

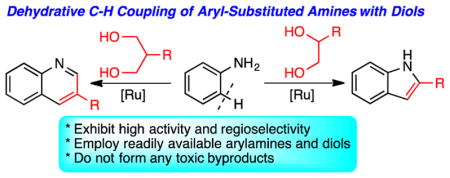
Abstract
The cationic ruthenium-hydride complex catalyzes dehydrative C-H coupling reaction of arylamines with 1,2-diols to form the indole products. The analogous coupling of arylamines with 1,3-diols afforded the substituted quinolines. The catalytic method directly forms these coupling products in a highly regioselective manner without generating any toxic byproducts.
Graphical Abstract
INTRODUCTION
To promote synthetic efficiency and to reduce environmental pollutions resulted from the formation of byproducts, concerted research efforts have been directed to devise transition metal catalyzed C–H coupling methods for the synthesis of biologically active nitrogen heterocyclic compounds.1 Catalytic C–H coupling methods have been shown to provide step efficient routes to indole and related nitrogen heterocyclic compounds, while alleviating inherent problems associated with the classical methods on requiring prefunctionalized substrates and wasteful byproduct formation.2 Most notably, a number of oxidative C–H coupling methods of arylamines with unsaturated hydrocarbon substrates have been successfully developed for the synthesis of indole and quinoline compounds,3 and very recently, the analogous C–H coupling reactions have been achieved by using earth-abundant Co and Ni catalysts.4 Since Watanabe’s seminal report on the ruthenium-catalyzed annulation of aniline with diols,5 the dehydrative C–H coupling methods of arylamines with diols have emerged as a sustainable green catalytic protocol for the synthesis of nitrogen heterocycles. A number of groups employed Ru catalytic systems to effect the intermolecular couplings of anilines with diols and glycols in combination with trialkylammonium salts to synthesize indole and quinoline derivatives.6 Larock’s Pd-catalyzed coupling and annulation methods have been successfully utilized to synthesize a variety of substituted indole products.7 Using IrCl3/BINAP catalyst system, Ishii achieved a highly regioselective coupling of naphthylamines with 1,2- and 1,3-diols to give indole and quinoline derivatives.8 Recently, a number of atom-economical C–H cross coupling methods have been used to promote intramolecular annulation of N-arylimines and enamines.9 So far, synthetic utility of these catalytic coupling methods has been greatly limited because of the requirement of excess amount of prefunctionalized nitrogen substrates, difficulty in controlling regioselectivity as well as the lack of functional group tolerance, and relatively harsh reaction conditions (>170 °C).
We recently reported that a well-defined cationic ruthenium-hydride complex [(C6H6)(PCy3)(CO)RuH]+BF4− (1) is a highly effective catalyst for the dehydrative C–H coupling reaction of phenol with diols to afford benzofuran derivatives.10 We reasoned that the analogous coupling of arylamines with diols might lead to the formation of indoles. Herein, we disclose an effective catalytic synthesis of substituted indoles and quinolines from the dehydrative coupling reactions of arylamines with diols. The catalytic method employs readily available amine and diol substrates, and gives the coupling products without using any reactive reagents or forming any wasteful byproducts.
RESULTS AND DISCUSSION
Catalyst Screening and Optimization Studies
We initially screened suitability of the coupling reaction between aniline and 1,2-diols by using the Ru catalyst 1. The treatment of aniline (0.5 mmol) with 1-phenyl-1,2-ethandiol (0.75 mmol) in the presence of 1 (3 mol %) and cyclopentene (1.5 mmol) in 1,4-dioxane (2 mL) was heated at 110 °C for 14 h (eq 1). The product 3a was cleanly formed, albeit with a low conversion (ca. 20%), as analyzed by both GC and NMR methods.
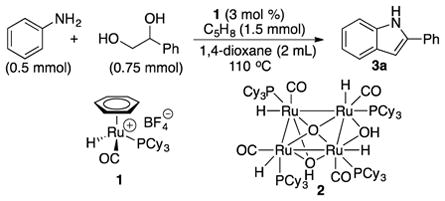 |
(1) |
To attain optimized reaction conditions, we next screened both Ru catalysts and additive effects for the coupling reaction (Table 1). The cationic Ru-H complex, either in isolated form of the complex 1 or in-situ generated from the tetranuclear Ru complex [(PCy3)(CO)RuH]4(μ-O)(μ-OH)2 (2), was found to be most effective among the screened ruthenium catalysts (entries 2, 5).11 We also found that the addition of catalytic amount of HBF4·OEt2 to the Ru catalyst 1 led to a dramatic improvement on the product yield (entry 2), although the promotional effect of HBF4·OEt2 is not entirely clear at this point. Both 1,4-dioxane and chlorobenzene were found to be suitable solvents for the coupling reaction.
