overcomes many of the discussed challenges. The high resolution offered by MS allows to discriminate between modifications with nearly the same nominal mass shift such as acetylation (ΔM = 42.0106 Da) and trimethylation (ΔM = 42.0470 Da) [57]. MS is also the most suitable technique to identify novel histone PTMs. There are three different strategies for the analysis of histone proteins via MS: “bottom- up”, “top-down” and “middle-down” (reviewed in [58]). These strategies differ by the length of the amino acid sequence being analyzed, type of chromatography and MS acquisition method (Figure 3).
Figure 3: MS methods for the analysis of histone PTMs and their respective precursor metabolites.
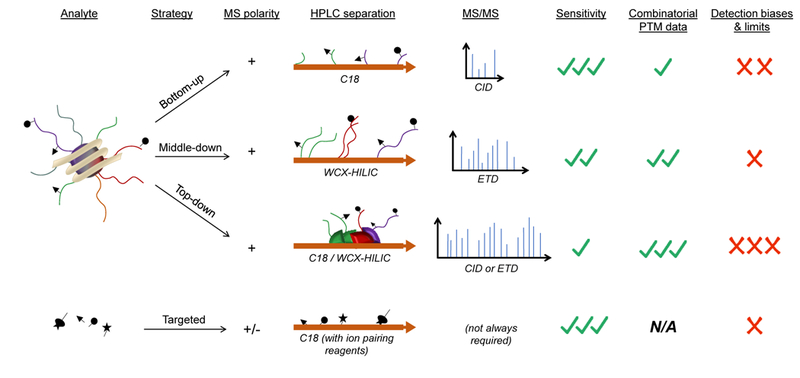
Histone analysis can be performed with three different strategies: bottom-up (short peptides), middle-down (long intact tails) and top-down (no digestion, full protein). Histone sequences always ionize preferentially in positive mode. Separation and MS detection varies depending on the strategy; in particular, for middle-down and top-down, chromatography has a critical role due to the large variety of isobaric forms that must be at least partially separated. Because of the number of isobaric forms, and the charge state distribution (wider for larger analytes), the sensitivity is lower for middle-down and top-down as compared to bottom-up. However, middle-down and top-down have fewer biases in ionization efficiency, as the presence of a PTM does not affect the ionization of a long polypeptide as much as compared to short peptides. Despite lower biases, the enormous variety of isobaric forms makes top-down MS still a semi-quantitative strategy. Metabolites can be analyzed in a targeted mode, mostly accompanied by synthetic and isotopically labeled standards to enhance confidence. Because of this, MS/MS is not always required. Separation is commonly performed with C18 chromatography aided by ion pairing reagents to improve LC binding and resolution.
MS strategies for the analysis of individual and combinatorial histone PTMs
“Bottom-up” is the most conventional MS strategy for the study of histone PTMs. Histones are cleaved into short (4–20 aa) peptides by trypsin (cleaves after KR) or ArgC (cleaves after R) digestion. Due to the high frequency of K and R residues on histones, the use of trypsin is combined with derivatization of lysine residues with propionylation or heavy labeled acetylation [59–61]. Chemical derivatization prevents trypsin from digesting after lysine residues, but also leads to longer and more hydrophobic peptides, more suitable for reversed-phase chromatography. Data-independent acquisition (DIA) is becoming more and more popular for acquiring histone peptides [62]. This is because histone peptides have numerous isobaric forms, where MS/MS fragmentation is required for differential quantification. The main advantage of bottom-up MS is the ease of use and the high sensitivity achieved; i.e. in a recent publication, we proved that the analysis of common PTMs can be performed with as low as 50,000 cells as starting material [63]. Bottom-up MS has been proven successful in identifying and quantifying many novel histone PTMs including crotonylation, malonylation, succinylation and O-GlcN-acylation [22,64,65]. One drawback of the bottom-up MS approach is its limitation in discriminating histone variants, as several PTMs belong to peptides that have the same exact sequence between isotypes. In addition, the physical linkage among PTMs on a given histone is lost, allowing only the analysis of short-range PTM combinations (e.g., H3K27 and H3K36) [66]. However, the high sensitivity can compensate for these limitations when performing MS analysis with specific enrichment methods. For instance, in combination with PTM-specific antibodies for enrichment of distinct histone modifications (H3K27me2/3 and H4K20me1), Voigt et al. demostrated the application of bottom-up MS for the study of PTM patterns on single and separate histone copies within a nuclesome [67].