Table 1.
Catalyst Screening for the Dehydrative Coupling of Aniline with 1-Phenyl-1,2-ethandiola
| entry | catalyst | additive | yd (%)b |
|---|---|---|---|
| 1 | [RuH(C6H6)(CO)(PCy3)]+BF4− (1) | 19 | |
| 2 | [RuH(C6H6)(CO)(PCy3)]+BF4− (1) | HBF4·OEt2 | 83 |
| 3 | [RuH(CO)(PCy3)]4(O)(OH)2 (2) | 0 | |
| 4 | [RuH(CO)(PCy3)]4(O)(OH)2 (2) | NH4PF6 | 18 |
| 5 | [RuH(CO)(PCy3)]4(O)(OH)2 (2) | HBF4·OEt2 | 96 |
| 6 | RuHCl(CO)(PCy3)2 | 0 | |
| 7 | RuHCl(CO)(PCy3)2 | HBF4·OEt2 | 23 |
| 8 | RuCl2(PPh3)3 | <3 | |
| 9 | RuCl2(PPh3)3 | HBF4·OEt2 | 42 |
| 10 | [RuCl2(p-cymene)]2 | 0 | |
| 11 | [RuCl2(p-cymene)]2 | HBF4·OEt2 | 0 |
| 12 | Ru3(CO)12 | 0 | |
| 13 | Ru3(CO)12 | NH4PF6 | 0 |
| 14 | RuH2(CO)(PPh3)3 | 0 | |
| 15 | [RuH(CO)(CH3CN)2(PCy3)2]+BF4− | 0 | |
| 16 | RuCl3·3H2O | 0 | |
| 17 | [Ru(COD)Cl2]x | HBF4·OEt2 | <3 |
| 18 | Cy3PH+BF4− | 0 | |
| 19 | HBF4·OEt2 | 0 |
Reaction conditions: aniline (0.5 mmol), 1-phenyl-1,2-ethandiol (0.75 mmol), catalyst (3 mol % Ru), additive (7 mol %), cyclopentene (1.5 mmol), 1,4-dioxane (2 mL), 110 °C, 14 h.
The product yield was determined by 1H NMR using hexamethylbenzene as an internal standard.
Reaction Scope
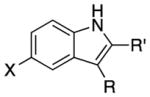
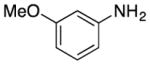
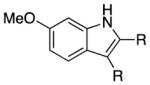
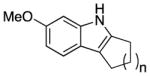
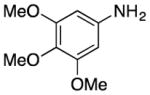
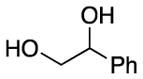
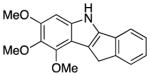
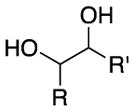
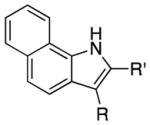
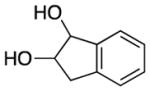
We explored the scope of the coupling reaction of anilines with 1,2-diol substrates (Table 2). Both aliphatic and aryl-substituted 1,2-diols readily reacted with aniline to give the regioselective 2-substituted indole products 3a–3c (entries 1–3). The secondary diol 2,3-butanediol gave the 2,3-disubstituted indole product 3d (entry 4), but with a considerably lower yield than the primary-secondary diols. The coupling with unsymmetric secondary1,2-diols generally gave a mixture of indole regioisomers. For the coupling of 3-methoxyaniline with 1,2-diols, 6-methoxyindole products 3e and 3f were obtained predominantly with a trace amount of 4-methoxyindole products (entries 5, 6), while the reaction with cyclic 1,2-diols smoothly formed the tricyclic indole products 3g–3i (entries 7–9). The coupling of electron-rich 3,4,5-trimethoxyaniline with 1,2-diols also gave the indole product 3j and 3k (entries 11, 12). The coupling reaction of 1-naphthylamine with 1,2-diols predictively led to the formation of polycyclic indole derivatives 3l–3q (entries 12–17). Both primary-secondary and secondary-secondary 1,2-diols afforded the corresponding 2-substituted and 2,3-disubstituted 1H-benzo[g]indoles 3l–3o in moderate to good yields (entries 12–15). The coupling reaction of 1-naphthylamine with 1,2-indandiol and 1,2-cyclooctanediol gave polycyclic indole products, 3p and 3q, respectively (entries 16, 17). The catalytic method achieves regioselective synthesis of indoles from the direct coupling of arylamines with 1,2-diols without employing any reactive reagents or forming harmful byproducts.
Table 2.
Dehydrative Coupling of Arylamines with 1,2-Diolsa
| entry | amine | diol | product(s) | temp (°C) | yield (%) | |
|---|---|---|---|---|---|---|
|
|
|
||||
| 1 | R = H | R′ = Ph | 3a | 110 | 94 | |
| 2 | R = H | R′ = n-Bu | 3b | 120 | 74 | |
| 3 | R = H | R′ = Me | 3c | 140 | 61 | |
| 4 | R = Me | R′ = Me | 3d | 130 | 60 | |
|
|
|
||||
| 5 | R = n-Bu | 3e | 110 | 80 | ||
| 6 | R = Ph | 3f | 130 | 91 | ||
|
|
|||||
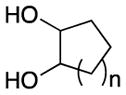
| 7 | n = 1 | 3g | 110 | 62 | ||
| 8 | n = 2 | 3h | 110 | 85 | ||
| 9 | n = 4 | 3i | 140 | 57b | ||
| 10 |
|
|
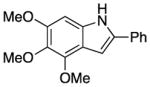 3j |
135 | 94 | |
| 11 |
|
 3k |
140 | 62 | ||
|
|
|
||||
| 12 | R = H | R′ = n-Bu | 3l | 130 | 63 | |
| 13 | R = H | R′ = Ph | 3m | 100 | 74 | |
| 14 | R = Me | R′ = Me | 3n | 130 | 72 | |
| 15 | R = Ph | R′ = Ph | 3o | 130 | 42 | |
| 16 |
|
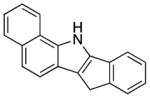 3p |
130 | 63 | ||
| 17 |
|
 3q |
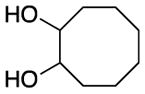
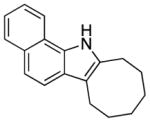
||||
Reaction conditions: amine (1.0 mmol), 1,2-diol (1.5 mol), 2 (0.75 mol %), cyclopentene (3.0 mmol), HBF4·OEt2 (7 mol %), 1,4-dioxane (3 mL), 14–16 h.
The product yield was determined by 1H NMR.
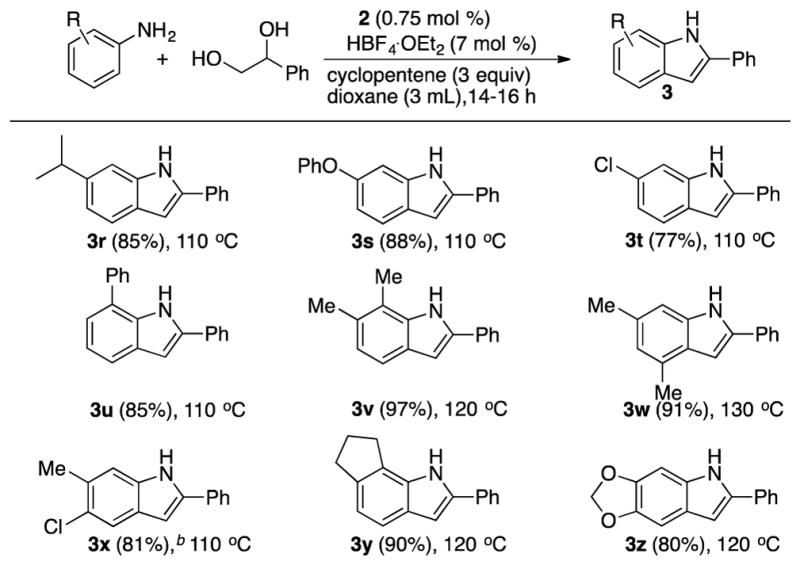
We next explored the coupling of substituted anilines with 1-phenyl-1,2-ethanediol to further illustrate its synthetic versatility (Table 3). Mono- and disubstituted anilines readily reacted with the diol substrate to give the indole products in good to excellent yields. Anilines with electron-releasing group were found to promote the coupling reaction in yielding the indole products 3r and 3s. As indicated in the coupling with 3-chloroaniline, anilines with electron-withdrawing group generally gave lower product yield than ones with electron-donating group. Sterically demanding 2-substituted anilines and 4-aminoindan also gave the corresponding indole products 3u, 3v and 3y, respectively. In all cases, the indole products were formed predictively in a high regioselective fashion.