In “top-down” MS no proteolytic digestion is performed prior to MS analysis. Hence, combinatorial patterns of PTMs and the exact histone variant are potentially identified unambiguously due to the unique signal they produce [68]. Core histones are more suitable for this approach due to their low molecular weight (11–15 kDa) and their high abundance. This analysis necessarily requires high resolution MS and MS/MS due to the high charge state of the analytes. Moreover, electron capture dissociation or electron transfer dissociation (ECD/ETD) [69] are more suitable fagmentation methods than typical collision based methods used in bottom-up MS. Despite the intuitive advantages, top-down MS still remains an elitary approach;due to its limitations, it requires highly specialized analysts [70]. The main challenge is the number of potential isobaric forms when considering the intact protein. For example, H4 alone has over 26 billion theoretical proteoforms [71] which hardly separate by chromatography, and many of them end up populating the same MS/MS spectrum. Typically, offline two-dimentional liquid chromatography (2D LC) separation of histones has been carried out. In the first dimension, histones are separated by family members (H4, H2B, H2A and H3) based on their order of hydrophobicity using reversed-phase (RP) chromatography [72]. The second dimension involves separation of modified states (mostly acetylation or methylation) using weak cation exchange-hydrophilic interaction liquid chromatography (WCX-HILIC) [73]. Using this combined approach, Pesavento et al. were able to identify 42 unique combinatorial PTMs on histone H4 and their dynamic changes during the cell cycle in HeLa cells [74]. Even though the entire workflow remains challenging, most histones and histone variants have been characterized using top-down MS [75–79]. Top-down MS is also computationally challenging. DiMaggio et al. proposed an algorithm based on a mixed integer linear optimization using ETD LC-MS/MS data [80]. Commercial software adopted to the analysis of heavily modified histone proteins are ProSightPC (Thermo), developed from the free version ProSightPTM [81], and BIG Mascot [82], a version of Mascot (MatrixScience) that can analyze polypeptides and intact proteins >16 kDa. More recently, LeDuc et al. developed the C-score [83] to enhance the confidence in the correct mapping of PTMs on the identified intact protein sequences. In fact, the real challenge of identifying a top-down MS/MS spectrum is not the identification of the histone sequence, but the unambiguous mapping of the modified amino acid residues.
The “middle-down” MS strategy is a compromise between top-down and bottom-up. In middle-down MS, proteins are digested into longer peptides (3–9 kDa) with proteolytic enzymes that have less frequent cleavage sites in histones such as AspN or GluC. Digestion by these proteases generates polypeptides that consist of intact histone N-terminal tails (40–50 aa residues). Thus, this approach allows for the analysis of co-existing PTMs on the histone tail, where most PTMs reside, without the burdensome technical challenges of top-down MS. This approach proved its high throughput feasibility in a publication of Young et al. [84]. It was then optimized in 2014 [85], and today it can be effectively combined with metabolic labeling of the protein sequence or the PTMs, allowing turnover studies [86]. This last study showed highly comparable results between the bottom-up and the middle-down MS quantification of single histone PTM, further improving previous optimization [87]. Like top-down though, middle-down MS requires different LC separation, MS acquisition and informatics analysis than bottom-up MS. Histone N-terminal tails are commonly separated by “saltless” pH gradient using a WCX-HILIC type of chromatography developed by Young et al. [84]. Detection is performed using high resolution MS and MS/MS, and ideally ETD fragmentation. More recently, separation of isobaric histone tails was achieved by using ion mobility [88], paving the way to potential new orthogonal ways to resolve complex mixtures and improve sensitivity. The challenges of data analysis are also mostly based on the accuracy by which a software is able to unambiguously map the localization of PTMs. Frankly, there is no software developed on purpose for middle-down MS. Most of the analyses are performed with abovementioned software tools intended for top-down MS. To date, the only workflow which proved to provide a comparable quantification to bottom-up MS [86,87] is the following: MS/MS ETD spectra deconvolution using Xtract (Thermo), database searching using Mascot (MatrixScience) and data filtering and quantification using isoScale [85].