Table 3.
Dehydrative Coupling of Anilines with 1-Phenyl-1,2-ethanediola
|
Reaction conditions: aniline (1.0 mmol), 1-phenyl-1,2-ethanediol (1.5 mol), 2 (0.75 mol %), HBF4·OEt2 (7 mol %), cyclopentene (3.0 mmol), 1,4-dioxane (3 mL), 14–16 h.
Combined yield of two regioisomers.
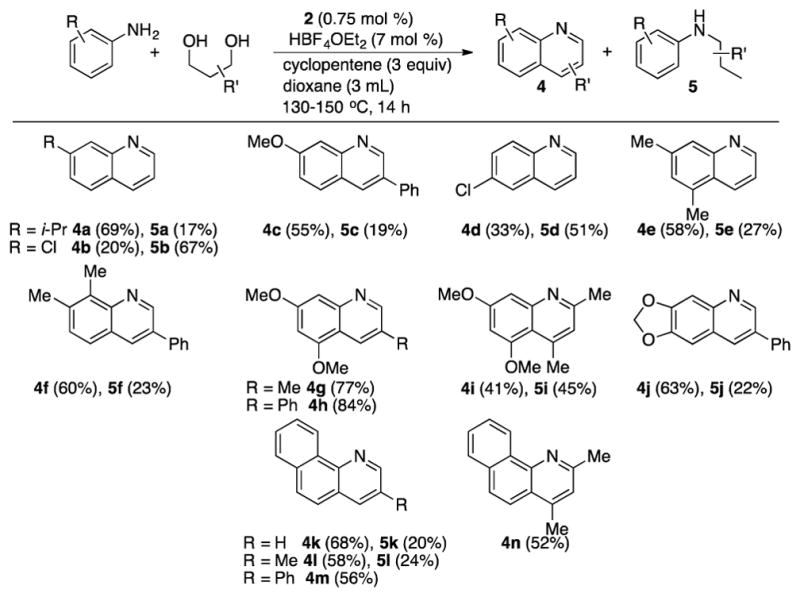
We have been able to extend the scope of catalytic method to the coupling between arylamines with 1,3-diols to produce the quinoline derivatives (Table 4). The coupling reaction of mono- and disubstituted anilines with 1,3-diols directly afforded the quinoline products 4. Generally, a higher temperature was needed to attain an optimal conversion and selectivity in forming the quinoline products (130–150 °C). In most cases, significant amount of the byproducts N-alkylanilines 5 was formed, but both quinoline and N-alkylaniline products 4 and 5 were readily separated by silica gel column chromatography. The byproduct 5 is likely resulted from the dehydrative hydrogenolysis of the alcoholic group over the requisite ortho-C–H coupling and annulation of the diol substrate.
Table 4.
Dehydrative Coupling of Arylamines with 1,3-Diolsa
|
Reaction conditions: amine (1.0 mmol), 1,3-diol (1.5 mol), 2 (0.75 mol %), HBF4·OEt2 (7 mol %), cyclopentene (3.0 mmol), 1,4-dioxane (3 mL), 130–150 °C, 14 h.
The N-alkylaniline 5 was formed as the major product with electron-deficient 3- and 4-chloroanilines, apparently from the preferential alcohol hydrogenolysis over the ortho-C–H coupling path. In contrast, anilines with electron-donating group reacted efficiently with 1,3-diols to form the quinoline products 4g–4j with a trace amount of the byproducts 5. The coupling with secondary 1,3-diols was sluggish, leading to a complex mixture of coupling products, but the coupling of 3,5-dimethoxyaniline with 2,4-pentanediol led to a 1:1 mixture of the quinoline product 4i and the N-alkylated aniline product 5i. The analogous treatment of 1-naphthylamine with 1,3-diols formed the corresponding polycyclic quinoline derivatives 4k–4n.