MS strategies for the analysis of metabolites precursors of histone PTMs
In recent years, metabolomics has made enormous steps towards untargeted identification and more accurate quantification of metabolites using MS. The Human Metabolome Database (HMDB) was released in 2007 [89], and it currently contains >100,000 metabolites. Identifying the entire metabolome of an organism is still very much a challenge, as metabolites have an extremely dynamic regulation, and they span between a wide range of abundances. Specifically, metabolites are considered abundant if > 1 μM, or relatively rare if < 1 nM. Luckily, the list of metabolites precursors of histone PTMs is confined and it can be performed with targeted acquisition. It can also be performed with low resolution MS, as long as MS/MS is performed to increase the confidence in depicting the proper signal [90]. When using high resolution MS, it is possible to profile metabolites even without using necessarily MS/MS [91]. Metabolite extraction is generally a straightforward protocol, especially if the interest is limited to glycolysis, the tricarboxylic acid (TCA) cycle, the pentose-phosphate pathway, and metabolism of amino acids. Differently from proteomics, acquisition is normally performed by switching between positive and negative polarity, as selected metabolites are more easily ionized in negative mode, e.g. ATP. The type of chromatography may vary depending on the class of metabolites, although a general good starting point is the use of C18 reversed phase chromatography with the addition of ion pairing reagents, e.g. tributylamine [91] (Figure 3). Feeding cell cultures using stable isotope labeled metabolites opens up for exciting experiments where the turnover of protein PTMs can be investigated based on the ratio heavy/light of the quantified modification on a peptide. In a recent publication, Kori et al. estimated the turnover of acetylations on proteins by labeling either glucose or acetate with heavy isotopes [92]. Results showed a faster acetylation turnover for all proteins when glucose was labeled. However, quantification of heavy labeled acetyl-CoA proved that the cell uses more glucose than acetate to produce acetyl-CoA, implying that the calculated turnover rate was biased by which metabolite was labeled as reference. This, once again, confirmed that monitoring the relative levels of metabolites can reveal much about the regulation of PTMs occurring on the chromatin proteome. Several other publications present excellent quality analysis of targeted metabolites. In our personal experience, we found that C18 reversed phase chromatography (with ion pairing reagents) running in the high microliter flow-rate (100–200 μL/min) combined with targeted MS acquisition is the safest approach for combined sensitivity, confidence and reproducibility.
Histone modifications and metabolism: co-dependency in health and disease
As most chromatin-modifying enzymes demand the use of metabolic intermediates, it is expected that the proper functioning of chromatin to regulate physiological processes and to maintain homeostasis requires an intimate coordination with cell metabolism. The relationship between histone modifications and the cellular metabolic state is a theme that has sparked the interest of many researchers over the last few years [93–96]. For instance, by employing MS and [13C] glucose-labeling in cell culture, Evertts et al., monitored the rate of the incorporation of histone acetylations by regulation of glucose flux [97]. This study showed once again that glucose is the predominant source for nuclear acetyl-CoA, as near complete acetylation turnovers were observed when using isotopically labeled glucose, followed by glutamine and acetate in cultured HEK293 cells. Similarly, other studies have investigated protein PTM turnover, including O-GlcNAc [98] (Figure 4), providing an important timescale perspective on how frequently modifications are exchanged on histones and other proteins.
Sabari et al. showed that manipulations of the levels of crotonyl-CoA through the addition of crotonate to cell culture can shift the levels of histone lysine crotonylation (Kcr) [56]. Similarly, starvation-induced ketogenesis marked by an increase in the concentration of β-hydroxybutyrate in the liver and kidneys was found to dramatically induce the levels of histone lysine β-hydroxybutyrylation (Kbhb) [99]. Further evidence suggesting that the levels of precursor metabolites directly affect the levels of histone PTMs has also been shown in vitro. Through in nucleo experiments, we were able to demonstrate that the availability of acyl-CoA co-substrates has a direct impact on the levels of nuclear histone acylations [50]. Strikingly, this study also showed a strong positive correlation between the levels of various acyl-CoA donors and their corresponding histone acylations in proliferative and differentiated myogenic cells in vivo. Although the mechanisms for the catalysis of histone marks, in particular histone acylations, has proven to be both enzymatic and non-enzymatic in nature [50,100,101], the existence of a quantitative link between intracellular metabolite concentrations and chromatin modifications, and possibly epigenetic regulation, has become more concrete.
Metabolic regulation of chromatin has already been recognized in both healthy and diseased conditions. For example, multiple studies have investigated the implication of nutritional challenge through a high-fat diet in the reprogramming of genes regulating the circadian clock in the liver [93,102,103]. This mechanism involves large oscillation of transcripts mediated by chromatin remodeling via acetylation of histone H3 at positions K9 and K14 [93,104]. Similarly, the implication of epigenetic regulation in cell senescence and aging has been extensively discussed [93]. While further investigation is required to ascertain the involvement of cell metabolism in normal epigenetic traits, most research has focused on elucidating the implications of altered metabolism in the development of disease, specifically cancer. The proposed mechanisms through which dysfunctional metabolic states can elicit epigenetic changes contributing to the formation of cancer could be summarized as follows: first, extensive metabolic rewiring has been recognized as a hallmark of cancer cells [105,106]. As such, tumorigenesis-associated metabolic alterations can influence the availability of cofactors required for chromatin-modifying enzymes, directly affecting the epigenome and in turn, the transcriptome [1,94,107,108]. Secondly, the altered epigenetic landscape in cancer cells can affect the expression of genes involved in cell metabolism, mostly through aberrant DNA methylations and histone modifications, and deregulation of metabolic signaling pathways by microRNAs (miRNAs) [108–110]. While the mechanisms driving tumorigenesis are still not fully understood, it is intuitive that integrating metabolomics and proteomics approaches could significantly increase our understanding of the bigger picture and, in particular, the interplay between epigenetics and metabolism.