 |
(2) |
Mechanistic Studies
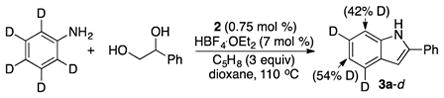
We performed a deuterium labeling experiment to probe the H/D exchange pattern on the coupling products and to discern its mechanistic implications. The treatment of deuterium-labeled C6D5NH2 (0.5 mmol) with 1-phenyl-1,2-ethanediol (0.75 mmol) in the presence of 2 (0.75 mol %), HBF4·OEt2 (7 mol %) and cyclopentene (3 equiv) in 1,4-dioxane was heated at 110 °C for 14 h (eq 2). The deuterium content of the isolated product 3a-d showed a selective deuterium incorporation to both ortho (42% D) and para (54% D) arene positions as analyzed by 1H and 2H NMR (Figure S1, SI). The observed H/D exchange pattern suggests of a rapid and reversible ortho- and para-C–H bond activation process that is mediated by the Ru catalyst. Such rapid and reversible ortho-arene C–H activation process has been commonly observed in chelate assisted C–H coupling reactions.12 We previously observed a similar H/D exchange pattern on the para-arene C–H position from the Ru-catalyzed coupling reaction of arylamines with terminal alkynes, and we attributed the result by invoking an arene C–H metallation process mediated by an electrophilic Ru catalyst.13
 |
(3) |
 |
(4) |
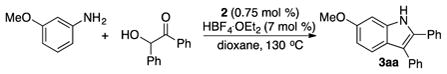
We conducted a series of control experiments to distinguish possible reaction pathways. We reasoned that the observed regioselectivity of the indole products could be explained via an initial dehydrogenation of diol substrate and the formation of an imine intermediate. To test this hypothesis, the reaction of 3-methoxyaniline with benzoin was carried out in the absence of the hydrogen scavenger (cyclopentene) under otherwise similar reaction conditions, which formed the indole product 3aa in 82% yield (eq 3). In support of the initial formation of α-hydroxyketone, 2-hydroxyacetophenone was cleanly formed when 1-phenyl-1,2-ethanediol was treated with 2 and cyclopentene (24% conversion in 3 h). A number of late transition metal catalysts have been found to mediate chemoselective oxidation of secondary alcohols over the primary ones.14
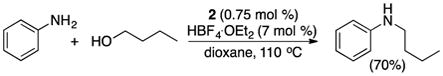
In a separate experiment, the coupling reaction of aniline with n-butanol selectively formed N-butylaniline in 70% yield (eq 4). In this case, N-alkylation product was formed preferentially over the ortho-alkylated aniline product. This result is in sharp contrast to the previously reported coupling reaction of phenol with alcohols, where the ortho-alkylphenol product was exclusively formed.10 These experimental results support a reaction sequence involving the initial dehydrogenation of diol to α-hydroxyketone, and the subsequent ortho-arene C–H activation and annulation processes.
On the basis of these results, we present a plausible mechanistic hypothesis for the coupling reaction of aniline with a 1,2-diol (Scheme 1). As observed in the control experiments, we propose the initial dehydrogenation of diol substrate followed by the dehydrative coupling of aniline with the resulting α-hydroxyketone, which would lead to the formation of a α-hydroxyimine intermediate product 6. The subsequent ortho-arene C–H metallation followed by the dehydrative C–O bond cleavage and the reductive annulation steps would form the indole product 3, in a similar fashion as the dehydrative C–H insertion reactions of phenols.10 The requisite ortho-metallation and dehydration steps would be promoted by an electrophilic Ru-hydroxo species 7. We recently showed that Ru-hydroxo complexes are a key intermediate species in ketone hydrogenolysis reaction.15 Late metal-hydroxo and -phenoxo complexes have also been found to mediate a number of C–O cleavage reactions.16 In addition, the formation of N-alkylated product 5 can be readily explained by invoking a competitive hydrogenolysis pathway especially in case for the coupling of an electron-poor aniline with a 1,3-diol substrate. Our mechanistic hypothesis can not only explain the observed regioselectivity pattern on the indole product 3, but also is supported by both control experiments as well as the literature precedents on the alcohol dehydrogenation reaction.14,17
Scheme 1.