Acknowledgments
The authors gratefully acknowledge the NIH grants CA196539, GM110174 and AI118891. We apologize to all authors whose contribution to the field was not mentioned due to space limitation.
Footnotes
Conflicts of interest
The authors declare no financial conflict of interest.
References
- [1].Suvà ML, Riggi N, Bernstein BE, Epigenetic reprogramming in cancer. Science 2013, 339, 1567–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Jones PA, Baylin SB, The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415–428. [DOI] [PubMed] [Google Scholar]
- [3].Kornberg RD, Chromatin structure: a repeating unit of histones and DNA. Science 1974, 184, 868–871. [DOI] [PubMed] [Google Scholar]
- [4].Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ, Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260. [DOI] [PubMed] [Google Scholar]
- [5].Luger K, in:, eLS, John Wiley & Sons, Ltd, 2001. [Google Scholar]
- [6].Margueron R, Reinberg D, Chromatin structure and the inheritance of epigenetic information. Nat. Rev. Genet. 2010, 11, 285–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Arya G, Schlick T, Role of histone tails in chromatin folding revealed by a mesoscopic oligonucleosome model. Proc. Natl. Acad. Sci. 2006, 103, 16236–16241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Chi P, Allis CD, Wang GG, Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers. Nat. Rev. Cancer 2010, 10, 457–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Murr R, Interplay between different epigenetic modifications and mechanisms. Adv. Genet. 2010, 70, 101–141. [DOI] [PubMed] [Google Scholar]
- [10].Allfrey VG, Faulkner R, Mirsky AE, ACETYLATION AND METHYLATION OF HISTONES AND THEIR POSSIBLE ROLE IN THE REGULATION OF RNA SYNTHESIS*. Proc. Natl. Acad. Sci. U. S. A. 1964, 51, 786–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Struhl K, Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998, 12, 599–606. [DOI] [PubMed] [Google Scholar]
- [12].Megee PC, Morgan BA, Mittman BA, Smith MM, Genetic analysis of histone H4: essential role of lysines subject to reversible acetylation. Science 1990, 247, 841–845. [DOI] [PubMed] [Google Scholar]
- [13].Durrin LK, Mann RK, Kayne PS, Grunstein M, Yeast histone H4 N-terminal sequence is required for promoter activation in vivo. Cell 1991, 65, 1023–1031. [DOI] [PubMed] [Google Scholar]
- [14].Miranda TB, Jones PA, DNA methylation: the nuts and bolts of repression. J. Cell. Physiol. 2007, 213, 384–390. [DOI] [PubMed] [Google Scholar]
- [15].Zhang Y, Reinberg D, Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev. 2001, 15, 2343–2360. [DOI] [PubMed] [Google Scholar]
- [16].Goldknopf IL, Taylor CW, Baum RM, Yeoman LC, et al. , Isolation and characterization of protein A24, a “histone-like” non-histone chromosomal protein. J. Biol. Chem. 1975, 250, 7182–7187. [PubMed] [Google Scholar]
- [17].Stanley JS, Griffin JB, Zempleni J, Biotinylation of histones in human cells. Effects of cell proliferation. Eur. J. Biochem. 2001, 268, 5424–5429. [DOI] [PubMed] [Google Scholar]
- [18].Hassa PO, Haenni SS, Elser M, Hottiger MO, Nuclear ADP-ribosylation reactions in mammalian cells: where are we today and where are we going? Microbiol. Mol. Biol. Rev. MMBR 2006, 70, 789–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Shoemaker CB, Chalkley R, An H3 histone-specific kinase isolated from bovine thymus chromatin. J. Biol. Chem. 1978, 253, 5802–5807. [PubMed] [Google Scholar]
- [20].Peng C, Lu Z, Xie Z, Cheng Z, et al. , The first identification of lysine malonylation substrates and its regulatory enzyme. Mol. Cell. Proteomics MCP 2011, 10, M111.012658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zhang Z, Tan M, Xie Z, Dai L, et al. , Identification of lysine succinylation as a new post-translational modification. Nat. Chem. Biol. 2011, 7, 58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Tan M, Luo H, Lee S, Jin F, et al. , Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 2011, 146, 1016–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Chen Y, Sprung R, Tang Y, Ball H, et al. , Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol. Cell. Proteomics MCP 2007, 6, 812–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Tan M, Peng C, Anderson KA, Chhoy P, et al. , Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab. 2014, 19, 605–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Dai L, Peng C, Montellier E, Lu Z, et al. , Lysine 2-hydroxyisobutyrylation is a widely distributed active histone mark. Nat. Chem. Biol. 2014, 10, 365–370. [DOI] [PubMed] [Google Scholar]