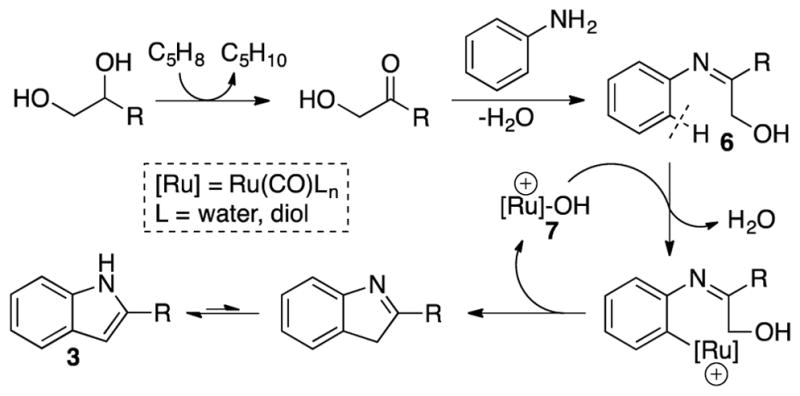
A Plausible Mechanistic Pathway for the Dehydrative Coupling of Aniline with a 1,2-Diol
CONCLUSION
In summary, the cationic ruthenium-hydride complex was found to be an effective catalyst precursor for the dehydrative coupling of anilines with 1,2- and 1,3-diols to form substituted indole and quinoline products. The catalytic method employs readily available arylamine and diol substrates, exhibits high activity toward substituted anilines, and does not require any reactive reagents or generate any toxic byproducts.
EXPERIMENTAL SECTION
General Information
All operations were carried out in a nitrogen-filled glove box or by using standard high vacuum and Schlenk techniques unless otherwise noted. Solvents were freshly distilled over appropriate drying reagents. All organic substrates were received from commercial sources and were used without further purification. The 1H, 2H, 13C and 31P NMR spectra were recorded on a Varian 300 or 400 MHz FT-NMR spectrometer. Mass spectra were recorded from Agilent 6850 GC-MS spectrometer with a HP-5 (5% phenylmethylpolysiloxane) column (30 m, 0.32 mm, 0.25 μm). High resolution mass spectra were obtained at the Mass Spectrometry/ICP Lab, Department of Chemistry and Biochemistry, University of Wisconsin-Milwaukee, Milwaukee, WI. Elemental analyses were performed at the Midwest Microlab, Indianapolis, IN.
General Procedure for the Catalytic Synthesis of Indole and Quinoline Products
In a glove box, complex 2 (13 mg, 0.75 mol %) and HBF4·OEt2 (12 mg, 7 mol %) were dissolved in 1,4-dioxane (1 mL) in a 25 mL Schlenk tube equipped with a Teflon stopcock and a magnetic stirring bar. The resulting mixture was stirred for 5 to 10 min until the solution turned to a pale green color. In an alternative procedure, the complex 1 (17 mg, 3 mol %) and HBF4·OEt2 (12 mg, 7 mol %) were dissolved in 1,4-dioxane (1 mL). An arylamine (1.0 mmol), a diol (1.5 mmol), cyclopentene (204 mg, 3 equiv) and 1,4-dioxane (2 mL) were added to the reaction tube. After the tube was sealed, it was brought out of the glove box, and was stirred in an oil bath set at 110–130 °C (130–150 °C for the quinoline products) for 14 h. The reaction tube was taken out of the oil bath, and was cooled to room temperature. After the tube was open to air, the solution was filtered through a short silica gel column by eluting with CH2Cl2 (10 mL), and the filtrate was analyzed by GC-MS. Analytically pure product was isolated by a simple column chromatography on silica gel (280–400 mesh, hexanes/EtOAc).
Supplementary Material
Acknowledgments
The authors gratefully acknowledge financial support from the National Science of Foundation (CHE-1358439) and National Institute of Health General Medical Sciences (R15 GM109273).
Footnotes
Notes
The authors declare no competing financial interest.
Experimental procedures and NMR spectra of organic products (PDF)
References
- 1.Recent reviews on the catalytic C–H coupling methods directed to the synthesis of nitrogen heterocycles: Yeung CS, Dong VM. Chem Rev. 2011;111:1215–1292. doi: 10.1021/cr100280d.Girard SA, Knauber T, Li CJ. Angew Chem, Int Ed. 2014;53:74–100. doi: 10.1002/anie.201304268.Ye B, Cramer N. Acc Chem Res. 2015;48:1308–1318. doi: 10.1021/acs.accounts.5b00092.
- 2.Selected reviews on the synthesis of indole and related nitrogen heterocycles: Ranu BC. Eur J Org Chem. 2000:2347–2356.Cacchi S, Fabrizi G. Chem Rev. 2005;105:2873–2920. doi: 10.1021/cr040639b.Krüger K, Tillack A, Beller M. Adv Synth Catal. 2008;350:2153–2167.Inman M, Moody CJ. Chem Sci. 2013;4:29–41.
- 3.(a) Zeni G, Larock RC. Chem Rev. 2006;106:4644–4680. doi: 10.1021/cr0683966. [DOI] [PubMed] [Google Scholar]; (b) Shi Z, Zhang C, Li S, Pan D, Ding S, Cui Y, Jiao N. Angew Chem, Int Ed. 2009;48:4572–4576. doi: 10.1002/anie.200901484. [DOI] [PubMed] [Google Scholar]; (c) Shi Z, Glorius F. Angew Chem, Int Ed. 2012;51:9220–9222. doi: 10.1002/anie.201205079. [DOI] [PubMed] [Google Scholar]; (d) Platon M, Amardeil R, Djakovitch L, Hierso JC. Chem Soc Rev. 2012;41:3929–3968. doi: 10.1039/c2cs15350e. [DOI] [PubMed] [Google Scholar]
- 4.(a) Lerchen A, Vásquez-Céspedes S, Glorius F. Angew Chem, Int Ed. 2016;55:3208–3211. doi: 10.1002/anie.201510705. [DOI] [PubMed] [Google Scholar]; (b) Liang Y, Jiao N. Angew Chem, Int Ed. 2016;55:4035–4039. doi: 10.1002/anie.201511002. [DOI] [PubMed] [Google Scholar]; (c) Kong L, Yu S, Zhou X, Li X. Org Lett. 2016;18:588–591. doi: 10.1021/acs.orglett.5b03629. [DOI] [PubMed] [Google Scholar]; (d) Wang H, Moselage M, González MJ, Ackermann L. ACS Catal. 2016;6:2705–2709. [Google Scholar]; (e) Zhang ZZ, Liu B, Xu JW, Yan SY, Shi BF. Org Lett. 2016;18:1776–1779. doi: 10.1021/acs.orglett.6b00494. [DOI] [PubMed] [Google Scholar]
- 5.(a) Tsuji Y, Huh KT, Watanabe Y. Tetrahedron Lett. 1986;27:377–380. [Google Scholar]; (b) Tsuji Y, Huh KT, Watanabe Y. J Org Chem. 1987;52:1673–1680. [Google Scholar]
- 6.(a) Cho CS, Lim HK, Shim SC, Kim TJ, Choi HJ. Chem Commun. 1998:995–996. [Google Scholar]; (b) Cho CS, Oh BH, Kim JS, Kim TJ, Shim SC. Chem Commun. 2000:1885–1886. [Google Scholar]; (c) Cho CS, Kim JS, Oh BH, Kim TJ, Shim SC, Yoon NS. Tetrahedron. 2000;56:7747–7750. [Google Scholar]; (d) Tursky M, Lorentz-Petersen LLR, Olsen LB, Madsen R. Org Biomol Chem. 2010;8:5576–5582. doi: 10.1039/c0ob00106f. [DOI] [PubMed] [Google Scholar]; (e) Monrad RN, Madsen R. Org Biomol Chem. 2011;9:610–615. doi: 10.1039/c0ob00676a. [DOI] [PubMed] [Google Scholar]; (f) Zhang M, Xie F, Wang XT, Yan F, Wang T, Chen M, Ding Y. RSC Adv. 2013;3:6022–6029. [Google Scholar]
- 7.(a) Larock RC, Yum EK. J Am Chem Soc. 1991;113:6689–6690. [Google Scholar]; (b) Larock RC, Yum EK, Refvik MD. J Org Chem. 1998;63:7652–7662. [Google Scholar]; (c) Liu B, Song C, Sun C, Zhou S, Zhu J. J Am Chem Soc. 2013;135:16625–16631. doi: 10.1021/ja408541c. [DOI] [PubMed] [Google Scholar]; (d) Zhou B, Yang Y, Tang H, Du J, Feng H, Li Y. Org Lett. 2014;16:3900–3903. doi: 10.1021/ol501599j. [DOI] [PubMed] [Google Scholar]