- [26].Xie Z, Zhang D, Chung D, Tang Z, et al. , Metabolic Regulation of Gene Expression by Histone Lysine β-Hydroxybutyrylation. Mol. Cell 2016, 62, 194–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zhao Y, Garcia BA, Comprehensive Catalog of Currently Documented Histone Modifications. Cold Spring Harb. Perspect. Biol. 2015, 7, a025064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].He C, Sidoli S, Warneford-Thomson R, Tatomer DC, et al. , High-Resolution Mapping of RNA-Binding Regions in the Nuclear Proteome of Embryonic Stem Cells. Mol. Cell 2016, 64, 416–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Albaugh BN, Arnold KM, Denu JM, KAT(ching) metabolism by the tail: insight into the links between lysine acetyltransferases and metabolism. Chembiochem Eur. J. Chem. Biol. 2011, 12, 290–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Papanicolaou KN, O’Rourke B, Foster DB, Metabolism leaves its mark on the powerhouse: recent progress in post-translational modifications of lysine in mitochondria. Front. Physiol. 2014, 5, 301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, et al. , ATP-citrate lyase links cellular metabolism to histone acetylation. Science 2009, 324, 1076–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Shyh-Chang N, Locasale JW, Lyssiotis CA, Zheng Y, et al. , Influence of Threonine Metabolism on S-adenosyl-methionine and Histone Methylation. Science 2013, 339, 222–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Strahl BD, Allis CD, The language of covalent histone modifications. Nature 2000, 403, 41–45. [DOI] [PubMed] [Google Scholar]
- [34].Rando OJ, Combinatorial complexity in chromatin structure and function: revisiting the histone code. Curr. Opin. Genet. Dev. 2012, 22, 148–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Bhaumik SR, Smith E, Shilatifard A, Covalent modifications of histones during development and disease pathogenesis. Nat. Struct. Mol. Biol. 2007, 14, 1008–1016. [DOI] [PubMed] [Google Scholar]
- [36].Greer EL, Shi Y, Histone methylation: a dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Thompson LL, Guppy BJ, Sawchuk L, Davie JR, McManus KJ, Regulation of chromatin structure via histone post-translational modification and the link to carcinogenesis. Cancer Metastasis Rev. 2013, 32, 363–376. [DOI] [PubMed] [Google Scholar]
- [38].Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, et al. , Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400. [DOI] [PubMed] [Google Scholar]
- [39].Seligson DB, Horvath S, Shi T, Yu H, et al. , Global histone modification patterns predict risk of prostate cancer recurrence. Nature 2005, 435, 1262–1266. [DOI] [PubMed] [Google Scholar]
- [40].Chervona Y, Costa M, Histone modifications and cancer: biomarkers of prognosis? Am. J. Cancer Res. 2012, 2, 589–597. [PMC free article] [PubMed] [Google Scholar]
- [41].Walther TC, Mann M, Mass spectrometry–based proteomics in cell biology. J. Cell Biol. 2010, 190, 491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Steen H, Mann M, The ABC’s (and XYZ’s) of peptide sequencing. Nat. Rev. Mol. Cell Biol. 2004, 5, 699–711. [DOI] [PubMed] [Google Scholar]
- [43].Baldwin MA, Protein identification by mass spectrometry: issues to be considered. Mol. Cell. Proteomics MCP 2004, 3, 1–9. [DOI] [PubMed] [Google Scholar]
- [44].Smith LM, Kelleher NL, Consortium for Top Down Proteomics, Proteoform: a single term describing protein complexity. Nat. Methods 2013, 10, 186–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Schulz S, Häussler S, Chromatin immunoprecipitation for ChIP-chip and ChIP-seq. Methods Mol. Biol. Clifton NJ 2014, 1149, 591–605. [DOI] [PubMed] [Google Scholar]
- [46].Pillai S, Chellappan SP, ChIP on chip and ChIP-Seq assays: genome-wide analysis of transcription factor binding and histone modifications. Methods Mol. Biol. Clifton NJ 2015, 1288, 447–472. [DOI] [PubMed] [Google Scholar]
- [47].Britton L-MP, Gonzales-Cope M, Zee BM, Garcia BA, Breaking the histone code with quantitative mass spectrometry. Expert Rev. Proteomics 2011, 8, 631–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Clayton AL, Rose S, Barratt MJ, Mahadevan LC, Phosphoacetylation of histone H3 on c-fos- and c-jun-associated nucleosomes upon gene activation. EMBO J. 2000, 19, 3714–3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kungulovski G, Kycia I, Mauser R, Jeltsch A, Specificity Analysis of Histone Modification-Specific Antibodies or Reading Domains on Histone Peptide Arrays. Methods Mol. Biol. Clifton NJ 2015, 1348, 275–284. [DOI] [PubMed] [Google Scholar]
- [50].Simithy J, Sidoli S, Yuan Z-F, Coradin M, et al. , Characterization of histone acylations links chromatin modifications with metabolism. Nat. Commun. 2017, 8, 1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Bock I, Dhayalan A, Kudithipudi S, Brandt O, et al. , Detailed specificity analysis of antibodies binding to modified histone tails with peptide arrays. Epigenetics 2011, 6, 256–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Hattori T, Taft JM, Swist KM, Luo H, et al. , Recombinant antibodies to histone post-translational modifications. Nat. Methods 2013, 10, 992–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Fuchs SM, Krajewski K, Baker RW, Miller VL, Strahl BD, Influence of combinatorial histone modifications on antibody and effector protein recognition. Curr. Biol. CB 2011, 21, 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Egelhofer TA, Minoda A, Klugman S, Lee K, et al. , An assessment of histone-modification antibody quality. Nat. Struct. Mol. Biol. 2011, 18, 91–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Rothbart SB, Dickson BM, Raab JR, Grzybowski AT, et al. , An Interactive Database for the Assessment of Histone Antibody Specificity. Mol. Cell 2015, 59, 502–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Sabari BR, Tang Z, Huang H, Yong-Gonzalez V, et al. , Intracellular crotonyl-CoA stimulates transcription through p300-catalyzed histone crotonylation. Mol. Cell 2015, 58, 203–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Yang L, Tu S, Ren C, Bulloch EMM, et al. , Unambiguous determination of isobaric histone modifications by reversed-phase retention time and high-mass accuracy. Anal. Biochem. 2010, 396, 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Sidoli S, Cheng L, Jensen ON, Proteomics in chromatin biology and epigenetics: Elucidation of post-translational modifications of histone proteins by mass spectrometry. J. Proteomics 2012, 75, 3419–3433. [DOI] [PubMed] [Google Scholar]
- [59].Garcia BA, Mollah S, Ueberheide BM, Busby SA, et al. , Chemical derivatization of histones for facilitated analysis by mass spectrometry. Nat. Protoc. 2007, 2, 933–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Bonaldi T, Imhof A, Regula JT, A combination of different mass spectroscopic techniques for the analysis of dynamic changes of histone modifications. PROTEOMICS 2004, 4, 1382–1396. [DOI] [PubMed] [Google Scholar]
- [61].Arnaudo AM, Molden RC, Garcia BA, Revealing histone variant induced changes via quantitative proteomics. Crit. Rev. Biochem. Mol. Biol. 2011, 46, 284–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Sidoli S, Simithy J, Karch KR, Kulej K, Garcia BA, Low Resolution Data-Independent Acquisition in an LTQ-Orbitrap Allows for Simplified and Fully Untargeted Analysis of Histone Modifications. Anal. Chem. 2015, 87, 11448–11454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Guo Q, Sidoli S, Garcia BA, Zhao X, Assessment of quantification precision of histone post-translational modifications by using an ion trap and down to 50,000 cells as starting material. J. Proteome Res. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Sakabe K, Wang Z, Hart GW, β-N-acetylglucosamine (O-GlcNAc) is part of the histone code. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 19915–19920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Xie Z, Dai J, Dai L, Tan M, et al. , Lysine succinylation and lysine malonylation in histones. Mol. Cell. Proteomics MCP 2012, 11, 100–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Tian Z, Tolić N, Zhao R, Moore RJ, et al. , Enhanced top-down characterization of histone post-translational modifications. Genome Biol. 2012, 13, R86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Voigt P, LeRoy G, Drury WJ, Zee BM, et al. , Asymmetrically Modified Nucleosomes. Cell 2012, 151, 181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Thomas CE, Kelleher NL, Mizzen CA, Mass spectrometric characterization of human histone H3: a bird’s eye view. J. Proteome Res. 2006, 5, 240–247. [DOI] [PubMed] [Google Scholar]