- 8.Aramoto H, Obora Y, Ishii Y. J Org Chem. 2009;74:628–633. doi: 10.1021/jo801966u. [DOI] [PubMed] [Google Scholar]
- 9.(a) Würtz S, Rakshit S, Neumann JJ, Dröge T, Glorius F. Angew Chem, Int Ed. 2008;47:7230–7233. doi: 10.1002/anie.200802482. [DOI] [PubMed] [Google Scholar]; (b) Wei Y, Deb I, Yoshikai N. J Am Chem Soc. 2012;134:9098–9101. doi: 10.1021/ja3030824. [DOI] [PubMed] [Google Scholar]; (c) Huang F, Wu P, Wang L, Chen J, Sun C, Yu Z. J Org Chem. 2014;79:10553–10560. doi: 10.1021/jo5014542. [DOI] [PubMed] [Google Scholar]
- 10.Lee DH, Kwon KH, Yi CS. J Am Chem Soc. 2012;134:7325–7328. doi: 10.1021/ja302710v. [DOI] [PubMed] [Google Scholar]
- 11.Yi CS, Zeczycki TN, Guzei IA. Organometallics. 2006;25:1047–1051. doi: 10.1021/om0510674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Kakiuchi F, Murai S. Acc Chem Res. 2002;35:826–834. doi: 10.1021/ar960318p. [DOI] [PubMed] [Google Scholar]; (b) Albrecht M. Chem Rev. 2010;110:576–623. doi: 10.1021/cr900279a. [DOI] [PubMed] [Google Scholar]
- 13.Yi CS, Yun SY. J Am Chem Soc. 2005;127:17000–17006. doi: 10.1021/ja055608s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Chung K, Banik SM, De Crisci AG, Pearson DM, Blake TR, Olsson JV, Ingram AJ, Zare RN, Waymouth RM. J Am Chem Soc. 2013;135:7593–7602. doi: 10.1021/ja4008694. [DOI] [PubMed] [Google Scholar]; (b) Manzini S, Urbina-Blanco CA, Nolan SP. Organometallics. 2013;32:660–664. [Google Scholar]; (c) Tseng KNT, Kampf JW, Szymczak NK. Organometallics. 2013;32:2046–2049. [Google Scholar]; (d) Chakraborty S, Lagaditis PO, Förster M, Bielinski EA, Hazari N, Holthausen MC, Jones WD, Schneider S. ACS Catal. 2014;4:3994–4003. [Google Scholar]
- 15.Kalutharage N, Yi CS. J Am Chem Soc. 2015;137:11105–11114. doi: 10.1021/jacs.5b06097. [DOI] [PubMed] [Google Scholar]
- 16.(a) Arnold PL, Scarisbrick AC. Organometallics. 2004;23:2519–2521. [Google Scholar]; (b) Baratta W, Chelucci G, Gladiali S, Siega K, Toniutti M, Zanette M, Zangrando E, Rigo P. Angew Chem, Int Ed. 2005;44:6214–6219. doi: 10.1002/anie.200502118. [DOI] [PubMed] [Google Scholar]; (c) Hamilton RJ, Bergens SH. J Am Chem Soc. 2008;130:11979–11987. doi: 10.1021/ja8034812. [DOI] [PubMed] [Google Scholar]; (d) Takebayashi S, Dabral N, Miskolzie M, Bergens SH. J Am Chem Soc. 2011;133:9666–9669. doi: 10.1021/ja202732q. [DOI] [PubMed] [Google Scholar]
- 17.(a) de Graauw CF, Peters JA, van Bekkum H, Huskens J. Synthesis. 1994:1007–1017. [Google Scholar]; (b) Ito H, Noyori RJ. J Am Chem Soc. 2000;122:1466–1478. [Google Scholar]; (c) Casey CP, Singer SW, Powell DR, Hayashi RK, Kavana M. J Am Chem Soc. 2001;123:1090–1100. doi: 10.1021/ja002177z. [DOI] [PubMed] [Google Scholar]; (d) Johnson JB, Bäckvall JE. J Org Chem. 2003;68:7681–7684. doi: 10.1021/jo034634a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.