- [69].Shen Y, Tolić N, Xie F, Zhao R, et al. , Effectiveness of CID, HCD, and ETD with FT MS/MS for degradomic-peptidomic analysis: comparison of peptide identification methods. J. Proteome Res. 2011, 10, 3929–3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Liu X, Hengel S, Wu S, Tolić N, et al. , Identification of ultramodified proteins using top-down tandem mass spectra. J. Proteome Res. 2013, 12, 5830–5838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Consortium UniProt, Reorganizing the protein space at the Universal Protein Resource (UniProt). Nucleic Acids Res. 2012, 40, D71–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Tian Z, Zhao R, Tolić N, Moore RJ, et al. , Two-dimensional liquid chromatography system for online top-down mass spectrometry. Proteomics 2010, 10, 3610–3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Garcia BA, Pesavento JJ, Mizzen CA, Kelleher NL, Pervasive combinatorial modification of histone H3 in human cells. Nat. Methods 2007, 4, 487–489. [DOI] [PubMed] [Google Scholar]
- [74].Pesavento JJ, Bullock CR, LeDuc RD, Mizzen CA, Kelleher NL, Combinatorial Modification of Human Histone H4 Quantitated by Two-dimensional Liquid Chromatography Coupled with Top Down Mass Spectrometry. J. Biol. Chem. 2008, 283, 14927–14937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Boyne MT, Pesavento JJ, Mizzen CA, Kelleher NL, Precise characterization of human histones in the H2A gene family by top down mass spectrometry. J. Proteome Res. 2006, 5, 248–253. [DOI] [PubMed] [Google Scholar]
- [76].Pesavento JJ, Mizzen CA, Kelleher NL, Quantitative analysis of modified proteins and their positional isomers by tandem mass spectrometry: human histone H4. Anal. Chem. 2006, 78, 4271–4280. [DOI] [PubMed] [Google Scholar]
- [77].Chen Y, Hoover ME, Dang X, Shomo AA, et al. , Quantitative Mass Spectrometry Reveals that Intact Histone H1 Phosphorylations are Variant Specific and Exhibit Single Molecule Hierarchical Dependence. Mol. Cell. Proteomics MCP 2016, 15, 818–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Dang X, Singh A, Spetman BD, Nolan KD, et al. , Label-Free Relative Quantitation of Isobaric and Isomeric Human Histone H2A and H2B Variants by Fourier Transform Ion Cyclotron Resonance Top-Down MS/MS. J. Proteome Res. 2016, 15, 3196–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Siuti N, Roth MJ, Mizzen CA, Kelleher NL, Pesavento JJ, Gene-specific characterization of human histone H2B by electron capture dissociation. J. Proteome Res. 2006, 5, 233–239. [DOI] [PubMed] [Google Scholar]
- [80].DiMaggio PA, Young NL, Baliban RC, Garcia BA, Floudas CA, A mixed integer linear optimization framework for the identification and quantification of targeted post-translational modifications of highly modified proteins using multiplexed electron transfer dissociation tandem mass spectrometry. Mol. Cell. Proteomics MCP 2009, 8, 2527–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].LeDuc RD, Taylor GK, Kim Y-B, Januszyk TE, et al. , ProSight PTM: an integrated environment for protein identification and characterization by top-down mass spectrometry. Nucleic Acids Res. 2004, 32, W340–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Karabacak NM, Li L, Tiwari A, Hayward LJ, et al. , Sensitive and specific identification of wild type and variant proteins from 8 to 669 kDa using top-down mass spectrometry. Mol. Cell. Proteomics MCP 2009, 8, 846–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].LeDuc RD, Fellers RT, Early BP, Greer JB, et al. , The C-score: a Bayesian framework to sharply improve proteoform scoring in high-throughput top down proteomics. J. Proteome Res. 2014, 13, 3231–3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Young NL, DiMaggio PA, Plazas-Mayorca MD, Baliban RC, et al. , High throughput characterization of combinatorial histone codes. Mol. Cell. Proteomics MCP 2009, 8, 2266–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Sidoli S, Schwämmle V, Ruminowicz C, Hansen TA, et al. , Middle-down hybrid chromatography/tandem mass spectrometry workflow for characterization of combinatorial post-translational modifications in histones. Proteomics 2014, 14, 2200–2211. [DOI] [PubMed] [Google Scholar]
- [86].Sidoli S, Lu C, Coradin M, Wang X, et al. , Metabolic labeling in middle-down proteomics allows for investigation of the dynamics of the histone code. Epigenetics Chromatin 2017, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Sidoli S, Lin S, Karch KR, Garcia BA, Bottom-up and middle-down proteomics have comparable accuracies in defining histone post-translational modification relative abundance and stoichiometry. Anal. Chem. 2015, 87, 3129–3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Shliaha PV, Baird MA, Nielsen MM, Gorshkov V, et al. , Characterization of Complete Histone Tail Proteoforms Using Differential Ion Mobility Spectrometry. Anal. Chem. 2017, 89, 5461–5466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Wishart DS, Tzur D, Knox C, Eisner R, et al. , HMDB: the Human Metabolome Database. Nucleic Acids Res. 2007, 35, D521–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Yuan M, Breitkopf SB, Yang X, Asara JM, A positive/negative ion-switching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat. Protoc. 2012, 7, 872–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Lu W, Clasquin MF, Melamud E, Amador-Noguez D, et al. , Metabolomic analysis via reversed-phase ion-pairing liquid chromatography coupled to a stand alone orbitrap mass spectrometer. Anal. Chem. 2010, 82, 3212–3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Kori Y, Sidoli S, Yuan Z-F, Lund PJ, et al. , Proteome-wide acetylation dynamics in human cells. Sci. Rep. 2017, 7, 10296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Berger SL, Sassone-Corsi P, Metabolic Signaling to Chromatin. Cold Spring Harb. Perspect. Biol. 2016, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Kaochar S, Tu BP, Gatekeepers of Chromatin: Small Metabolites Elicit Big Changes in Gene Expression. Trends Biochem. Sci. 2012, 37, 477–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Fan J, Krautkramer KA, Feldman JL, Denu JM, Metabolic regulation of histone post-translational modifications. ACS Chem. Biol. 2015, 10, 95–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Lu C, Thompson CB, Metabolic Regulation of Epigenetics. Cell Metab. 2012, 16, 9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Evertts AG, Zee BM, Dimaggio PA, Gonzales-Cope M, et al. , Quantitative dynamics of the link between cellular metabolism and histone acetylation. J. Biol. Chem. 2013, 288, 12142–12151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Wang X, Yuan Z-F, Fan J, Karch KR, et al. , A Novel Quantitative Mass Spectrometry Platform for Determining Protein O-GlcNAcylation Dynamics. Mol. Cell. Proteomics MCP 2016, 15, 2462–2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Xie Z, Zhang D, Chung D, Tang Z, et al. , Metabolic Regulation of Gene Expression by Histone Lysine β-Hydroxybutyrylation. Mol. Cell 2016, 62, 194–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Baeza J, Smallegan MJ, Denu JM, Site-specific reactivity of nonenzymatic lysine acetylation. ACS Chem. Biol. 2015, 10, 122–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Kuo Y-M, Andrews AJ, Quantitating the Specificity and Selectivity of Gcn5-Mediated Acetylation of Histone H3. PLOS ONE 2013, 8, e54896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Eckel-Mahan KL, Patel VR, de Mateo S, Orozco-Solis R, et al. , Reprogramming of the Circadian Clock by Nutritional Challenge. Cell 2013, 155, 1464–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Eckel-Mahan K, Sassone-Corsi P, Metabolism and the circadian clock converge. Physiol. Rev. 2013, 93, 107–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Nakahata Y, Grimaldi B, Sahar S, Hirayama J, Sassone-Corsi P, Signaling to the circadian clock: plasticity by chromatin remodeling. Curr. Opin. Cell Biol. 2007, 19, 230–237. [DOI] [PubMed] [Google Scholar]
- [105].Vander Heiden MG, Cantley LC, Thompson CB, Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Ward PS, Thompson CB, Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate. Cancer Cell 2012, 21, 297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Carrer A, Wellen KE, Metabolism and epigenetics: a link cancer cells exploit. Curr. Opin. Biotechnol. 2015, 34, 23–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Wong CC, Yu J, Qian Y, Interplay between epigenetics and metabolism in oncogenesis: mechanisms and therapeutic approaches. Oncogene 2017, 36, 3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Pavlova NN, Thompson CB, THE EMERGING HALLMARKS OF CANCER METABOLISM. Cell Metab. 2016, 23, 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Chen B, Li H, Zeng X, Yang P, et al. , Roles of microRNA on cancer cell metabolism. J. Transl. Med. 2012, 10, 228. [DOI] [PMC free article] [PubMed] [Google Scholar]